Abstract
In a semi-review paper, we show that the hidden Jahn–Teller effect (JTE) and pseudo-JTE (PJTE) in molecular systems and solids, under certain conditions lead to the formation of two coexisting stable space configurations with different magnetic and dielectric properties, switchable by external perturbations. One of the stable configurations has a high space symmetry and a non-zero or higher spin (HS) (non-zero magnetic moment), the other being distorted, but with zero or lower spin (LS). The number of systems with hidden JTE or PJTE is innumerable; we demonstrate this on the (no exhaustible, too) group of systems with half-filed closed-shell degenerate electronic (orbital or band) configurations e2 and t3. The spin-crossover-change from the high symmetry HS arrangement to the low-symmetry LS geometry is accompanied by (driven by the PJTE) orbital disproportionation, in which the system prefers spins-paired states with two electrons on the same orbital (and lower symmetry charge distribution) over the Hund’s spin-parallel arrangement involving several orbitals. Ab initio calculations previously carried out on a series of molecular systems and clusters in crystals, including CuF3, Si3, Si4, Ge4, C4H4, Na4−, C603−, CuO6 (in two crystal environments, LiCuO2 and NaCuO2), etc., confirmed the general theory and allowed for estimates of the parameter values including relaxation times. The hidden JTE and PJTE are thus general tools for search and studies of polyatomic systems with bistabilities.
1. Introduction
The interest in polyatomic systems with coexisting and switchable magnetic-dielectric bistability properties is very strong and increasing in view of their virtual role in going-on continuous innovations in electronics and spintronics. This semi-review paper incorporates previous and novel achievements of the theory of the Jahn–Teller effect (JTE) and pseudo-JTE [1,2,3,4,5,6] to show that there are many (whole classes of) molecular and solid-state systems with two or more coexisting stable configurations (separated by not very large energy barriers), among which some are magnetic, but nonpolar, the other being dipolar, but nonmagnetic, or they differ by the magnetic moment, with possible transitions between them under external perturbations, and they are all controlled by the recently discovered hidden JTE or PJTE.
First, we notice that the two (or more) configurations in such bistability are different in the spin state, hence they cannot follow from the same electronic configuration of the system. On the other hand, the vibronic coupling in the Hamiltonian does not contain spin operators, meaning that neither the JTE, nor the PJTE may lead to the magnetic bistability under consideration: the equivalent minima of the multiminimum adiabatic potential energy surface (APES) produced by these effects have the same spin state. It follows that neither the JTE, nor the PJTE instability may lead to magnetic bistabilities. The prediction of the latter in such systems emerged only after the role of the so-called “hidden” JTE (h-JTE) and “hidden” PJTE (h-PJTE) was revealed [4]. These hidden effects originate in the excited states of the system and, being sufficiently strong, they penetrate the ground state to form a coexisting stable configuration with the parameters of the excited state (Section 2).
Obviously, the number of systems with possible h-JTE or h-PJTE is enumerable. We demonstrate it on the huge variety of systems with half-filled closed-shell electronic configurations e2 and t3, where e and t are, respectively, twofold and threefold degenerate molecular orbitals, or bands in the solid state. They form a practically inexhaustible group of molecular (organic and inorganic) and solid state systems. In the ground state of such systems, according to Hund’s rule, the two electrons in the e orbital and the three electrons in the t orbital occupy, respectively, the two and the three orbitals (bands) with parallel spins, resulting in a totally-symmetric charge distribution with nonzero spin. In these cases the dipolar-distorted configurations occurs via the h-PJTE due to orbital disproportionation [5], when, for instance, the two electrons in the e2 configuration are driven by the PJTE to occupy one of the two e orbitals with antiparallel spins.
The definition and basic features of the h-JTE and h-PJTE are introduced in Section 2, while the specific properties of systems with half-filled electronic shells of degenerate electronic configurations are outlined in Section 3. Section 4 considers in more detail magnetic-dielectric bistability of specific molecular systems. Section 5 describes such bistability in solid states, with conclusions in Section 6.
2. Hidden Jahn–Teller and Pseudo-Jahn–Teller Effects
First, we reiterate the obtained earlier conclusion that the JTE and the PJTE are the only source of instability and distortions of high-symmetry configurations of polyatomic systems [1,2,3]. This important conclusion has been challenged by examples of molecular systems, which at face seem to contradict to its statement. Indeed, there are molecules in high-symmetry configurations, which are unstable in nondegenerate states (no JTE), with no appropriate excited state to induce the PJTE, e.g., when the excited states of appropriate symmetry are too high in energy or have a different spin multiplicity (see below). We have shown that in such cases the distortions are still due to the JTE or PJTE which originate in the excited states; but, being sufficiently strong, they penetrate the ground state [4], producing additional lower (global) minima of the adiabatic potential energy surface (APES), in which the system is distorted. We call them ‘hidden’ JTE and PJTE (h-JTE and h-PJTE, respectively) because they are not seen directly as a feature of degeneracy and pseudodegeneracy in the reference high-symmetry configuration; they appear only when the special role of an excited state is recovered.
