
Paramagnetic Solid-State NMR Study of Solid Solutions of Cobaltocene with Ferrocene and Nickelocene


Abstract
1. Introduction
2. Results and Discussion
2.1. Ferrocene/Cobaltocene Mixtures in Solution
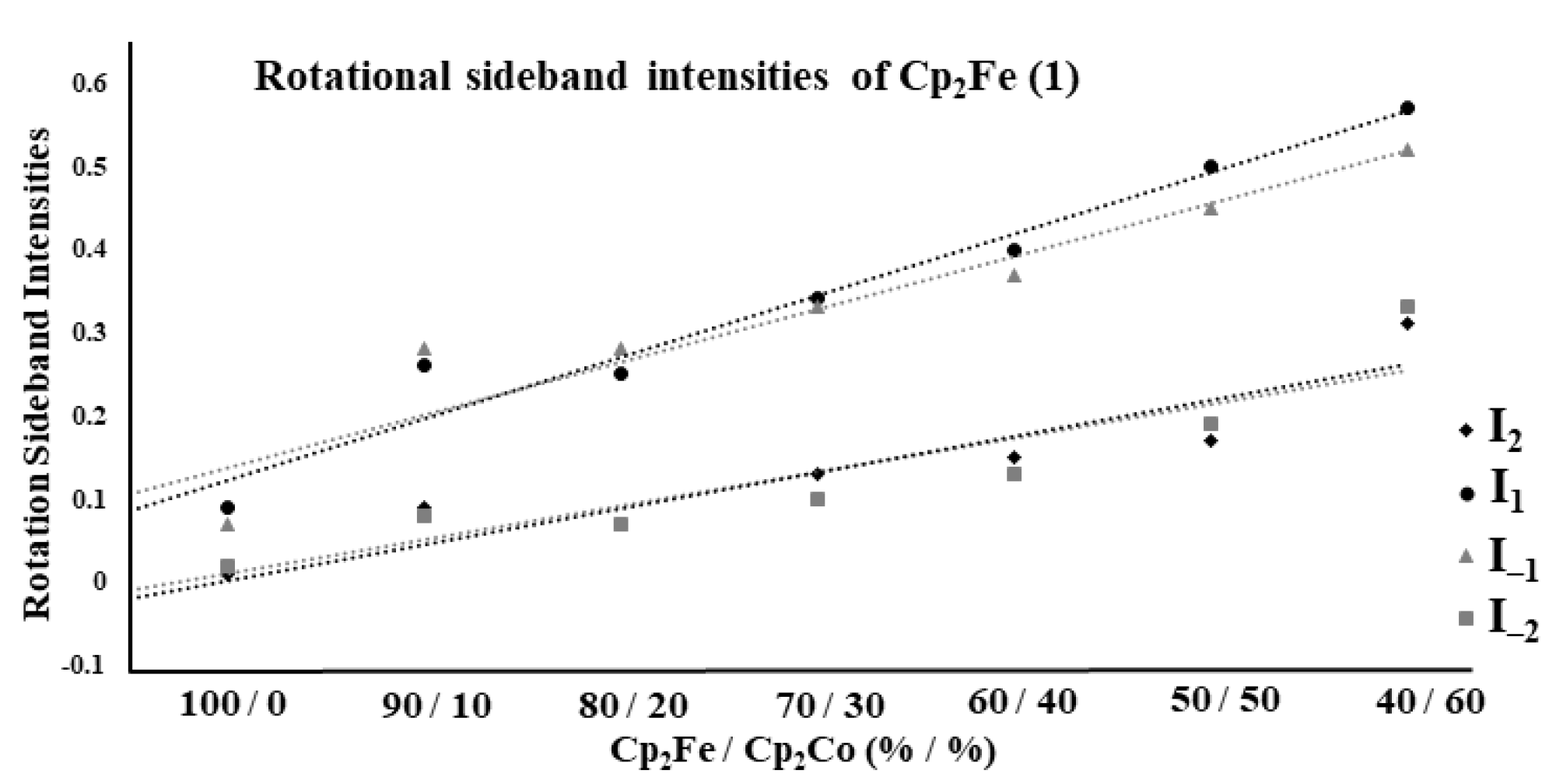
2.2. Ferrocene/Cobaltocene Solid Solutions
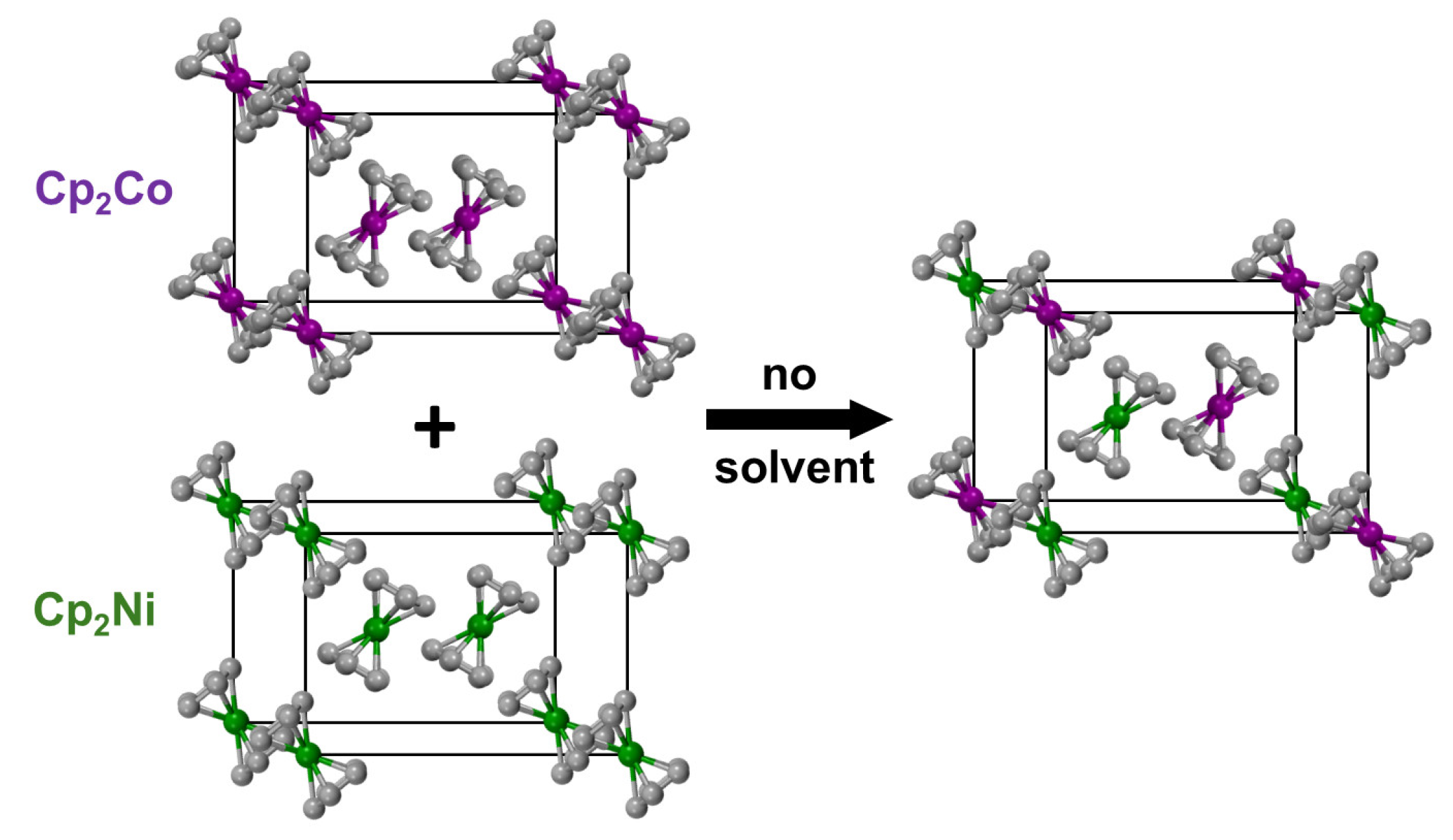
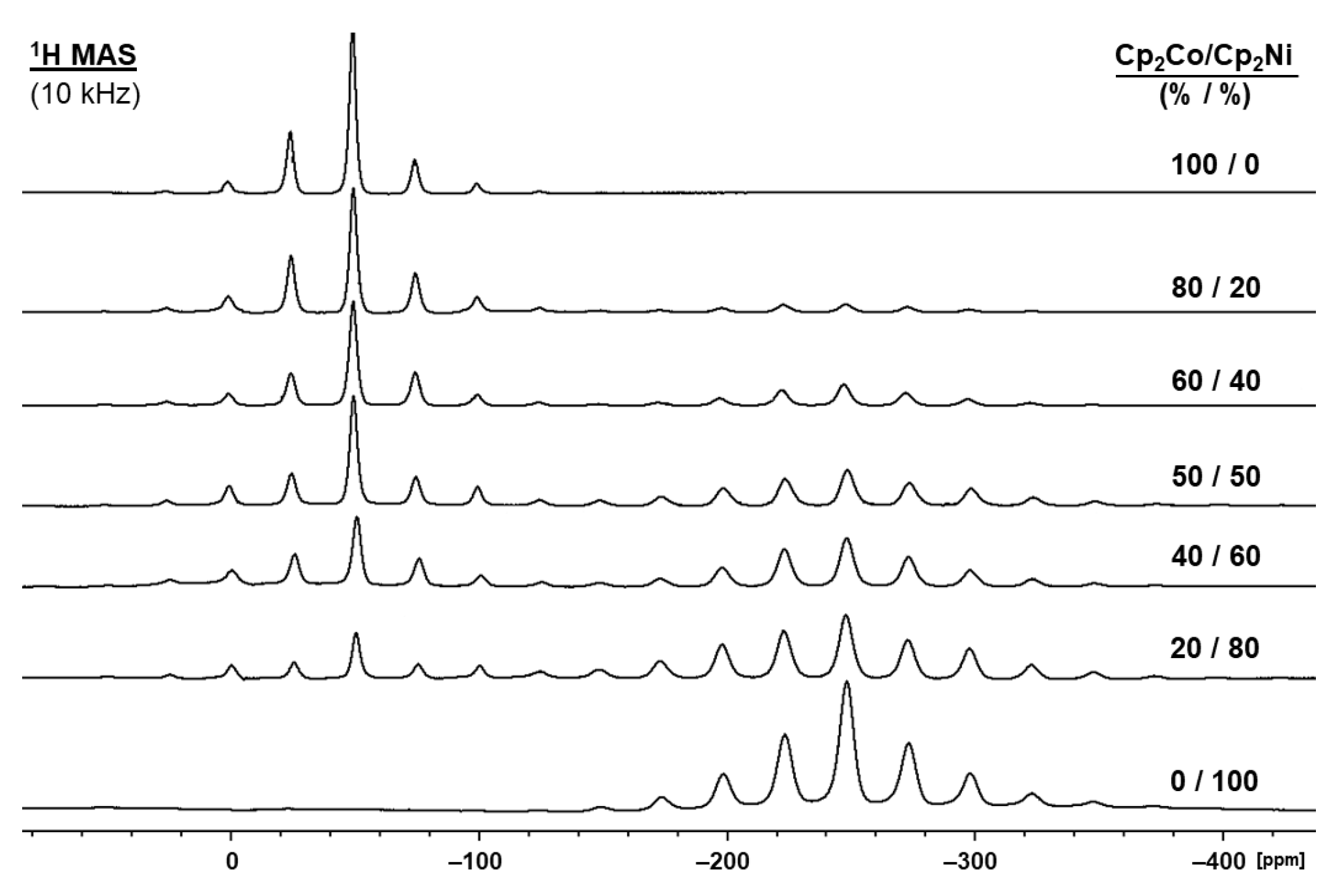
2.3. Nickelocene/Cobaltocene Solid Solutions
3. Conclusions
4. Experimental Section
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Callister, W.D., Jr. Materials Science and Engineering: An Introduction, 7th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 0-471-35446-5. [Google Scholar]
- Ter Horst, J.H.; Deij, M.A.; Cains, P.W. Discovering New Co-Crystals. Cryst. Growth Des. 2009, 9, 1531–1537. [Google Scholar] [CrossRef]
- Tilborg, A.; Norberg, B.; Wouters, J. Pharmaceutical salts and cocrystals involving amino acids: A brief structural overview of the state-of-art. Eur. J. Med. Chem. 2014, 74, 411–426. [Google Scholar] [CrossRef]
- Blagden, N.; Berry, D.J.; Parkin, A.; Javed, H.; Ibrahim, A.; Gavan, P.T.; De Matos, L.L.; Seaton, C.C. Current directions in co-crystal growth. New J. Chem. 2008, 32, 1659–1672. [Google Scholar] [CrossRef]
- Seiler, P.; Dunitz, J.D. A New Interpretation of the Disordered Crystal Structure of Ferrocene. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1979, 35, 1068–1074. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Takamizawa, S. Deformation twinning of ferrocene crystals assisted by the rotational mobility of cyclopentadienyl rings. Dalton Trans. 2015, 44, 5688–5691. [Google Scholar] [CrossRef]
