1. Introduction
RE2Me1Me2O6 double-perovskite oxides, where
Me1 and
Me2 are different 3D transition metals, are finding more and more applications. Over the past year, several excellent reviews on the study of various properties of double perovskites have been published. Recent advances in the characterization of the crystal structure, magnetic properties, and cryogenic magnetocaloric effect are summarized in [
1]. The halide double perovskite with structural flexibility, as well as lower toxicity and higher stability, attracted the attention of the scientific community and stimulated the research on halide double perovskites with a lead-free A
2B(I)B(III)X
6 composition [
2]. The problems arising in the design and synthesis of double perovskites and possible ways to overcome them are discussed in [
3]. Promising applications of double perovskites for the development of high-performance photodetectors are considered in [
4].
The double perovskite Sr
2FeNbO
6 compound is being actively studied in connection with its possible application for boosting the charge separation for efficient photocatalytic CO
2 reduction in the heterostructure of Cs
2AgBiBr
6/Sr
2FeNbO
6 [
5]. Electrochemical impedance spectroscopy measurements indicated that ZrO
2-stabilized Sr
2FeNbO
6-Y
2O
3 can be used as a hydrogen electrode in high-temperature steam electrolysis [
6]. Fe
3+ atoms in the cubic lattice of Sr
2FeNbO
6 play a crucial role in yielding high visible-light photocatalytic activity [
7]. For photocatalytic applications such as pure hydrogen production and solar-to-electrical energy conversion, compounds with an energy band gap (E
g) between 1.4 and 2 eV are needed. The Sr
2FeNbO
6 ceramic with an E
g of 1.9 eV has great potential in these applications [
8]. The possibility of its use in industrial applications was considered in [
9] as an alternative fuel electrode in solid oxide electrolysis cells due to their high chemical stability. The review [
10] summarizes recent studies on Sr
2Me1Me2O
6-based double perovskites for high-temperature thermoelectric power generation. It was shown that the decoupling of phonon-glass and electron-crystal behaviors in oxides is possible by reducing thermal conductivity due to the induced dipolar glassy state as a result of relaxor ferroelectricity [
10]. It should be noted that the ionic radii of iron and niobium in a sixfold environment almost coincide (0.67 Å and 0.66 Å, respectively), which is also realized for compounds with Mo and Fe [
11]. Therefore, the authors of [
11] suggest using thin films to obtain an ordered distribution of elements. The B-site ordering double perovskites has also been widely reported in room-temperature electrolysis, such as water splitting [
12]. Doping phosphorus into perovskites is an effective way to develop high-performance electrocatalysts for the oxygen reduction reaction [
13].
Double perovskites of Sr
2FeNbO
6 were studied in multiple works [
14,
15,
16]. However, these papers show different results of structural and magnetic studies. According to [
17], Sr
2FeNbO
6-δ prepared using a conventional solid-state method features the orthorhombic structure
Pnma (
a = 5.62 Å,
b = 7.96 Å, and
c = 5.61 Å). The tetragonal structure with space group I4/m (
a = 5.61 Å and
c = 7.97 Å) was obtained via the conventional ceramic method [
18]. The orthorhombic structure with space group
Pnma and lattice parameters a = 5.6082(9) Å, b = 7.9642(1) Å, and c = 5.6084(9) Å was reported in [
14]. The cubic structure with
a = 3.99 Å was found in [
19]. Sr
2FeNbO
6 has an orthorhombic unit cell with a
Pbnm space group, and their lattice constants are
a = 0.56077(9) nm,
b= 0.56038(9) nm, and
c = 0.78991(8) nm from neutron diffraction data [
16]. The crystal structure is presented in the paper [
8]. Different transition temperatures have been reported: in [
15], Sr
2FeNbO
6-δ was shown to experience a spin glass transition at
TG = 32.5 K; in [
16], a spin glass transition was observed at
TG = 20 K. The established phase transition temperatures for a double perovskite Sr
2FeNbO
6 differ strongly from the ordering temperatures of its constituent single perovskite. SrFeO
3 has a cubic structure with
a = 3.85 Å and an antiferromagnetic transition at
TN = 134 K [
20]. Bulk SrNbO
3 is not stable under ambient conditions [
21].