Figure 1 illustrates this statement. We see that the ground state of the system is stable in the high-symmetry configuration at Q = 0, but it is unstable in this configuration in the lowest excited double-degenerate state due to the JTE (Figure 1a), or due to the pseudodegenerate interaction with a higher state in the PJTE (Figure 1b). If these excited-state effects are sufficiently strong, their stabilization energies may offset the energy gap to the ground state (EJT > Δ in the JTE or EPJT > Δ in the PJTE case) and produce additional global minima, in which the equilibrium ground state is distorted [4]. In case of the h-JTE triggered by the E⊗e JTE problem in the excited state the additional configuration occurs when (F2/2K0) > Δ, where F is the vibronic coupling constant, and K0 is the primary force constant, while for the h-PJTE the condition of a possible additional stable configuration is more complicated [4],
where is the constant of vibronic coupling between the two excite states, and the constants Δ12 and Δ0 are shown in Figure 1b. In both cases, h-JTE and h-PJTE, under the above conditions, we get two stable configurations, distorted and undistorted, but distinguished from the JT case, where the possible distortions are restricted by the JT active modes, the PJT induced distortion may be of any kind, dependent on the symmetries of the mixing excited states. Another distinguished feature of the h-PJTE is that it leads to orbital disproportionation discussed in Section 3.
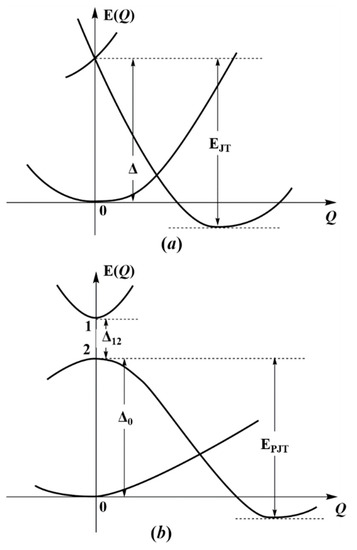
Figure 1.
Illustration to the origin of h-JTE and h-PJTE. In both cases the distortion of the system in the ground state global minimum of the APES is due to the JTE (a), or PJTE (b) in the excited state; it takes place when their stabilization energy (respectively, EJT or EPJT) is larger than the energy gap to the ground state of the high-symmetry configuration (Δ and Δ0, respectively); Δ12 is the energy gap between the two excited states producing the h-PJTE.
The h-JTE was first demonstrated on the structure and properties of the ozone molecule O3 [7] for which ab initio calculations [8,9] show that in the high-symmetry regular-triangular geometry the ground state 1A is nondegenerate, and its excited double degenerate 1E state is rather high, at the energy gap Δ ~8.5 eV, so no JTE or PJTE is expected in the reference high-symmetry configuration. Nevertheless, the regular-triangular configuration of O3 is not the ground-state one: its APES has lower-in-energy three equivalent minima, in which the configuration of the molecule is obtuse-triangular (with a nonzero dipole moment). The ab initio calculation of the cross section of the APES of this molecule along the distortion coordinate Qθ, carried out including the ground 1A state and first excited 1E state [7] follows the prediction of the theory for the h-JTE: the three equivalent obtuse-triangular configurations with lower energy emerge from the E⊗e problem of the JTE in the excited 1E state, yielding a strong stabilization energy EJT ~9 eV that penetrates the energy gap of ~8.5 eV (Figure 2).
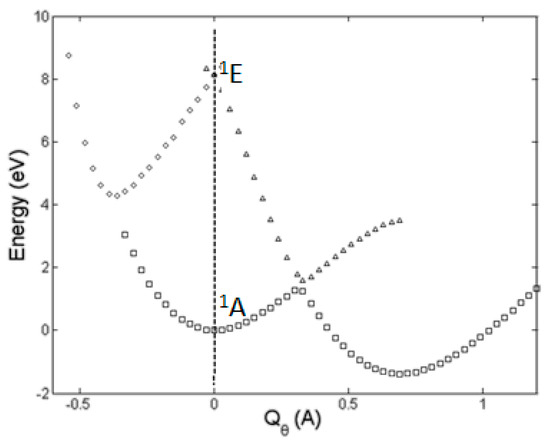
Figure 2.
Cross-section of the APES of the ozone molecule along the Qθ component of the double degenerate e mode obtained by numerical ab initio calculations of the ground 1A and highly excited 1E state, explicitly demonstrating that the ground state distorted configurations are due to the JTE in the excited state [7]. In the global minimum at Qθ = 0.69 Å the molecule has a significant dipole moment, and the E-A avoided crossing takes place at Qθ ~0.35 Å.
Note that in the case of the ozone molecule, both the ground state and the excited state responsible for the h-JTE have the same spin state. Therefore, the two stable configurations—distorted and undistorted—have the same magnetic properties, hence the bistability in this case is simply dielectric: dipolar-nonpolar. However, if the two electronic states, participating in the hidden effects possess different spin states, the bistability would be both polar and magnetic. There are many such possibilities among polyatomic systems. Below we demonstrate this by the examples from h-PJTE that incorporate a whole class of molecular systems and solids with possible bistabilities. There are also cases when both the h-JTE and h-PJTE take place simultaneously in the same system (see, e.g., the electronic structure of the CO3 molecule [10]).
3. Half-Filled Closed-Shell Degenerate Electronic Configurations with h-PJTE and Spin Crossover
With respect to the bistabilities discussed in this paper, the systems with half-filled closed-shell e2 or t3 electronic configurations, which occur in a wide range of molecular and solid-state systems, are of special interest. The JTE nature of possible distortions of the ground state of such systems is ad hoc most ‘hidden’ because their closed-shell charge distribution is totally symmetric. In addition, the ground states in these electronic configurations are no subjects of the PJTE either, since their excited states have a different spin multiplicity than that of the ground (Hund) state (see below). Nor can the ground state be formed by a strong JTE in the low-lying excited states (as in the O3 case). Nevertheless, almost all of the e2 and t3 systems have distorted geometries in the ground state due to the h-PJTE (Figure 1b), which is especially pronounced in these cases.
