2.1. Crystalline Phase Calculations
Experimentally, it has been observed that the spin transition of compound
3 is significantly different depending on whether the material is freshly prepared or dried after a cooling-heating cycle. In the first case, MeNO
2 molecules are present in the crystalline phase (
i.e.,
3[BF
4]
2·2MeNO
2), the transition is reproducible and occurs at around 320 K. After a cooling-heating cycle, the solvent molecules evaporate and the dried sample (
i.e.,
3[BF
4]
2) undergoes a transition centered at 266 K with a small hysteresis loop of
ca. 20 K. In order to investigate the effect of the solvent on the SCO transition of
3[BF
4]
2, we have performed solid state calculations of the HS and LS phases with and without MeNO
2 in the crystalline cell. It is reported in the experimental paper that the crystal of
3[BF
4]
2 is not isostructural to
3[BF
4]
2·2MeNO
2, but that it retains the terpyridine embrace structure [
27]. Given the absence of
X-ray data for
3[BF
4]
2, we have simply assumed that the MeNO
2 molecules evaporate without major changes in the crystal. Of course, this implies that those calculations do not exactly compare with the experimental measurements of the solvent-free phase but, indeed, they allow for a more direct comparison of the effect of the solvent in the energetics of
3[BF
4]
2. Therefore, the unit cells evaluated in this section contain four SCO molecules and eight BF
4− counterions for
3[BF
4]
2 and also eight MeNO
2 molecules for [BF
4]
2·2MeNO
2. Those unit cells have undergone a variable-cell optimization procedure to achieve the different HS and LS minima. Any major changes are observed in the optimized structures with respect to the starting ones, and the relative energy differences (
) are collected in
Table 1. First, one must notice that
is not comparable to the experimental
value, since the latter includes the vibrational contribution to enthalpy (
) that is not evaluated here. That said, the computed
is 28.5 kJ/mol for the dried sample
3[BF
4]
2 and 38.0 kJ/mol for the
3[BF
4]
2·2MeNO
2. This increase in
is in agreement with the fact that the solvent-containing sample undergoes the transition at higher temperatures and, thus, indicates that the presence of solvent has an effect of
ca. 10 kJ/mol on
(
i.e., 38.0–28.5 kJ/mol, see
Table 1).
Table 1.
Adiabatic energy gaps associated to the spin transition of compound 3 obtained in the crystalline phase with and without solvent. Contribution of the molecular geometries of the Spin Crossover (SCO) units to, obtained from calculations on isolated molecules. Contribution of the intermolecular interactions between SCO, counterion and solvent molecules to (). Negative (Positive) values for those contributions indicate that the High Spin (Low Spin) state is favored. Note that in the calculation of, only SCO are included in the calculation, even if their minima are computed in unit cells that include solvent molecules and counterions. Energies are given in kJ/mol.
Table 1.
Adiabatic energy gaps associated to the spin transition of compound 3 obtained in the crystalline phase with and without solvent. Contribution of the molecular geometries of the Spin Crossover (SCO) units to, obtained from calculations on isolated molecules. Contribution of the intermolecular interactions between SCO, counterion and solvent molecules to (). Negative (Positive) values for those contributions indicate that the High Spin (Low Spin) state is favored. Note that in the calculation of, only SCO are included in the calculation, even if their minima are computed in unit cells that include solvent molecules and counterions. Energies are given in kJ/mol.
| Unit cell | | | |
|---|
| 3[BF4]2 | 28.5 | 15.5 | 13.0 |
| 3[BF4]2·2MeNO2 | 38.0 | 20.5 | 17.5 |
At this point, one must notice that the stability of the different unit cells depends on (i) the geometry of the constituents and on (ii) the intermolecular interactions that arise between them. In order to evaluate both effects, we have first performed another set of calculations in which we have calculated the energy of the isolated SCO molecules in its solid state minima, but without any counterion or solvent molecules in the unit cell and, thus, being completely isolated (obtaining
). Notice that the
values account for all the effects based on the spin transiting units (such as their internal geometry) and excludes the influence of any sort of intermolecular interactions between SCO, counterions (CI) and solvent molecules. Those calculations predict quite different adiabatic energy gaps between the crystal and the isolated molecules for both the
3[BF
4]
2 and
3[BF
4]
2·2MeNO
2 minima (compare
and
, in
Table 1), which indicates that the molecular geometries are only partially responsible for the
ΔH values. The main structural parameters are given in
Table 2 (for unit cell parameters see
Table S1). In addition, by looking at the absolute energy of the isolated molecules in the
3[BF
4]
2 and
3[BF
4]
2·2MeNO
2 minima, one can see that the HS molecules are destabilized (have less absolute energy than in
3[BF
4]
2) in the presence of solvent, whereas the opposite is seen for the LS molecules. This explains the 5 kJ/mol difference between 15.5 and 20.5 kJ/mol. In the next section we analyze the effect that the ligand distortions have on the SCO behavior of those compounds.
Table 2.
Absolute energies (in Ry) and geometries of isolated molecules of compound
3 in the LS and HS minima obtained from the crystalline phase with (
3[BF
4]
2·2MeNO
2) and without (
3[BF
4]
2) solvent. It is shown the mean Fe-N distances (in Å), and the two angles that define the distortion of the ligands [
28]. Given that four non-identical SCO molecules are present in each unit cell, all shown values correspond to an average.
Table 2.