Mössbauer spectra of Sr
2FeNbO
6 were measured at 298 K and 6 K in [
14]. The authors concluded that the symmetry at the Fe site in the lattice is no longer cubic. The experimental spectra were reproduced via the single-doublet fitting at room temperature with δ = 0.390(1) mm/s, ε = 0.250(1) mm/s, Γ = 0.462(4) mm/s and δ = 0.481(10) mm/s, ε = 0.001(9) mm/s, Γ = 0.894(37) mm/s, and
Bhf (T) = 48.71(7) at 6 K. However, the temperature dynamics and fluctuations of the spin moment have not been studied. The purpose of the present research is to study magnetic and transport properties of the Sr
2FeNbO
6-δ ceramics in the temperature range of 4–300 K via the Mössbauer method and the specific heat data analysis.
2. Materials and Methods
Double perovskite Sr2FeNbO6 was obtained via pyrolysis of nitrate-organic mixtures of the following components: Sr(NO3)2 (strontium nitrate, chemically pure grade), Fe (carbonyl, ultra-pure grade), Nb2O5 (niobium oxide, chemically pure grade), NH4OH (chemically pure grade), HNO3 (chemically pure grade), and C5H7(OH)5 (chemically pure grade). The compound C5H7(OH)5 (pentanpentaol, xylitol) as an organic component promotes the dissolution of niobium oxide in an alkaline medium and simultaneously ensures the pyrolysis process in the self-ignition mode of the solution.
Iron nitrate (II) is obtained by dissolving a stoichiometric amount of carbonyl iron in dilute (1:1) nitric acid with low heating. Sr(NO3)2 was dissolved in distilled water. The resulting solutions were mixed. Next, a stoichiometric amount of niobium oxide was added to the reaction mixture. With constant stirring, the solution was neutralized to pH = 7 with ammonia, then the pH of the solution was slowly adjusted to 12.5. After that, with constant stirring, the compound C5H7(OH)5 was added to the mixture in the ratio Nb2O5:C5H7(OH)5 = 1:5 mol. The reaction mass was kept for 9–10 h until maturation. The next day, another 4–5 g of C5H7(OH)5 was added as an organic fuel, and the solution was evaporated before the combustion process started. The pyrolysis product is a black ultrafine powder, which was calcined at 950 °C for 1.5–2 h to remove carbon impurities. The resulting sample was pelletized and calcined at 1100–1150 °C for 8 hours. The total synthesis time was two days.
X-ray examination (
Figure 1) was performed using a Shimadzu XRD-7000 S automatic diffractometer with an exposure time of 3–5 s per point. X-ray pattern processing was performed with the FULLPROF-2018 software. According to X-ray diffraction data, the resulting product is the double perovskite Sr
2FeNbO
6-δ. We have not detected any foreign impurities and impurities of unreachable substances. The Sr
2FeNbO
6-δ phase diffraction pattern can be described based on two space groups. The first of which is the Pm3m space group with
a = 3.96748(8)Å, which is consistent with [
19]. X-ray diffraction pattern is presented for the cubic group in
Figure 1a. The Rietveld refined crystal structural parameters are summarized in
Table 1. The second possible description of the X-ray diffraction pattern is given within the Pbnm space group and lattice parameters
a =
b = 5.605(7);
c = 7.929(4)
V and Ǻ
3 = 250.28(2). An X-ray diffraction pattern is presented for the Pbnm group in
Figure 1b. The Rietveld refined crystal structural parameters are summarized in
Table 2.
XFA measurements performed via the Bruker S2 Ranger X-ray fluorescence spectrometer allowed us to determine the elements ratio as Fe/Sr/Nb = 0.17:0.55:0.28. Based on the electrical neutrality of the sample, we calculated the oxygen content as 5.4 for the Fe3+, Sr2+, and Nb5+ valences. The real formula of the sample is Sr2NbFe0.6O5.4. Oxygen deficiency is observed in the sample according to XFA measurements, so the Pbnm space group is more suitable for describing the structure Sr2NbFe0.6O5.4.