Half-filled closed-shell electronic e2 and t3 configurations may occur in any molecular or solid-state system with at least one threefold (or higher) symmetry axis. For example, the Cu3+ ion in octahedral environment in crystals and coordination systems has the configuration is (t2g)6e2 occupied by its eight d-electrons, two in the outer e2 configuration (Section 5). The latter remains also in the in the D3h symmetry of the CuF3 molecule (Section 4). The energy terms formed by the interelectron interactions in e2 configurations is shown in Figure 3. We see that in these cases the ground state of the undistorted configuration (triangular, octahedral, etc.) is a spin triplet, 3A2, but a strong PJTE mixing of two excited singlet states, 1A1 and 1E, may produce an additional lower-energy state with a distorted configuration (provided the stabilization energy EPJT is larger than the energy gap Δ to the ground state, as in Figure 1b). For a triangular system, the distorted configuration has a non-zero dipole moment, but a zero magnetic moment (zero spin from the 1A1 state), whereas the undistorted configuration in the 3A2 state has a zero dipole moment, but a nonzero magnetic moment.
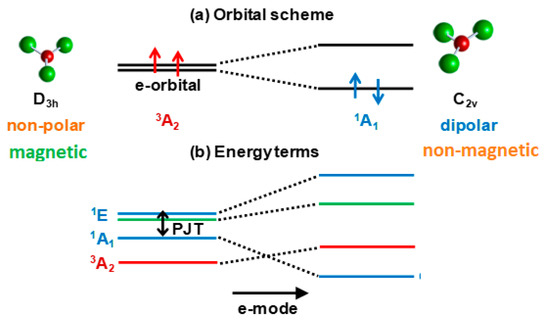
Figure 3.
Illustration to the formation of two coexisting configurations with different magnetic and structural properties: (a) In a typical orbital energy level scheme of the electronic e2 configuration the ground state 3A2 is formed by the two electrons with parallel spins, each of them occupying one e orbital, resulting in the eθ↑eε↑ configuration, which is totally symmetric but magnetic (the spin S = 1); the picture shows the example of a planar triangular molecule (e.g.,CuF3); (b) In the energy terms scheme of the same electronic e2 configuration the first excited singlet state 1A1 with the electron configuration (1/√2)(eθ↑eθ↓ + eε↑eε↓) is totally symmetric too, but due to the PJT interaction with the higher 1E1 state it undergoes the orbital disproportionation resulting in either eθ↑eθ↓ or eε↑eε↓ configurations, both having a lower symmetry of charge distribution that distorts the systems in the e mode direction, but a zero spin state.
The two different spin states of the two energy-minima configurations is a remarkable feature of systems with half-filled closed-shell e2 and t3 configurations. It is directly related to orbital disproportionation [5], when the triplet 3A state (formed by the mentioned above two electrons that occupy the two e orbitals with parallel spins), which is expected to be the ground state according to the Hund’s rule, is overridden by the 1A state with zero spin (with two electrons with antiparallel spins in one e orbital), the whole process being driven by the PJTE that lowers the energy.
Labeling the two e functions by ε and θ and the two spin states by arrows up and down, we can present the wavefunctions of the excited singlet terms 1A1 and 1E before their PJTE mixing (Figure 3) as
In all of these states, the charge distribution is symmetrical with respect to the θ and ε components. Due to the PJTE, the 1Eθ component mixes with the 1A1 function to result in their linear combination, which in the case of sufficiently strong vibronic coupling produces a disproportionate distribution of either or [5]
In either of these two cases, the charge distribution becomes non-totally symmetric and distorts the high-symmetry configuration of the system. In other words, if the h-PJTE conditions are met, it is more energetically convenient for the system to pair its electrons on the same orbital and distort the nuclear framework than to remain symmetrical in either the ground or excited state of the undistorted configuration (the gain of energy due to the added covalency by the PJTE distortion [1,3] is larger than the loss due to higher interelectron repulsion in the violation of the Hund’s rule).
A quite similar effect takes place in the case of electron configuration t3. In this case the interelectron interaction spans the states , , , and with the ground quadruplet state 4A2, which corresponds to the Hund’s rule distribution . A strong PJTE coupling between the two excited states 2T1 and 2T2 results in a lower orbital disproportionate component of the type with a doublet spin-state instead of the quadruplet in the high-symmetry configuration [5].
As follows from these results, orbital disproportionation in systems with half-filled closed-shell electronic configurations is necessarily accompanied by lowering the spin of the electronic ground state. For the e2 configuration it means transition from the high-spin (HS) triplet 3A state to the low-spin (LS) singlet state 1A, while for t3 this transition is from the quadruplet (HS = 3/2) to the doublet (LS = 1/2) state. Since this transition is induced by the PJTE distortion originating from an excited electronic state, the two states, HS undistorted and LS distorted, coexist in two minima of the APES which may have close energies (Figure 2). In between these two minima there is a crossing between the two states of different spin, a spin crossover. The results of ab initio calculations in Section 4 and Section 5 show explicitly the spin crossover that takes place in the specific molecules and solids under consideration. It is demonstrated above to take place in molecular systems with electronic e2 or t3 configurations, meaning molecules with at least one three-fold axis of symmetry and appropriate number of electrons, as well as in solids with corresponding bands (Section 5), but it may be expected in any systems with h-JTE or h-PJTE.