- Bünder, W.; Weiss, E. Verfeinerung der Kristallstruktur von Dicyclopentadienylkobalt, (η5-C5H5)2Co. J. Organomet. Chem. 1975, 92, 65–68. [Google Scholar] [CrossRef]
- Antipin, M.Y.; Boese, R.; Auga, N.; Schmid, G. Redetermination of the cobaltocene crystal structure at 100 K and 297 K: Comparison with ferrocene and nickelocene. Struct. Chem. 1993, 4, 91–101. [Google Scholar] [CrossRef]
- Ding, K.; Cullen, D.A.; Zhang, L.; Cao, Z.; Roy, A.D.; Ivanov, I.N.; Cao, D. A general synthesis approach for supported bimetallic nanoparticles via surface inorganometallic chemistry. Science 2018, 362, 560–564. [Google Scholar] [CrossRef]
- Harmon-Welch, G.E.; Hoefler, J.C.; Trujillo, M.R.; Bhuvanesh, N.; Bakhmutov, V.I.; Blümel, J. Creating Solid Solutions of Metallocenes: Migration of Nickelocene into the Ferrocene Crystal Lattice in the Absence of a Solvent. J. Phys. Chem. C 2023, 127, 3059–3066. [Google Scholar]
- Benzie, J.W.; Harmon-Welch, G.E.; Hoefler, J.C.; Bakhmutov, V.I.; Blümel, J. Molecular Dynamics and Surface Interactions of Nickelocene Adsorbed on Silica: A Paramagnetic Solid-State NMR Study. Langmuir 2022, 38, 7422–7432. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Limbach, H.-H. Solid State NMR for Nonexperts: An Overview of Simple but General Practical Methods. Solids 2021, 2, 139–154. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K.; Spiess, H.W. Multidimensional Solid-State NMR and Polymers; Academic Press: London, UK, 1994. [Google Scholar]