Mössbauer studies of the Sr
2FeNbO
6-δ sample were performed on a WissEl spectrometer (Germany) operating in the constant acceleration regime. As a source of resonant gamma radiation, we used
57Co in a rhodium matrix with an activity of ~ 50 mCi (Ritverc, St. Petersburg, Russia). For low-temperature Mössbauer measurements, a CFICEV flow cryostat (ICE Oxford, UK) with a CryoCon 32B temperature controller was used, which makes it possible to maintain the sample temperature with an accuracy of ±0.2 K. The absorber was made from a finely ground powder of the investigated compound mixed with boron nitride to obtain a sample uniform in thickness and to disorder the orientation of microcrystallites. For measurements, we used a sample with a surface density of 27 mg/cm
2. The velocity scale of the spectrometer was calibrated using the spectrum of a thin metallic iron foil. The values of isomer shifts are given relative to the gravity center of the metallic iron spectrum at room temperature. The spectra were processed using the SpectrRelax 2.1 software [
22].
Specific heat capacity measurements were performed using the PPMS-9 device in the 5–300 K temperature range at 0 and 9 T.
4. Discussion
The asymmetry of the quadrupole lines of Mössbauer spectra can be driven by several reasons, one of which can be related to the texture of the sample at room temperature. To test this assumption, we recorded the spectrum at two orientations of the sample, (i) when the wavevector of the resonant quanta is normal to the sample plane and (ii) when it makes a “magic” angle φ = 54.7
o with the normal to the sample plane, i.e., at an orientation that equalizes the amplitudes of the doublet lines, if the asymmetry of the lines is due to the texture of the sample. As can be seen from
Figure 8, a change in the orientation did not lead to a change in the amplitudes of doublet lines. Therefore, the observed asymmetry can be associated with the presence of centers of iron atoms with different local environments. This assumption is also consistent with the fact that the width of the more intense left doublet line (Γ
L = 0.47 mm/s) is greater than the width of the right line (Γ
R = 0.43 mm/s).
To determine the number of iron centers and calculate their relative content, the Mössbauer spectrum was fitted under the assumption that there are two doublets with a distribution of quadrupole shifts. The inset in
Figure 2a shows the distribution function reconstructed from the experimental spectrum using the SpectrRelax 2.1 software. As can be seen, the distribution function consists of two peaks corresponding to two centers of iron ions in the sample. The isomer shifts of these centers are quite close and correspond to Fe
3+ ions in an octahedral environment. The distribution peaks for the Fe
13+ at IS ~ 0.356 mm/s, while for the Fe
23+ center, it peaks at IS ~ 0.374 mm/s. Although the isomer shifts of these centers are almost equal, the quadrupole shifts are noticeably different (ε ~ 0.2 mm/s for Fe
23+ and ε ~ 0.3 mm/s for Fe
13+) and have a wide distribution.
Oxygen octahedral around iron is somewhat deformed from the ideal, according to the space group Pbnm. This leads to the appearance of groups of iron atoms with different local environments. This difference is small, however, in the Mössbauer spectra; it leads to an asymmetry of the experimental doublet spectrum and to the appearance of two Fe3+ centers. Of course, the peaks of these centers in the distribution function are blurred due to the influence of Nb5+ and non-stoichiometry of the studied sample with oxygen.
Figure 2b,c show the distribution functions of quadrupole shifts for the Fe
13+ and Fe
23+ centers obtained via fitting the experimental spectra using the two-doublet (i.e., two-center) model.
The fitting procedure yielded the values of the isomer shift and the average value of the quadrupole shift at room temperature for Fe13+ IS = 0.35(6) mm/s and <ε> = 0.19(8) mm/s, respectively. The hyperfine parameters for the Fe23+ center are IS = 0.37(4) mm/s and <ε> = 0.32(0) mm/s, respectively. The Fe13+ and Fe23+ centers account for 61% and 39% of the total area of the spectrum, respectively.