Absolute energies (in Ry) and geometries of isolated molecules of compound 3 in the LS and HS minima obtained from the crystalline phase with (3[BF4]2·2MeNO2) and without (3[BF4]2) solvent. It is shown the mean Fe-N distances (in Å), and the two angles that define the distortion of the ligands [28]. Given that four non-identical SCO molecules are present in each unit cell, all shown values correspond to an average.
| Minima | Absolute Energy | d(Fe–N) | φ (°) | θ (°) |
|---|
| 3HS [BF4]2 | −914.29032 | 2.17 | 176.20 | 82.71 |
| 3HS [BF4]2·2MeNO2 | −914.28873 | 2.16 | 168.47 | 81.55 |
| 3LS [BF4]2 | −914.30215 | 1.94 | 178.37 | 89.17 |
| 3LS [BF4]2·2MeNO2 | −914.30440 | 1.93 | 177.08 | 88.35 |
Apart from the effect of the solvent in the geometries, the calculated
values also depend on the amount of intermolecular interactions that are established in the LS and HS minima. The magnitude and sign of this effect corresponds to the difference between
and
, and has been also collected in
Table 1 as
. Those values mean that the intermolecular interactions are 13.0 and 17.5 kJ/mol more attractive in the LS than in the HS state for the dried and solvated crystals, respectively. In other words, the solvent molecules give rise to larger amount of intermolecular interactions in the LS crystals, effectively over-stabilizing them. This is probably due to a more-densely packed crystal structure as a result of the compression of the FeN
6 core of the SCO units. Notice that these intermolecular interactions can be originated between the different constituents of the unit cell, that is, between the SCO, the solvent and the CI molecules. We have attempted to obtain an approximate quantification of each of those interactions separately, but it has not been possible due to the difficulty to define appropriate cluster models. So, in summary, the presence of solvent molecules effectively contributes to the stabilization of the LS phase for two reasons: (1) a distortion of the molecular structures from their minima, which penalizes (stabilizes) the HS (LS) molecules (
ca. 5 kJ/mol), and (2) the creation of larger intermolecular interactions in the LT phase (
ca. 4.5 kJ/mol).
2.2. Gas Phase Calculations
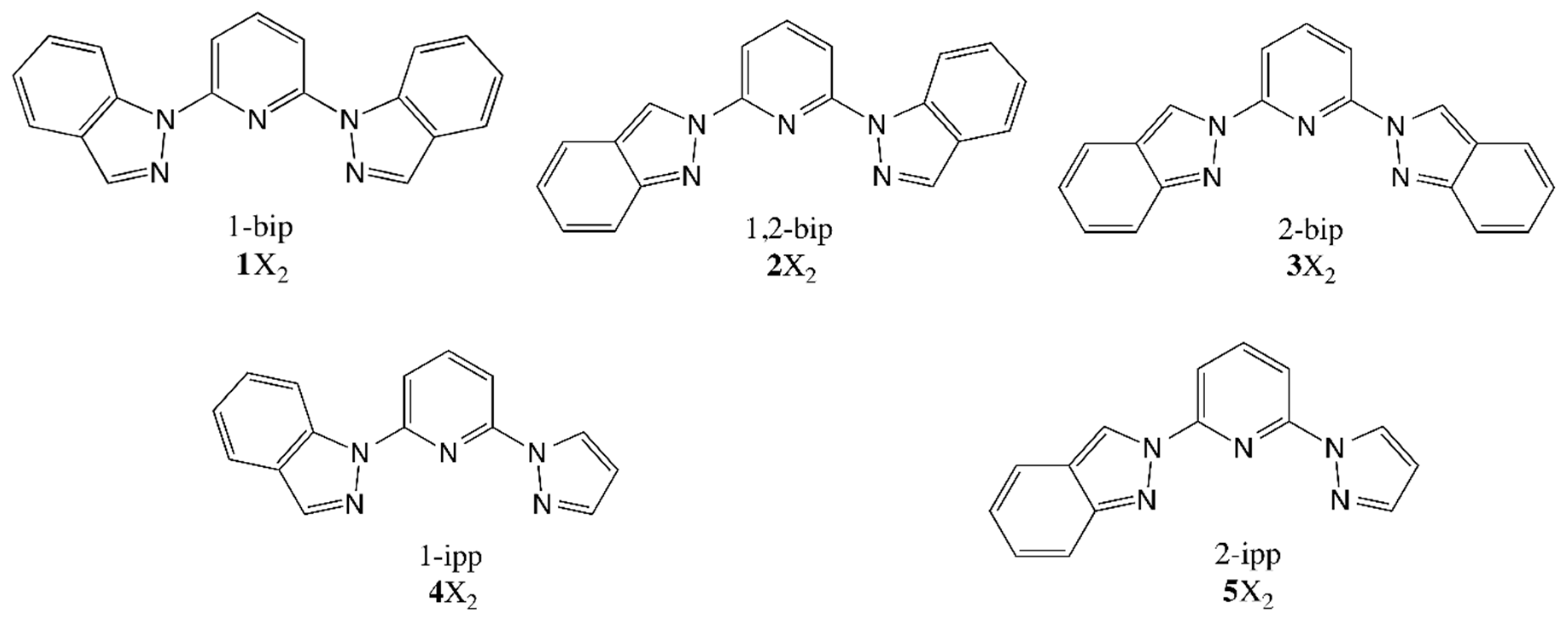
In addition to compound
3, other ligands were investigated in solvent conditions when forming a complex with iron(II) [
27]. Compounds
2,
3,
4 and
5 have different ligands based on a central pyridine and two lateral aromatic groups attached to it (see
Scheme 1). In the case of the ligands employed for
2 (1,2-bip) and
3 (2-bip), those groups are indazol-1-yl and indazol-2-yl, respectively. In turn, the ones used for
4 (1-ipp) and
5 (2-ipp) hold one pyrazol-1-yl moiety and either an indazol-1-yl (
4) or an indazol-2-yl (
5) moiety. Therefore, ligands
2 and
3 (and also
4 and
5) have the same chemical formula and, thus, can be easily compared. As we will show in the following paragraphs, the structural difference between the ligands 1,2-bip and 2-bip is responsible for a notable change in the SCO properties of
2 and
3 and, interestingly, this effect is also experienced by compounds
4 and
5 as a result of a change in their constituent 1-ipp and 2-ipp ligands, respectively.