- Samudrala, K.K.; Huynh, W.; Dorn, R.W.; Rossini, A.J.; Conley, M.P. Formation of a Strong Heterogeneous Aluminum Lewis Acid on Silica. Angew. Chem. Int. Ed. 2022, 61, e202205745. [Google Scholar] [CrossRef]
- Duncan, T.M. A Compilation of Chemical Shift Anisotropies; Farragut Press: Chicago, IL, USA, 1990. [Google Scholar]
- Pell, A.J.; Pintacuda, G.; Grey, C.P. Paramagnetic NMR in solution and the solid state. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 111, 1–271. [Google Scholar] [CrossRef] [PubMed]
- Bakhmutov, V.I. Strategies for solid-state NMR studies of materials: From diamagnetic to paramagnetic porous solids. Chem. Rev. 2011, 111, 530–562. [Google Scholar] [CrossRef]
- Fischer, R.A.; Nlate, S.; Hoffmann, H.; Herdtweck, E.; Blümel, J. Two Types of Intramolecular Lewis-Base Adducts with the [2-(Dimethyl-amino)ethyl]cyclopentadienyl Ligand: Synthesis and Crystal Structures of {η5:η1-C5H4[(CH2)2NMe2]}Ni-I and {η5-μ-C5H4[(CH2)2NMe2]}(Me3P)Ni-InI2. Organometallics 1996, 15, 5746–5752. [Google Scholar] [CrossRef]
- Blümel, J.; Hofmann, P.; Köhler, F.H. NMR Spectroscopy of Paramagnetic Complexes. Magn. Reson. Chem. 1993, 31, 2–6. [Google Scholar] [CrossRef]
- Blümel, J.; Hiller, W.; Herker, M.; Köhler, F.H. Solid-State Paramagnetic NMR Spectroscopy of Chromocenes. Organometallics 1996, 15, 3474–3476. [Google Scholar] [CrossRef]
- Heise, H.; Köhler, F.H.; Xie, X. Solid-state NMR spectroscopy of paramagnetic metallocenes. J. Magn. Reson. 2001, 150, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Schnellbach, M.; Blümel, J.; Köhler, F.H. The Union Carbide catalyst (Cp2Cr + SiO2), studied by solid-state NMR. J. Organomet. Chem. 1996, 520, 227–230. [Google Scholar] [CrossRef]