In the e2 and t3 systems the spin of the two nuclear configurations, distorted and undistorted, is different, with a higher spin in the undistorted configuration. This leads directly to magnetic moment μ bistability (μ1;μ2). For e2 systems the magnetic moment of the undistorted HS configuration with the spin S = 1 is μ1 ≅ 2.82 MB, while for the distorted LS geometry S = 0 and μ = 0, realizing a (2.82 MB; 0) magnetic bistability. On the other hand, the two geometries have different dielectric properties, e.g., dipole moments p, leading to dipolar bistability (p1; p2). Often the distorted LS configuration has a non-zero dipole moment, whereas p = 0 in the undistorted HS one, resulting in (0; p) bistability. Thus, in such systems we have two coexisting bistabilities, magnetic (2.82 MB; 0) and dipolar (0; p), with interesting (and peculiar) properties of either magnetic moment, but no dipole moment in one of the coexisting states, or dipole moment, but no magnetic moment in the other state [6]. Following the often-used terminology such systems can be termed single-molecule multiferroics.
Another spin crossover {XE “spin crossover”} phenomenon is well known to take place in cubic coordination systems of transition metal compounds (TMC) with electronic configurations d4-d7 that may produce either HS or LS complexes, subject to the strength of the ligand field [11,12]. For some values of the latter the two electronic configurations, high-spin and low-spin, may be close in energy, so they can crossover as a function of the breathing mode of the system (involving the metal-ligand distance). This spin crossover is known for a long time, and has been subjected to intensive studies during more than three decades; systems with two spin-states may serve as molecular materials for electronics [12]. However, the observation of the two states and transitions between them under perturbations (required for such materials) encounters essential difficulties because of fast radiationless transitions between them (very short lifetime of the higher in energy state due to its fast relaxation to the lower one). The two spin states in TMC cannot be observed beyond low temperatures because of their poor separation in space and fast relaxation due to the relatively high spin-orbital interaction in the metal [12]. So far, they have been observed only for some compounds in optical LSHS excitation at low temperatures <50 K and mostly as a cooperative effect in solids [12].
The crossover induced by the h-PJTE involving orbital disproportionation {XE “orbital disproportionation”} is essentially different from the spin crossover in TMC produced by the strength of the ligand field. Indeed, (1) the PJTE-induced spin crossover takes place in a variety of molecular systems, small to moderate, organic, and inorganic, as well as in metal-containing molecules, as illustrated below on a series of molecular systems taken as examples; (2) the HS—LS intersystem relaxation rate in the PJTE case is expected to be much lower than in TMC because the two spin states have different nuclear configurations, distorted and undistorted, producing a significant barrier between them and a small Franck–Condon factor, and the spin-orbital interaction in light-atom molecules is smaller by orders of magnitude than in TMC; (3) based on these considerations (followed by numerical estimates) it can be assumed that in the h-PJTE-induced spin-crossover the switch between the two states (in both directions) under perturbations can be observed as a single-molecule phenomenon and at relatively higher temperatures. The ab initio calculations (Section 4 and Section 5) confirm the conclusions of the h-PJTE theory.
4. Molecular Systems with h-PJTE-Induced Spin Crossover and Magnetic-Dielectric Bistability
In ab initio calculations of a series of molecular system, the main features of the outlined above h-PJTE-induced bistabilities with spin crossover and switchable magnetic-dielectric properties were confirmed, and numerical estimates of the parameters were obtained [6]. Consider first trivalent Cu compounds, for instance, the molecule CuF3 or the local center in the crystal LiCuO2 (see more in Section 5). As mentioned above, in both systems with symmetries D3h and Oh, respectively, the Cu3+ ion has the valence electronic d8 configuration, the outer two d electrons of which occupy the e orbitals forming the e2 configuration. The latter produce the noted above three terms (Figure 3), which in the D3h symmetry of CuF3 are labeled as 3A2′ (ground state), 1A1′, and 1E′. The results of full scale (high-level) ab initio calculations of all these terms in CuF3 as a function of the e type distortions (labeled by the angle α(F-Cu-F)) are shown in Figure 4 [6]. We see that, in accordance with the general theory, there is a strong PJTE interaction between the two excited 1A1′, and 1E′ states, due to which the lower 1A1′ state goes sharply down, crosses the HS triplet ground state of the undistorted configuration, and produces a global minimum with a distorted (α ≈ 93°) LS geometry. The difference between the zero point energies of these two configurations is 0.190 eV (all energies read-off the zero-point vibrational levels), while their excitation energies to the point of spin crossover (barrier heights) are δHS = 0.541 eV and δLS = 0.712 eV, respectively. In the distorted configuration the magnetic moment μ = 0 and the dipole moment p = 1.67 D, and in the undistorted state, as mentioned above, μ = 2.82 MB with p = 0. We thus get a perfect case of bistability, (2.82 MB, 0) vs. (0, 1.67 D), in which the two stable configurations are separated by both potential and magnetic (spin) barriers.