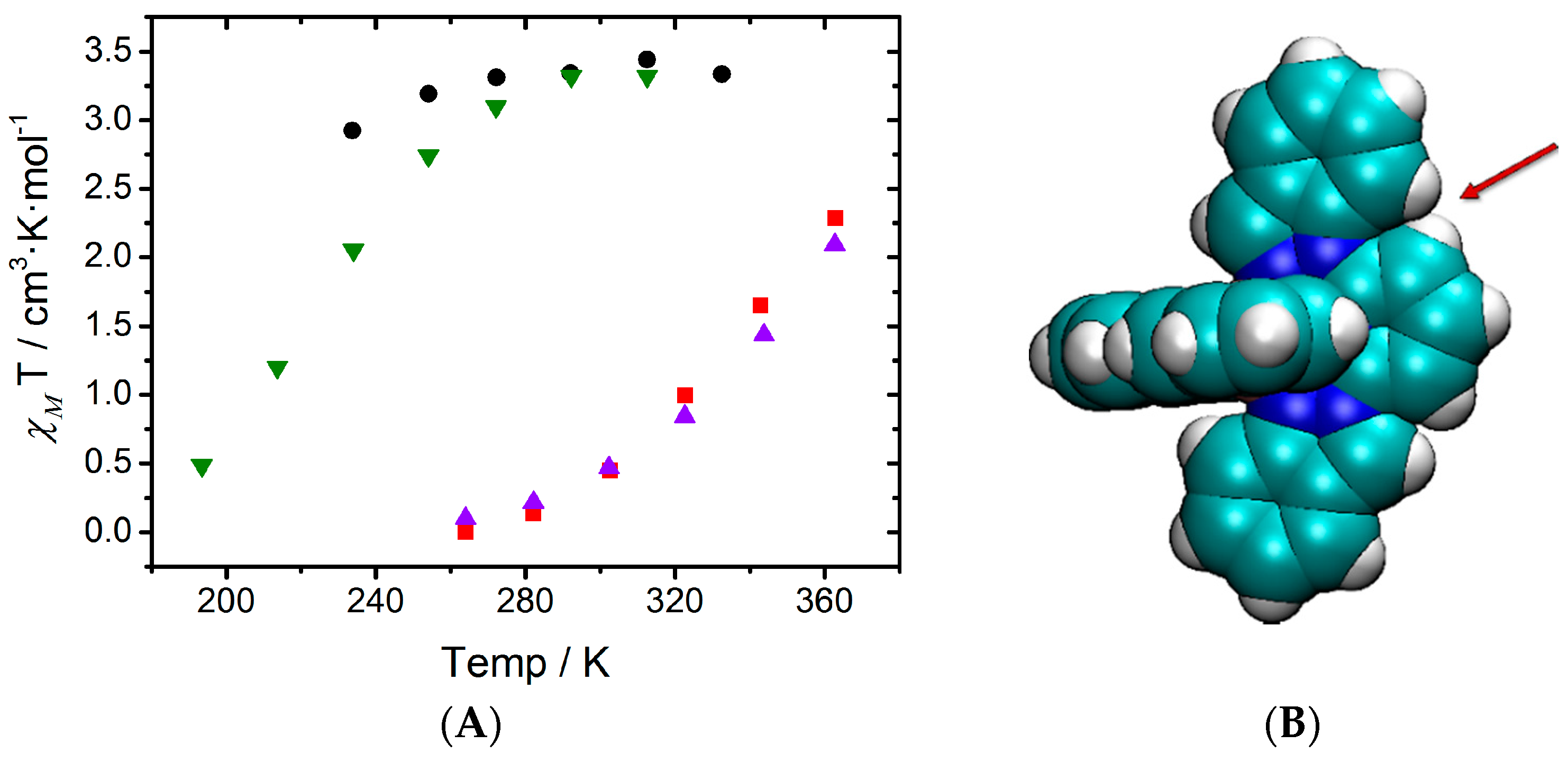
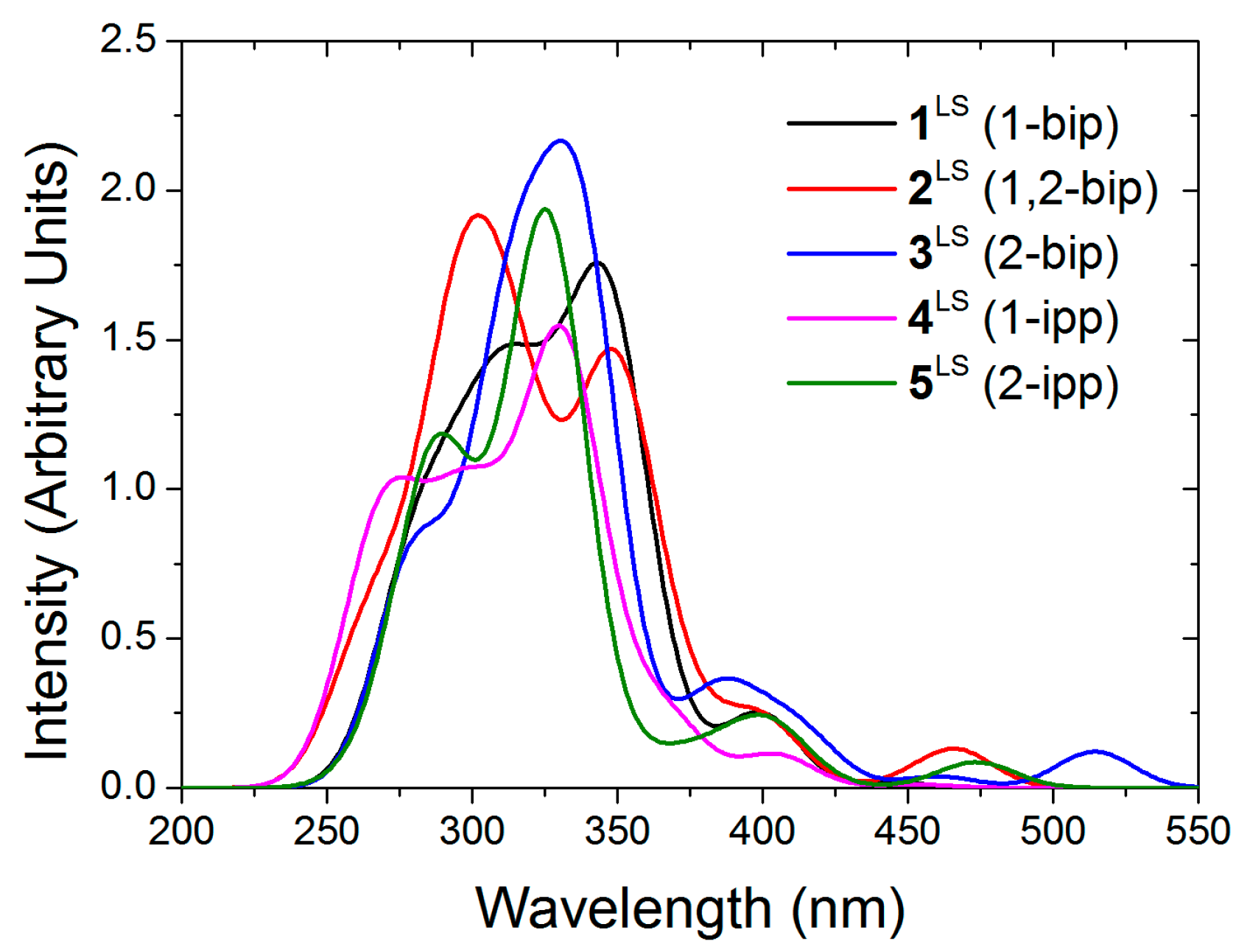
It has been experimentally observed that solutions of
2 and
4 at room temperature are mostly in the LS state, whereas
3 and
5 are fully HS (see
Figure 1A). These results were ascribed to conformational rigidity, imposed as a consequence of intra-ligand H···H contacts (see
Figure 1) involving the central pyridine and the indazol-1-yl group, the latter present only in
2 and
4. According to this explanation, the expansion of the FeN
6 first-coordination sphere as a result of the LS-to-HS transition would imply a distortion of the ligand, that would penalize
2HS and
4HS, but not
3HS and
5HS, since the aforementioned H···H contact would not be relevant when indazol-2-yl groups are present, such as in those compounds. Note that this argument implies that
2LS and
4LS do not suffer from an energy penalty, and that a double penalty would be present in
2 than in
4, since the former has two indazol-1-yl groups while the latter has only one. In the following lines we will demonstrate that this is, indeed, not the case, and that the different LS/HS relative stability in
2 and
4 with respect to
3 and
5 arises from the inter-ligand interactions that penalize the stability of
3LS and
5LS FeN