- Cluff, K.J.; Schnellbach, M.; Hilliard, C.R.; Blümel, J. The adsorption of chromocene and ferrocene on silica: A solid-state NMR study. J. Organomet. Chem. 2013, 744, 119–124. [Google Scholar] [CrossRef]
- Cluff, K.J.; Blümel, J. Adsorption of Metallocenes on Silica. Chem. Eur. J. 2016, 22, 16562–16575. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Autschbach, J. Temperature dependence of contact and dipolar NMR chemical shifts in paramagnetic molecules. J. Chem. Phys. 2015, 142, 054108. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 86th ed.; CRC Press: Boca Raton, FL, USA, 2005; p. 3257. ISBN 0-8493-0486-5. [Google Scholar]
- Reinhard, S.; Blümel, J. 31P CP/MAS NMR of polycrystalline and immobilized phosphines and catalysts with fast sample spinning. Magn. Reson. Chem. 2003, 41, 406–416. [Google Scholar] [CrossRef]
- Rettig, M.F.; Drago, R.S. Electron delocalization in paramagnetic metallocenes. I. Nuclear magnetic resonance contact shifts. J. Am. Chem. Soc. 1969, 91, 1361–1370. [Google Scholar] [CrossRef]
- Rajasekharan, M.V.; Bucher, R.; Deiss, E.; Zoller, L.; Salzer, A.K.; Moser, E.; Weber, J.; Ammeter, J.H. ESR Study of the Electronic Structure and Dynamic Jahn-Teller Effect in Nickelocenium Cation. J. Am. Chem. Soc. 1983, 105, 7516–7522. [Google Scholar] [CrossRef]
- Prins, R.; Reinders, F.J. Electron Spin Resonance of the Cation of Ferrocene. J. Am. Chem. Soc. 1969, 91, 4929–4931. [Google Scholar] [CrossRef]