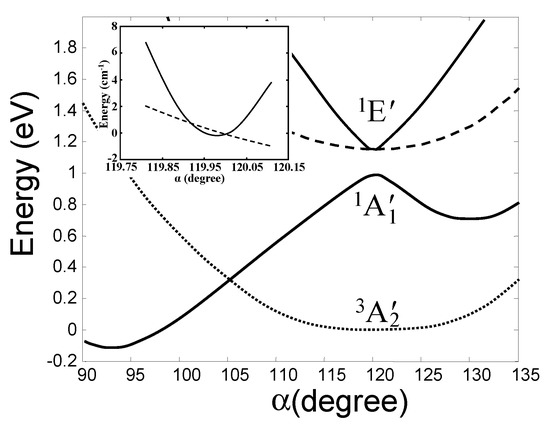
Figure 4.
Calculated cross-section of the APES of the planar CuF3 molecule along the angle α(F-Cu-F) (using the CASSCF method) showing the very weak JTE in the excited E state and a strong PJTE between the E and A states, resulting in the h-PJTE and magnetic-dielectric bistability [6]. The inset shows in more detail the two conical intersections in the E state in this direction.
In another example of e2-molecules, triangular Si3 (Figure 5) the energy terms, as expected, follow the energy level scheme in Figure 3, leading to the h-PJTE-induced two coexisting configurations with magnetic-dielectric bistability. It is similar to the CuF3 case, but with a lower potential barrier between the two configurations. On the contrary, for square-planar Si4 the potential barrier between the two coexisting configurations created by the h-PJTE is very high (Figure 6).
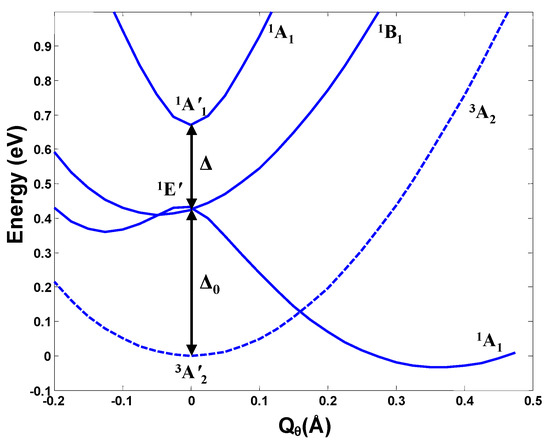
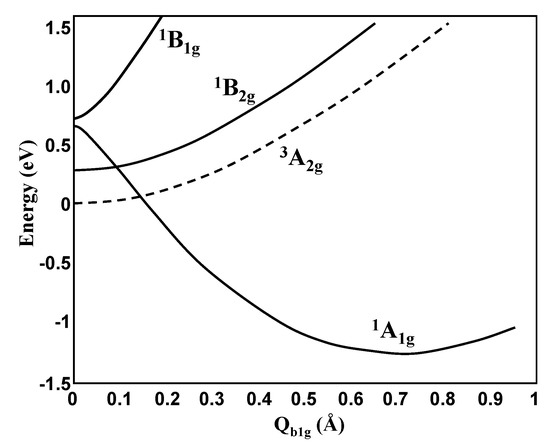
Figure 6.
Cross-section of the APES of the Si4 molecule along the b1-mode that distorts the system from square-planar to rhombic geometry due to the (1A1 + 1B1)⊗e PJT coupling between two excited states (cf. Figure 3).
Calculations for possible bistabilities in systems with electronic e2 configurations were also performed for clusters Ge4, C4H4, CuO6 (for the latter see Section 5), etc. Table 1 lists the estimated parameters of interest for these systems [6]. The coordinates Qa1 (totally symmetric), Qϑ and Qε (e-type) describe the symmetrized displacements of the system in the distorted configurations (read-off the undistorted one), while β is the energy barrier between them, read off the zero-point vibrations, and τ is the lifetime of the higher energy configuration in relaxation to the lower one via spin-orbital coupling and thermalization.

Table 1.
h-PJTE-induced HS-LS e-mode Qϑ, Qε, and totally symmetric Qa1 distortions, the energy barrier β (read off the vibrational zero-point energies), and lifetimes τ at low temperatures (T = 5K) for several systems with electronic e2 configurations (the CuO6 cluster is assumed to be in the crystal environment, see Section 5).
The data for τ in this table were obtained by employing an approach [11,12], which starts from the time-dependent Schrödinger equation and follows the evolution of the system in the presence of a dissipation factor, that describes the thermalization time of the vibrational levels of the system in the singlet state, and estimating the temperature dependence of τ by summation of the contribution of all the Boltzmann populated ones. The numerical estimates obtained in this way are strongly dependent on the influence of the environment of the cluster, which varies significantly. Being rather approximate, these data may be taken into account as a first view on the subject, which shows that, as expected, τ is the largest in clusters with solid state environment, and for free molecules it is the largest in Ge4. Si3 has the shortest lifetime (comparable to that of most TMC systems), which increases in systems with larger distortions, and reaches macroscopic values in Si4, Ge4, and LiCuO2 at low temperatures (but in Ge4 there are low-lying triplet excited states that may reduce the lifetime). In the crystals, LiCuO2 and NaCuO2 (Section 5), the estimates of the lifetime of the HS configuration are carried out for a separate cluster unit CuO6; it is extremely large at low temperatures (due to the large distortions in the diamagnetic phase) and remains macroscopic at room temperature. Since the cooperative interactions between the complexes in the condensed phases obviously increase the lifetimes, the values in Table 1 show the lower limit of the expected lifetimes. This means that the crystals LiCuO2 and NaCuO2 are quite feasible for practical use as bistability materials, and their long lifetime of both states in this bistability indicates that they can be used in the form of clusters, quantum dots, or thin films.