6 first-coordination sphere. In order to investigate this issue, we have first computed the adiabatic energy difference (
) associated to the four compounds both in gas phase and in their solvent, simulated using a self-consistent continuum solvation (SCCS) approach (see
Table 3). It is observed that
is larger for
2 and
4 than for
3 and
5, in agreement with experiment, both in gas phase and in solution. Notably interesting is that in all cases the LS state is largely stabilized by the solvent conditions, whose general effect on
is not negligible (8 kJ·mol
−1 in average, see
Table 3).
Figure 1.
(adapted from [
27]): (
A) Solution-phase magnetic susceptibility data of
2[ClO
4]
2 in CD
3NO
2 (red),
3[ClO
4]
2 in CD
3CN (black),
4[ClO
4]
2 in CD
3NO
2 (violet) and
5[ClO
4]
2 in (CD
3)
2CO (green); (
B) view of compound
4 highlighting the intra-ligand H···H contact.
Figure 1.
(adapted from [
27]): (
A) Solution-phase magnetic susceptibility data of
2[ClO
4]
2 in CD
3NO
2 (red),
3[ClO
4]
2 in CD
3CN (black),
4[ClO
4]
2 in CD
3NO
2 (violet) and
5[ClO
4]
2 in (CD
3)
2CO (green); (
B) view of compound
4 highlighting the intra-ligand H···H contact.
Table 3.
Adiabatic energy differences associated to the spin transition of compounds 2–5 in gas- and solvent-phase, simulated using the PCM approach. Energies are given in kJ·mol−1.
Table 3.