- Guan, X.; Stark, R.E. A general protocol for temperature calibration of MAS NMR probes at arbitrary spinning speeds. Solid State NMR 2010, 38, 74–76. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molar Ratio Cp2Fe/Cp2Co (%/%) | δ(1H) (ppm) | |
|---|---|---|
| Cp2Fe | Cp2Co | |
| 0/100 | – | −50.51 |
| 10/90 | 3.68 | −50.72 |
| 20/80 | 3.70 | −50.77 |
| 30/70 | 3.73 | −50.77 |
| 40/60 | 3.82 | −50.80 |
| 50/50 | 3.84 | −50.90 |
| 60/40 | 3.90 | −50.92 |
| 70/30 | 3.86 | −50.90 |
| 80/20 | 3.90 | −50.97 |
| 90/10 | 4.11 | −50.85 |
| 100/0 | 4.22 | – |
| Molar Ratio Cp2Fe/Cp2Co (%/%) | Δν1/2 (kHz) | |
|---|---|---|
| Cp2Fe | Cp2Co | |
| 0/100 | – | 1.18 |
| 10/90 | 1.85 | 1.30 |
| 20/80 | 1.25 | 1.16 |
| 30/70 | 1.03 | 1.25 |
| 40/60 | 0.92 | 1.21 |
| 50/50 | 0.91 | 1.20 |
| 60/40 | 0.90 | 1.23 |
| 70/30 | 0.86 | 1.26 |
| 80/20 | 0.81 | 1.22 |
| 90/10 | 0.81 | 1.21 |
| 100/0 | 0.81 | – |
| Molar Ratio Cp2Fe/Cp2Co (%/%) | 1H T1 (β) (ms) | |
|---|---|---|
| Cp2Fe | Cp2Co | |
| 0/100 | – | 1.3 (0.46) |
| 20/80 | 4.2 (0.36) | 2.0 (0.85) |
| 30/70 | 5.9 (0.20) | 2.4 (0.54) |
| 40/60 | 7.3 (0.46) | 2.0 (0.50) |
| 50/50 | 8.4 (0.40) | 2.5 (0.55) |
| 60/40 | 14 (0.62) | 2.3 (0.63) |
| 70/30 | 23 (0.69) | 2.1 (0.47) |
| 80/20 | 31 (0.73) | 2.4 (0.52) |
| 100/0 | 14,000 | – |
| Molar Ratio Cp2Co/Cp2Ni (%/%) | δ(1H) (ppm) | |
|---|---|---|
| Cp2Co | Cp2Ni | |
| 100/0 | −50.51 | – |
| 90/10 | −50.78 | −245.9 |
| 80/20 | −50.73 | −245.1 |
| 70/30 | −50.82 | −245.4 |