The change in the position of the center of gravity of the absorption lines with temperature is determined in the Debye approximation using the function.
where the first term
ISs(T) depends on the density of s-electrons in the
57Fe ion, the second term is due to the second-order Doppler effect,
m is the mass of the
57Fe nuclide,
k is the Boltzmann constant, and
c is the speed of light in a vacuum. The first term, which is determined only with the composition and almost independent on temperature, can be considered constant and denoted as
ISs (0). Therefore, the temperature dependence of the absorption-line position, i.e., the isomer shift, is completely determined with the second-order Doppler shift. In
Figure 5a, the solid line represents the result of fitting the experimental data using Formula (4). For the identified Fe
3+ centers, the best fit is achieved with
θD = 445 ± 6 K and
ISs(0) = 0.59 ± 0.01 mm/s. The
θD = 445 ± 6 K value is close to the Einstein temperature ~490 K obtained via fitting the temperature dependence of the specific heat capacity.
As can be seen from
Figure 6, a decrease in the sample temperature leads to the fact that for some paramagnetic ions the magnetic correlation energy becomes greater than thermal energy. Also, regions of magnetic ordering appear, leading to a decrease in both the intensity and area of doublets and forming a sextet structure of absorption lines for magnetically ordered ions.
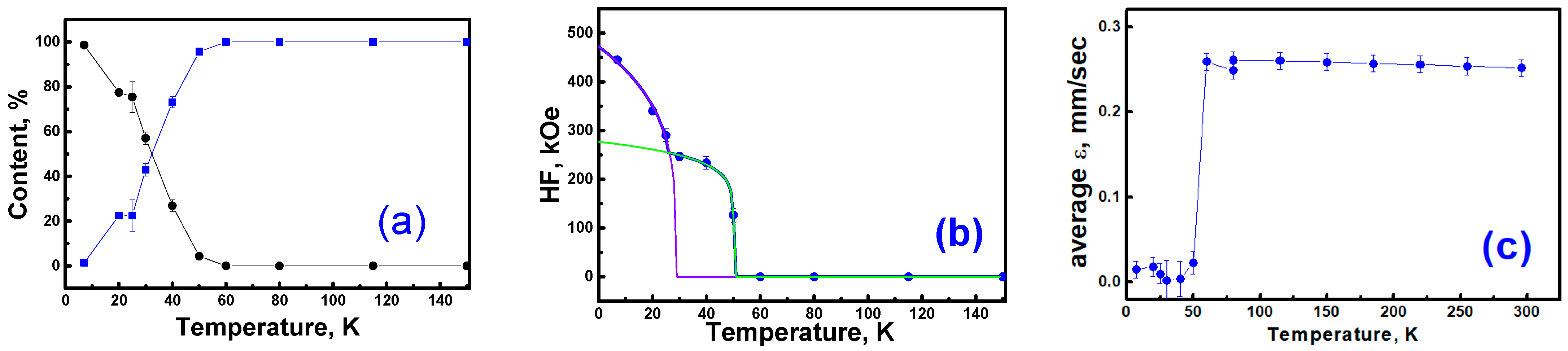
Figure 9a shows the temperature dependence of the percentage of paramagnetic and magnetically ordered ions in the sample at various temperatures. As can be seen, this process occurs in a temperature range below 40 K due to the existence of magnetic clusters with varied sizes of magnetic correlations.
At 40 K, this component is shown by the blue line (
Figure 6). As the temperature decreases, the area of this partial component increases, and already at 25 K and below, it acquires a clear sextet structure whose lines narrow with decreasing temperature (
Figure 6). The insets in
Figure 6 show the distributions of hyperfine fields, which provide the best fits of the experimental spectra. Along with this magnetic component, even at a temperature of 7 K (
Figure 6), the spectra of the Sr
2FeNbO
6-δ sample retain the doublet structure due to paramagnetic Fe
3+ ions, which are observed at higher temperatures.