Adiabatic energy differences associated to the spin transition of compounds 2–5 in gas- and solvent-phase, simulated using the PCM approach. Energies are given in kJ·mol−1.
| Compound | (Gas Phase) | (Solvent Phase) | Effect of Solvent |
|---|
| 2 | 25.6 | 33.9 | 8.3 |
| 3 | 14.2 | 17.4 | 3.2 |
| 4 | 25.6 | 37.6 | 12.0 |
| 5 | 13.4 | 22.8 | 9.4 |
At this point, we shall now determine the origin of the different spin-state stability between
2 and
3 (and between
4 and
5). To do so, we have first excised the nuclear coordinates of the ligand from the optimized structures of the HS and LS minima of the studied compounds and then evaluated their absolute energy (see
Table 4). It can be seen that, for each pair of comparable ligands: 1,2-bip (
2)
vs. 2-bip (
3), and 1-ipp (
4)
vs. 2-ipp (
5), the ligands tend to be destabilized when its structure “points” to the metal coordination center, that is, it is destabilized in compounds
3 and
5 with respect their analogues
2 and
4, respectively (for instance: −330.884
vs. −330.873 a.u. for the HS structures of
2 and
3, see
Table 4). However, once we compare the energy difference within the same ligand in its geometry extracted from the HS and LS minima of the corresponding SCO molecule, it can be seen that for both comparable pairs of ligands the energy difference is essentially the same (
ca. −24 kJ/mol for 1,2-bip
vs. 2-bip and
ca. −26 kJ/mol for 1-ipp and 2-ipp). If the adiabatic gap of these compounds was governed by the structure of the ligand,
2 and
3 should have equivalent energy differences, and, analogously, should
4 and
5 (see
Table 3). Therefore, the different LS/HS relative stability found between
2 and
3 (or
4 and
5) cannot be originated in a larger energy penalty of the ligands in the HS state of
2 (
4) with respect to
3 (
5), but in its effect on the FeN
6 coordination sphere.