| 60/40 | −50.85 | −245.8 |
| 50/50 | −50.92 | −245.9 |
| 40/60 | −50.87 | −245.8 |
| 30/70 | −51.07 | −245.4 |
| 20/80 | −51.86 | −245.8 |
| 10/90 | −51.88 | −245.9 |
| 0/100 | – | −245.4 |
| Molar Ratio Cp2Co/Cp2Ni (%/%) | Δν1/2 (kHz) | |
|---|---|---|
| Cp2Co | Cp2Ni | |
| 100/0 | 1.38 | – |
| 90/10 | 1.39 | 2.57 |
| 80/20 | 1.37 | 2.49 |
| 70/30 | 1.39 | 2.46 |
| 60/40 | 1.45 | 2.42 |
| 50/50 | 1.39 | 2.41 |
| 40/60 | 1.36 | 2.55 |
| 30/70 | 1.40 | 2.69 |
| 20/80 | 1.39 | 2.54 |
| 10/90 | 1.56 | 2.59 |
| 0/100 | – | 2.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harmon-Welch, G.E.; Bakhmutov, V.I.; Blümel, J. Paramagnetic Solid-State NMR Study of Solid Solutions of Cobaltocene with Ferrocene and Nickelocene. Magnetochemistry 2024, 10, 58. https://doi.org/10.3390/magnetochemistry10080058
Harmon-Welch GE, Bakhmutov VI, Blümel J. Paramagnetic Solid-State NMR Study of Solid Solutions of Cobaltocene with Ferrocene and Nickelocene. Magnetochemistry. 2024; 10(8):58. https://doi.org/10.3390/magnetochemistry10080058
Chicago/Turabian StyleHarmon-Welch, Gabrielle E., Vladimir I. Bakhmutov, and Janet Blümel. 2024. "Paramagnetic Solid-State NMR Study of Solid Solutions of Cobaltocene with Ferrocene and Nickelocene" Magnetochemistry 10, no. 8: 58. https://doi.org/10.3390/magnetochemistry10080058
APA StyleHarmon-Welch, G. E., Bakhmutov, V. I., & Blümel, J. (2024). Paramagnetic Solid-State NMR Study of Solid Solutions of Cobaltocene with Ferrocene and Nickelocene. Magnetochemistry, 10(8), 58. https://doi.org/10.3390/magnetochemistry10080058