Note that, distinguished from the case of TMC crossover, where the two configurations are displaced along just the totally symmetric coordinate, the relaxation between the two configurations in the bistability under consideration is strongly reduced because of their different symmetry, distorted and undistorted, and the spin-orbital coupling is much lower in the light atoms than in transition metal compounds.
As a demonstration of the t3 half-filed closed-shell h-PJTE-induced magnetic-dielectric bistability we show here examples from two different kinds of molecular systems: Na4− and C603− [6]. In the Na4− cluster in the high-symmetry configuration the four sodium atoms are arranged in a tetrahedron. The four 3s valence orbitals in this conformation form a1 and t2 symmetrized orbitals with the electronic configuration is a12t23 producing electronic terms , , , and (from the t23 configuration). The complete active space self-consistent (CASSCF) calculations of the electronic structure of this system in the ground and excited states as a function of the tetragonal e displacements using the cc-pvtz basis set and the s valence orbitals of Na as the active space are illustrated in Figure 7 [6].
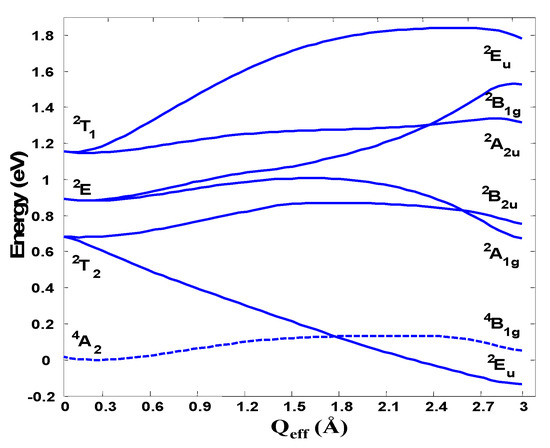
Figure 7.
Cross-section of the APES of Na4− along the e-mode distortion transforming the system from tetrahedral (Qeff = 0) to square-planar geometry due to the (T1 + T2)⊗e PJT coupling.
As expected from the general theory [5], there is no significant JTE distortions in any of the states formed by the t23 configuration, but there is a strong PJTE of the type (2T1 + 2T2)⊗e that pushes down one of the components of the 2T2 term making it the absolute minimum in which the tetrahedron is distorted in the e direction. We have thus a spin-quadruplet ground state in the undistorted tetrahedral configuration and a spin-doublet state in the distorted global minimum with the shape of a rhombus. Although the energies of the two minima of the APES in this case are very close, the energy barrier between them is rather high, β ~3000 cm−1 [6].
For the t1u3 configuration in the fullerene anions C603− the orbital disproportionation was first revealed by direct estimation of the electron interactions in the distorted configuration in order to explain the origin of conductivity in the alkaline doped fullerides A3C60 [13,14]. In this case the PJTE problem is (Hu + T1u)⊗hg, and there are many modes of hg symmetry that contribute to the distortion of the fullerene molecule [15]. Hence the problem is a multimode one [1,2,16], and the solution in Figure 8 is shown along the effective mode (often called “interaction mode” [1,2]).
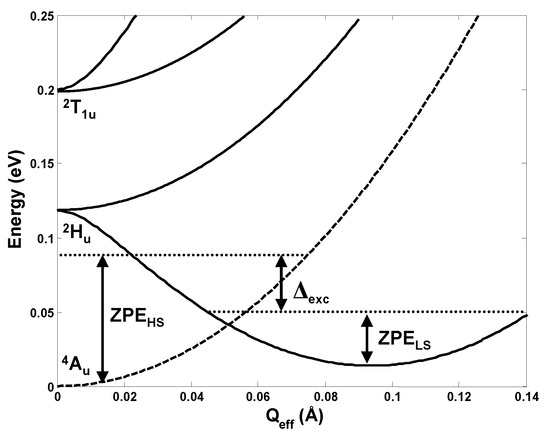
Figure 8.
Cross-section of the APES of C603− along the effective mode that takes the system from the undistorted high-spin minimum to the distorted low-spin minimum due to the multimode (Hu + T1u)⊗hg PJT coupling [15]. The zero-point energies (ZPE) along this mode (which are different from the global ZPE) show that the lowest vibronic state associated with the electronic spin-doublet state is lower than that for the spin-quadruplet.
5. Magnetic-Dielectric Bistability in Solid States Induced by h-PJTE
Half-filled closed-shell local formations in solid states may be even more important in applications because of their larger variety and better parameters. They include virtually all the cubic symmetry coordination compounds, ionic crystals, and impurity centers in crystals containing transition metal ions with the electronic configuration e2 (d8 ions in octahedral coordination and d2 ions in tetrahedral or cubic environment), and t3 (d3 in octahedral and d7 in tetrahedral coordination), as well as any other systems with h-JTE or h-PJTE involving an excited state with different multiplicity. Remarkably, the transition metal systems considered below are not the same as the mentioned above transition metal spin crossover ones. Experimentally, there is evidence of magnetic bistability in Ni2+(d8) complexes, in which a reversible equilibrium between octahedral magnetic and square-planar diamagnetic complexes has been observed [17].