Table 4.
Absolute energy and energy difference (ΔE) between the geometries of (left) each ligand, and (right) that of the FeN6 first coordination sphere, both extracted from the HS and LS minima of each complex. Absolute energies in Ry. Energy differences in kJ·mol−1.
Table 4.
Absolute energy and energy difference (ΔE) between the geometries of (left) each ligand, and (right) that of the FeN6 first coordination sphere, both extracted from the HS and LS minima of each complex. Absolute energies in Ry. Energy differences in kJ·mol−1.
| Minima | Ligand Structures | ΔE | [Fe(NCH)6]2+ Structures | ΔE |
|---|
| 1,2-bip | 2HS | −330.884 | 2LS | −330.865 | −24.89 | 2HS | −446.975 | 2LS | −447.001 | 34.63 |
| 2-bip | 3HS | −330.873 | 3LS | −330.855 | −24.10 | 3HS | −446.975 | 3LS | −446.994 | 25.14 |
| 1-ipp | 4HS | −282.999 | 4LS | −282.979 | −26.27 | 4HS | −446.977 | 4LS | −447.001 | 31.92 |
| 2-ipp | 5HS | −282.984 | 5LS | −282.963 | −27.40 | 5HS | −446.961 | 5LS | −446.983 | 28.84 |
In order to investigate this issue, we have again taken the nuclear coordinates of the LS and HS minima of compounds
2–
5 and removed all but the FeN
6 coordination sphere. Then, we have attached a –CH group to each N atom, yielding eight [Fe(NCH)
6]
2+ complexes with different geometries (
i.e., four from HS geometries and four from LS) whose energy has been then evaluated (
Table 4). Note that the –CH groups were positioned at 180° of the Fe–N bond, being all C–N distances 1.15 Å and all C-H distances 1.07 Å. In these results an important difference can be seen in the adiabatic gaps that result from the
2- and
3-derived [Fe(NCH)
6]
2+ compounds (34.6
vs. 25.1 kcal/mol), and the same between those related to
4 and
5 (31.9
vs. 28.8 kcal/mol). This means that the shape of the FeN
6 first-sphere of coordination, which results from the use of the four different ligands, has an important influence in the adiabatic gap of compounds
2–
5. In particular, it can be seen in
Table 4 that, whereas the absolute energy of the HS [Fe(NCH)
6]
2+ complexes is very similar, that of the LS derivatives is more different. As a result, we believe that the spin state stability of the [Fe(NCH)
6]
2+ complexes (and, thus, also of compounds
2–
5) is mostly dictated by the stability of the FeN
6 core when it is in the LS state.
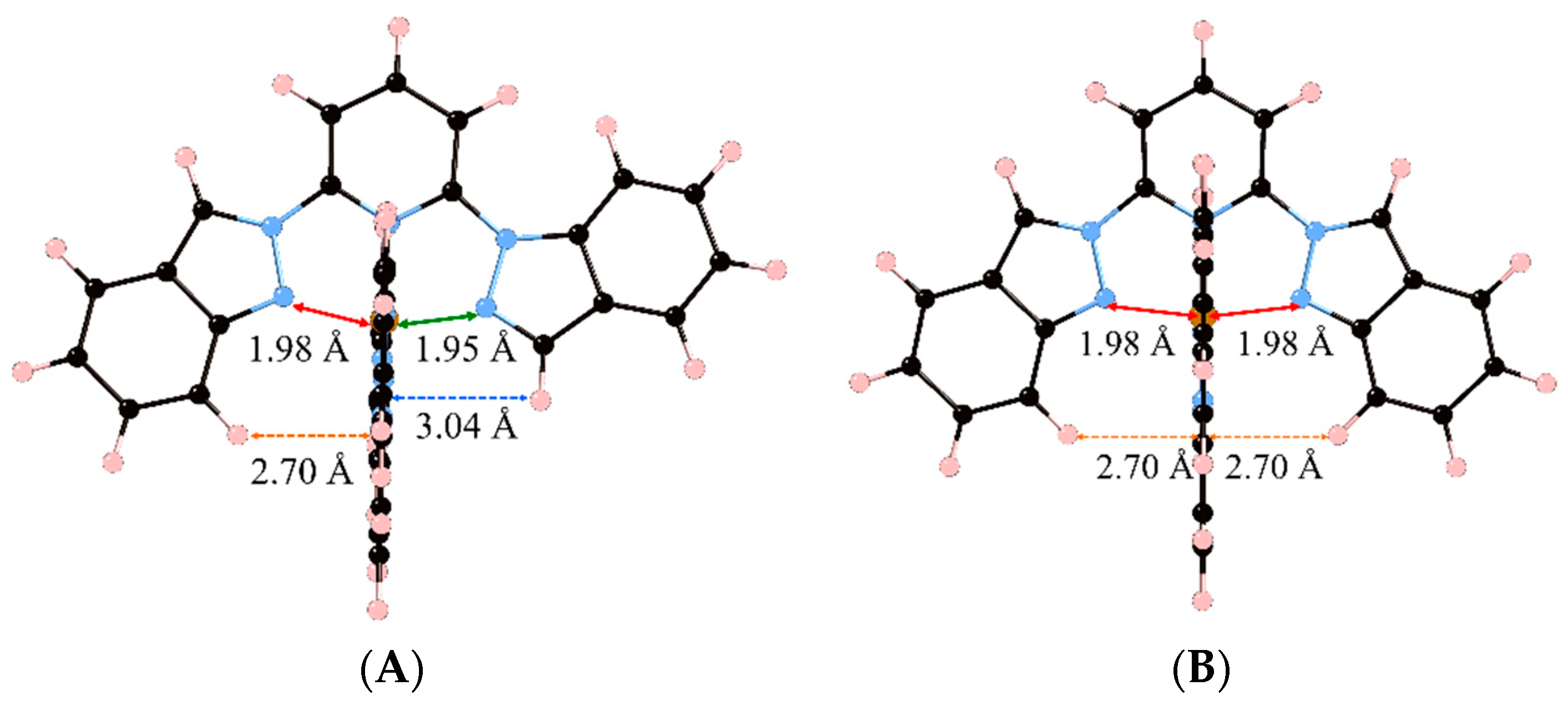
At this point, it is worth analyzing which structural constraints are responsible for this observation. To do so, we must compare the LS minima of compounds
2 and
3 (again, the same conclusions can be extracted for the pair of compounds
4 and
5), which are shown in
Figure 2. First, one must keep in mind that compounds
2 and
3 only differ by one of the groups that is attached to the central pyridine in each of the two tridentate ligands. As mentioned before, the ligand used in compound
2 (1,2-bip) has both an indazol-1-yl and an indazol-2-yl group, whereas the ligand used in compound
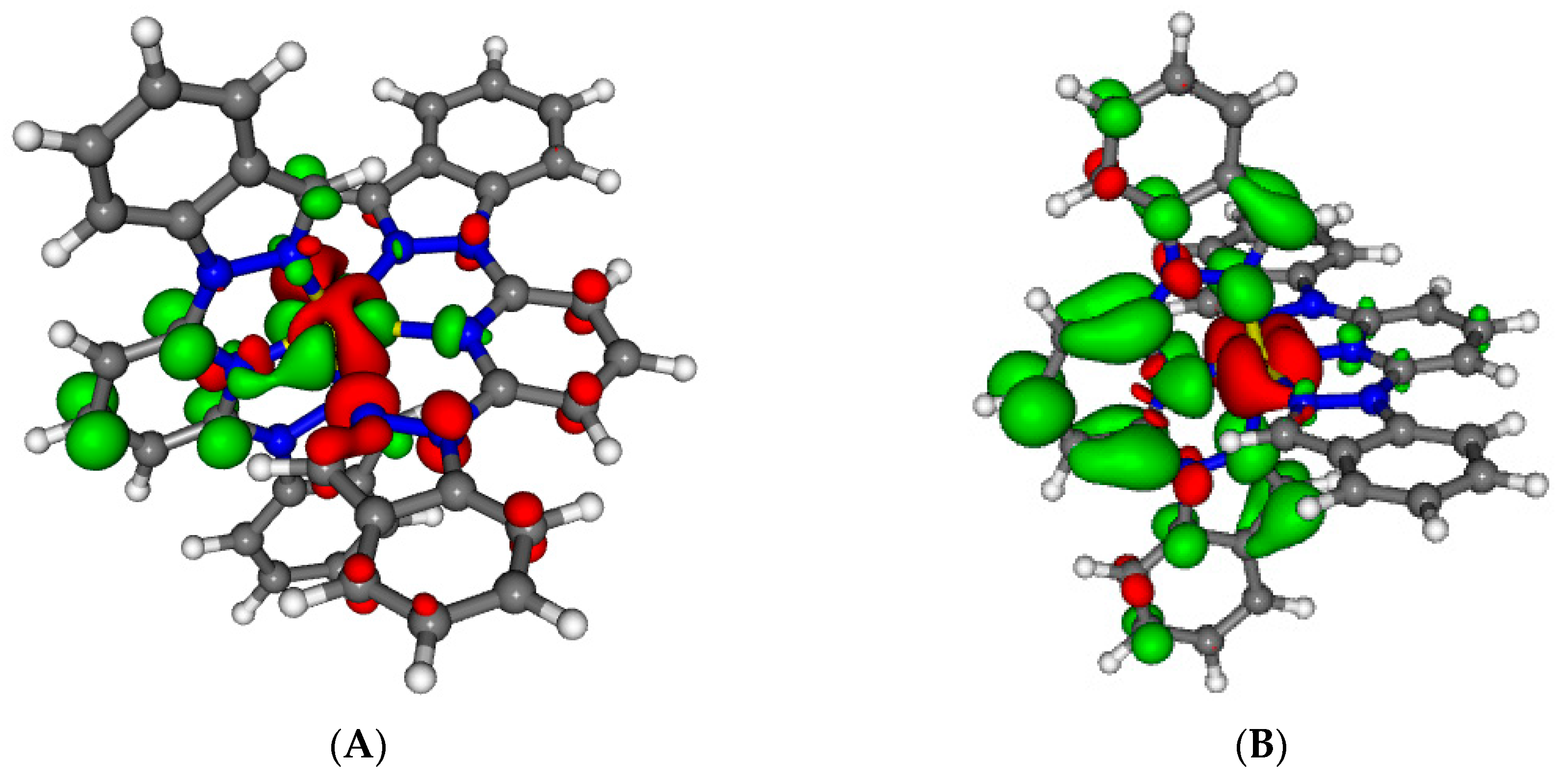
3 (2-bip) has two symmetric indazol-2-yl moieties. By looking at the two minima (
2LS and
3LS), it can be seen that the indazol-1-yl group is associated with an Fe-N distance of 1.95 Å (green line) and an inter-ligand distance of 3.04 Å (blue dashed line), whereas the indazol-2-yl group is associated with an Fe-N distance of 1.98 Å (red line) and an inter-ligand distance of
ca. 2.70 Å (orange dashed line). Our hypothesis is that compound
3LS suffers from a double energy penalty with respect to
2LS due to its structural arrangement. For the [Fe(NCH)
6]
2+ model complex, the optimum Fe–N distances are about 1.90 Å in the LS state due to its bonding character [
29]. However, the presence of two indazol-2-yl moieties in the ligands in compound