Electronic structure calculations for two solids from this class of compounds, the crystals LiCuO2 and NaCuO2 were carried out in the DFT-calculations with the hybrid B1WC functional [6]. The structure of these crystals is formed by alternating layers of vertex-sharing Cu3+(O2−)6 octahedrons and Li+ or Na+ ions, respectively. The Cu3+ ion has a (d8)e2 electronic configuration (as mentioned above, e denotes molecular orbital or symmetrized band states).
As predicted by the general theory and shown above for molecular cases, the HS configuration of these crystals is undistorted, while the LS state is diamagnetic with a very strong elongation of the CuO6 octahedrons. The LiCuO2 and NaCuO2 crystal structures were optimized in both the HS (ferromagnetic and antiferromagnetic) and LS (diamagnetic) states [6]. A summary of the numerical results is shown in Table 2. As expected, the ferromagnetic and antiferromagnetic phases have very similar geometries with regular octahedral coordination of the oxygen atoms around Cu, whereas the diamagnetic phase is strongly distorted to almost square-planar coordination: in LiCuO2 the metal-ligand distance in the magnetic phase is 2.01 Å, while in the diamagnetic phase the equatorial and axial ligands are at 1.86 Å and 2.76 Å, respectively (Figure 9). In NaCuO2 the same pattern is found, although the lattice parameter and distortions are by about 10% larger than in LiCuO2. The results obtained for the geometry of the diamagnetic phase of these crystals agree well (within 2%) with the experimental data [18,19]; the interatomic distance changes are here 3–4 times larger than in TMC crossover systems.
Table 2.
Experimental (in parenthesis) and calculated by DFT B1WC lattice parameters A, B, C (in Å), lattice angle γ (degrees), interatomic equatorial d1(Cu-Oeq) and axial d2(Cu-Oax) distances in the diamagnetic (DM), ferromagnetic (FM), and antiferromagnetic (AF) phases of LiCuO2 and NaCuO2 and their relative energies ΔE (in cm−1). Reproduced with permission from [6].
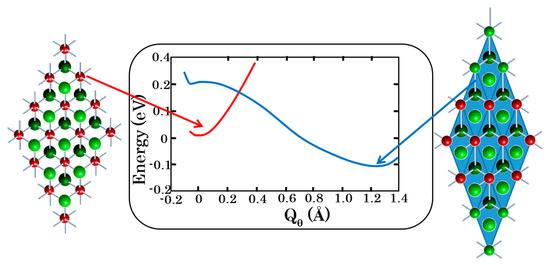
Figure 9.
PJTE-induced bistability in the crystal LiCuO2: the undistorted spin-triplet magnetic configuration (left) is by ~0.17 eV higher in energy than the strongly tetragonal-distorted zero-spin, diamagnetic phase (right). The Cu3+ ions are shown in red, while the oxygens are painted green (face plane) and dark green (rear plane).
The magnetic to diamagnetic phase transition has a strong repercussion on the crystal structure along the c-axis enlarging the unit cell by ~10% (≈0.5 Å) but the geometrical difference between the two bistability states does not translate in a large energy difference between them. Indeed, as shown in Table 2 and Figure 9 the energy difference between the phases is only of a few hundred cm−1. In the case of LiCuO2 the calculations yield that the magnetic phase (at the minimum of the APES) is lower by 150 cm−1 than the diamagnetic one, but with the vibrational zero-point-energy correction the two phases are almost degenerate. In NaCuO2 the non-magnetic phase is more stable. Due to these small energy differences between the phases it should be relatively easy to trigger a phase transition between them by external perturbation. Indeed, for LiCuO2 a phase transition is observed at T = 180 K [20]. For NaCuO2 the ab initio calculations show that a diamagnetic to magnetic phase transition is achievable at a pressure of ~6 GPa [6].
Following the results of Section 3, the distortion of the high-symmetry (magnetic) configuration toward the diamagnetic one by the PJTE in two excited states is accompanied by orbital disproportionation [5], due to which the two electrons in two e orbitals with parallel spins (and symmetric charge distribution) prefer to occupy one orbital with anti-parallel spins lowering the symmetry of the system, but gaining additional covalent metal-ligand bonding. This explains also the insulating properties of the diamagnetic NaCuO2 or LiCuO2 crystals that are subject to discussion in the literature [21]: the orbital disproportionation splits the two-fold degenerate e level or band (3z2-r2 and x2-y2 of Cu) and fully occupies the lower one.
6. Conclusions
It is shown that the previously revealed hidden JTE and PJTE are very instrumental in discovering novel molecular and solid-state systems with two coexisting stable configurations that differ in magnetic and dielectric properties, switchable under external perturbations. The existence of such system is illustrated by previously carried out ab initio calculations of several specific systems, but the importance of this result is in its generality: the number of molecular and solid state systems with h-JTE or h-PJTE is innumerable, and in any of them the two states, involved in these effects, may have different spin multiplicity, leading to magnetic-dielectric bistability. We demonstrated this statement on two classes of compounds with e2 and t3 electronic configurations, respectively (which are inexhaustible, too), but they are just a part of the infinite number of molecular and solid state systems with h-JTE or h-PJTE, leading to bistability.
With regard to possible applications, the direct calculation of the lifetime τ (relaxation time) of the induced higher in energy state in the bistability with respect to its thermal relaxation to the lower state is not trivial and needs further improvements taking into account the environmental conditions in molecular cases, but it is less a problem for solids where the cooperative interactions stabilize the two states in the bistability wells.
We conclude that h-JTE and h-PJTE provide for a new tool for search and study of systems with magnetic and dielectric bistabilities within a wide (unlimited) range of molecular systems and solids. Future studies should be focused on revealing specific molecular and solid-state systems with such h-JTE- or h-PJTE-induced bistabilities with bigger barriers between the two states and larger relaxation time for transitions between them.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Bersuker, I.B. The Jahn-Teller Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Bersuker, I.B.; Polinger, V.Z. Vibronic interactions in Molecules and Crystals; Springer: Berlin/Heidelberg, Germany, 1989. [Google Scholar]
- Bersuker, I.B. The Pseudo Jahn-Teller Effect—A Two-State Paradigm in Formation, Deformation, and Transformation of Molecular Systems and Solids. Chem. Rev. 2013, 113, 1351–1390. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, I.B. Recent Developments in the Jahn-Teller Effect theory. The Hidden Jahn-Teller Effect. In The Jahn-Teller Effect. Fundamentals and Implications for Physics and Chemistry; Springer Series of Chemical Physics; Köppel, H., Yarkony, D.R., Barentzen, H., Eds.; Springer: Heidelberg, Germany, 2009; Volume 97, Chapter 1; pp. 3–23. [Google Scholar]
- Garcia-Fernandez, P.; Bersuker, I.B.; Boggs, J.E. Orbital disproportionation and spin crossover as a pseudo Jahn-Teller effect. J. Chem. Phys. 2006, 125, 104102. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, P.; Bersuker, I.B. A class of molecular and solid state systems with correlated magnetic and dielectric bistabilities induced by the pseudo Jahn-Teller effect. Phys. Rev. Lett. 2011, 106, 246406. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, P.; Bersuker, I.B.; Boggs, J.E. Lost topological (Berry) phase factor in electronic structure calculations. Example: The ozone molecule. Phys. Rev. Lett. 2006, 96, 163005. [Google Scholar] [CrossRef] [PubMed]
- Siebert, R.; Fleurat-Lessard, P.; Schinke, R.; Bittererová, M.; Farantos, S.C. The vibrational energies of ozone up to the dissociation threshold: Dynamics calculations on an accurate potential energy surface. J. Chem. Phys. 2002, 116, 9749. [Google Scholar] [CrossRef]
- Babikov, D.; Kendrick, B.K.; Walker, R.B.; Pack, R.T.; Fleurat-Lesard, P.R.; Schinke, R. Metastable states of ozone calculated on an accurate potential energy surface. J. Chem. Phys. 2003, 118, 6298. [Google Scholar] [CrossRef]
- Liu, Y.; Bersuker, I.B.; Zou, W.; Boggs, J.E. Combined Jahn-Teller and pseudo Jahn-Teller effects in the CO3 molecule: A seven-state six-mode problem. J. Chem. Theory Comp. 2009, 5, 2679–2686. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.; Enachescu, M.L.; Daku, V.A.; Amstutz, N. Low-temperature lifetimes of metastable high-spin states in spin-crossover and in low-spin iron(II) compounds: The rule and exceptions to the rule. Coord. Chem. Rev. 2009, 250, 1642–1652. [Google Scholar] [CrossRef]
- Renz, F.; Oshio, H.; Ksenofontov, V.; Waldeck, M.; Spiering, H.; Gütlich, P. Strong Field Iron(II) complex converted by light into a long-lived high-spin state. Angew. Chem. Int. Ed. 2000, 39, 3699–3700. [Google Scholar] [CrossRef]
- Chibotaru, L.F.; Ceulemans, A. Orbital disproportionation of conduction-electron density in cubic lattices with threefold degenerate site orbitals. Phys. Rev. B 1996, 53, 15522. [Google Scholar] [CrossRef]
- Ceulemans, A.; Chibotaru, L.F.; Cimpoesu, F. Intramolecular Charge Disproportionation and the Band Structure of A3C60 Superconductors. Phys. Rev. Lett. 1997, 78, 3725. [Google Scholar] [CrossRef]
- Kosaka, M.; Tanigaki, K.; Hirosawa, I.; Shimakawu, Y.; Kuroshima, S.; Ebbesen, T.W.; Hizuke, J.; Kubo, Y. ESR studies of K-doped C60. Chem. Phys. Lett. 1993, 203, 429–432. [Google Scholar] [CrossRef]
- Köppel, M.; Domcke, W.; Cederbaum, L.S. Multimode molecular dynamics beyond the Born-Oppenheimer approximation. Adv. Chem. Phys. 1984, 57, 59–246. [Google Scholar]
- Venkataramani, S.; Jana, U.; Dommaschk, M.; Sönnichsen, F.D.; Tuczek, F.; Herges, R. Magnetic bistability of molecules in homogeneous solution at room temperature. Science 2011, 331, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Tergenius, L.-E. Room temperature synthesis and structural characterization on monoclinic LiCuO2 by X-ray and neutron diffraction. J. Alloys Comp. 1994, 203, 203–207. [Google Scholar] [CrossRef]
- Pickardt, J.; Paulus, W.; Schmalz, M.; Schöllhorn, C. Crystal growth and structure refinement of NaCuO2 by X-ray and neutron diffraction. J. Sol. State Chem. 1990, 89, 308–314. [Google Scholar] [CrossRef]
- Owens, F.J. Evidence of a phase transition in Cu-O chains of LiCuO2. Phys. C 1999, 313, 65–69. [Google Scholar] [CrossRef]
- Singh, D.J. Electronic structure of NaCuO2. Phys. Rev. B 1994, 49, 1580–1585. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).