3 leads to a minimum energy structure that shows four Fe–N distances of about 1.98 Å (
Figure 2B) while the distance between the iron and pyridine is ca. 1.91 Å. This large structural difference between the LS of the model complex and
3LS arises from the tridentate ligand rigidity but also from the inter-ligand interactions within the complex. The later effect can be demonstrated by analyzing the optimum geometry of
2LS, and is crucial for understanding their different LS stability. In
2LS, the optimum Fe–N distances between the iron(II) and the indazol-1-yl moieties are reduced until 1.95 Å (
Figure 2A). This decrease in the Fe-N distance can only be originated in the lack of steric repulsion between ligands in the complex, and only limited by the rigidity of the tridentate ligand. It is worth mentioning that, although these decrease in the Fe-N distance is extremely small, about 0.03 Å, its effect when evaluating the relative stability of the spin states can be highly significant in these complexes (note that 0.008 Ry correspond to 10 kJ/mol, which can lead to a significant shift in
T1/2 [
29,
30]). Notice that structural distortions in the coordination geometries have also led to remarkable changes in the SCO behavior of other families of compounds [
31].
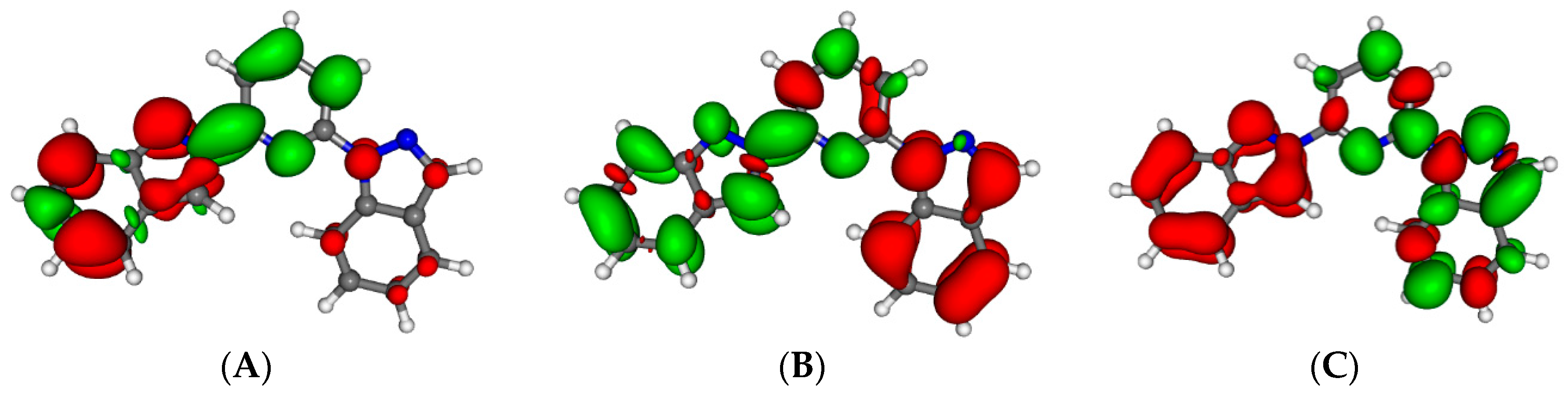
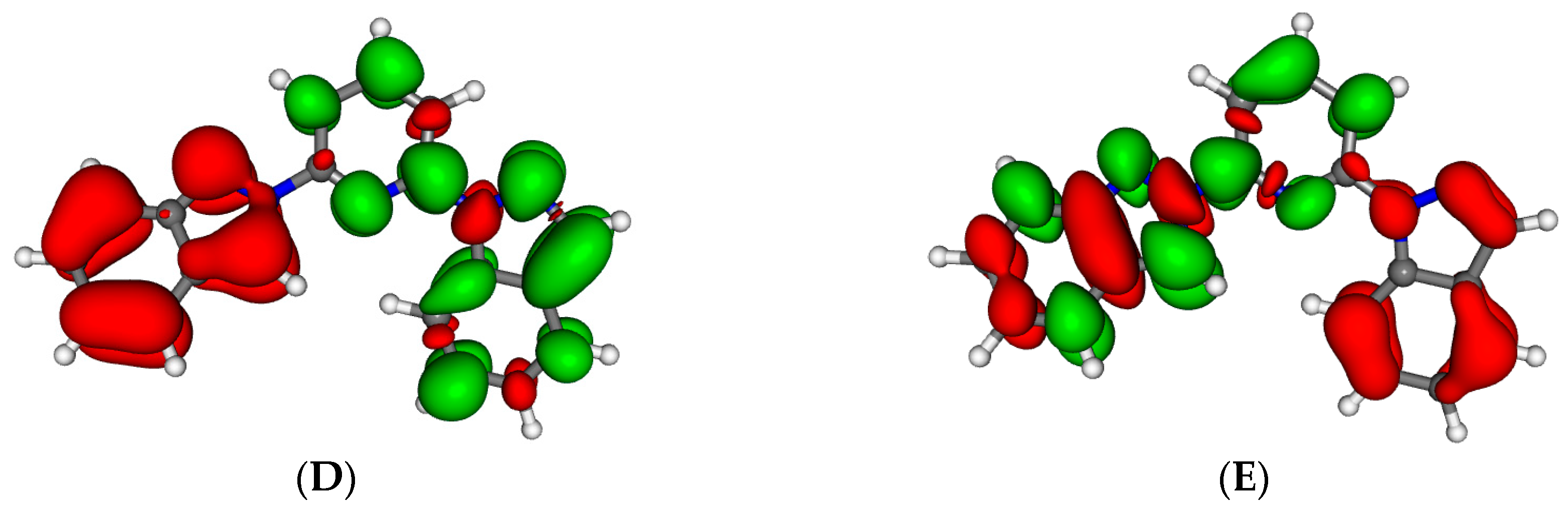
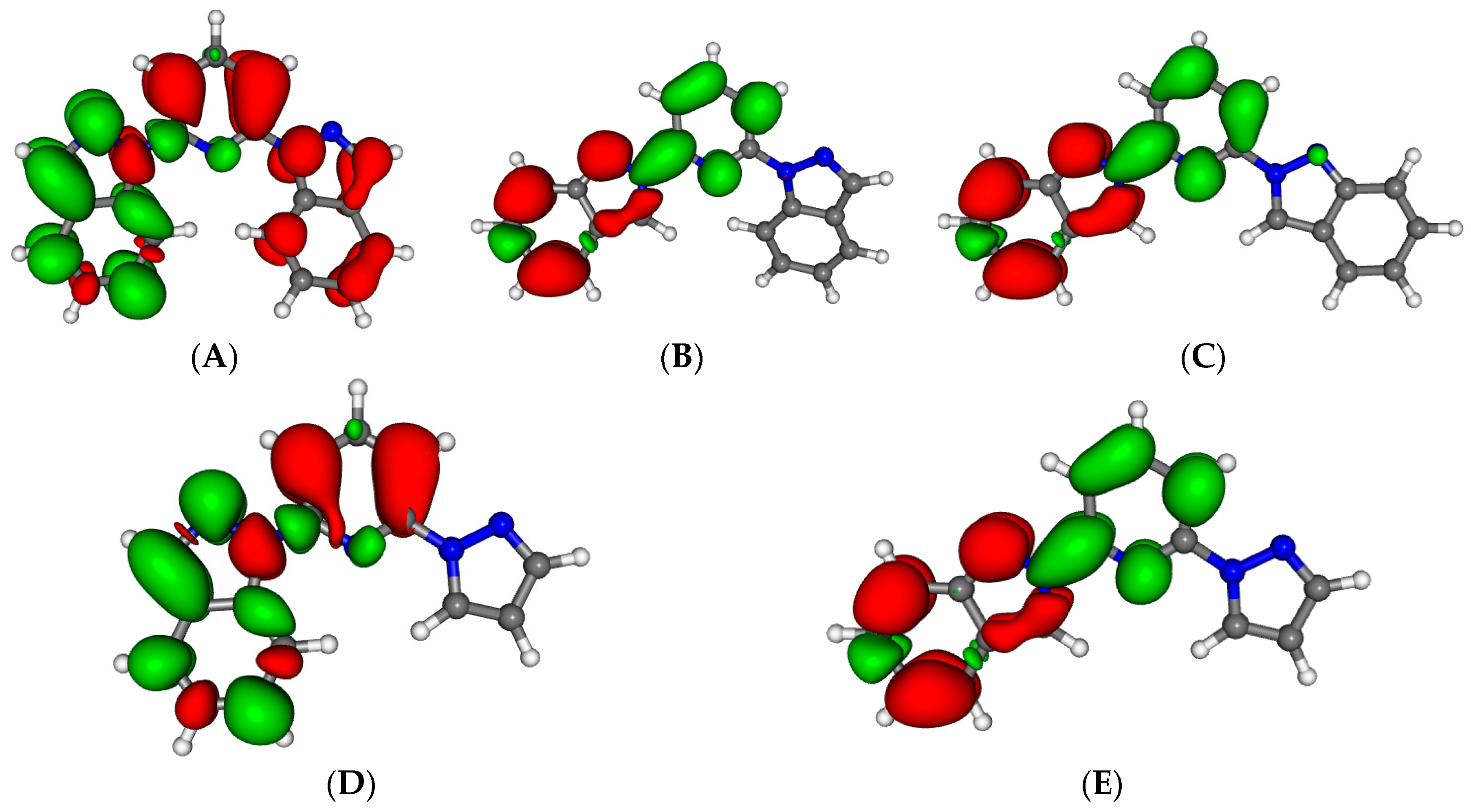
Figure 2.
Minimum energy structures of compounds 2 (A) and 3 (B) in its Low Spin state. Important Fe–N distances and inter-Ligand distances have been highlighted (see main text).
Figure 2.
Minimum energy structures of compounds 2 (A) and 3 (B) in its Low Spin state. Important Fe–N distances and inter-Ligand distances have been highlighted (see main text).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