Due to this feature, the Mössbauer spectra in the temperature range under consideration demonstrate a complex profile, which is the sum of the contributions from already magnetically ordered ions with a sextet structure and the contributions from ions characterized by a relaxation line shape. In such cases, the method of restoring the distribution of hyperfine parameters is often used to determine the averaged hyperfine parameters from the experimental spectra. The insets in
Figure 6 show the distribution functions of hyperfine fields on
57Fe nuclei, which provide the best values of χ
2 for spectra taken at 7, 20, 25, 30, and 40 K.
The uncertainty of the distribution function values is denoted by error bars in
Figure 6. As can be seen, the hyperfine magnetic field distribution function is determined with the highest accuracy at 7 K. It features a Gaussian-like shape with asymmetric wings towards low and high magnetic-field values.
Figure 9b shows the temperature dependence of the average hyperfine magnetic field on
57Fe nuclei. Unfortunately, the insufficient number of experimental points in the temperature dependence of the average hyperfine magnetic field does not allow us to unambiguously determine the details of the mechanism of antiferromagnetic ordering of iron atoms in this sample. Nevertheless, it follows from Mössbauer measurements at
T = 7 K that the sample contains two types of magnetic particles that differ in length and strength of magnetic correlations.
In addition, the magnetic moment of magnetically ordered atoms will depend on the number of nearest Fe neighbors involved in the indirect exchange interaction. As this number decreases, the hyperfine magnetic field on the nucleus decreases, leading to the appearance of contributions to the resonant absorption in the inner part of the spectrum. This feature is characterized in the distribution function of hyperfine fields by the extension of the wing of the Gaussian-like distribution toward weaker magnetic fields. The shape of the temperature dependence of the observed average hyperfine field on Fe
3+ nuclei suggests the presence of two types of magnetic clusters with different critical ordering temperatures.
Figure 9b shows the result of the approximation of the experimental data using the following power function:
where
H1 and
H2 are the values of hyperfine fields at saturation,
T1 and
T2 are the critical temperatures for a given type of magnetic clusters, and
b1 and
b2 are the exponents. The best fits of the experimental data yielded the following values:
H1 = 472 kOe,
H2 = 277 kOe,
T1 = 29 K,
T2 = 50 K,
b1 = 0.25, and
b2 = 0.12. In
Figure 9b, these components are shown as solid violet and green lines.
Another feature in low-temperature Mössbauer spectra is the symmetrical shape of the experimental spectra without a clear manifestation of quadrupole splitting. At higher temperatures (
T > 50 K), the observed average quadrupole shift is ~0.25 mm/s (
Figure 9c). However, with the onset of the magnetic ordering process, i.e., at temperatures below 40 K, the average value of the quadrupole splitting of magnetically ordered ions becomes negligibly small (
Figure 9c). As is known, in the presence of a hyperfine magnetic field, the changes caused by the quadrupole splitting strongly depend on both the magnitude and the mutual orientation of the axis of the electric field gradient tensor (EFG) on the nucleus with respect to the direction of the hyperfine magnetic field. When the quadrupole interaction is much weaker than the magnetic hyperfine interaction, the quadrupole splitting can be expressed using the following formula:
where
θ and
φ are the angles between the direction of the magnetic hyperfine field and the main axis of the EFG tensor. In the case of the random orientation of these vectors, the line shifts caused by the QS quadrupole interaction will be zero on average. Consequently, in our sample, upon the onset of magnetic ordering, the directions of the magnetic moments of the iron ions, and hence the directions of hyperfine magnetic fields on the Fe nuclei, are randomly distributed with respect to the directions determined with the principal axes of the EFG tensor. Mössbauer spectroscopy showed three different environments around iron [
24] for Sr
2FeMo
1−xNb
xO
6 compounds at room temperature.
The blurring of the phase transition temperature is confirmed by the specific heat measured in the whole temperature range, which shows no λ-type anomaly that can indicate a transition to a magnetically ordered state.
Figure 7a shows the Cp(T)/T dependence at
B = 0 and 9 T in temperature range of 4–300 K, where the red line corresponds to the lattice contribution to the specific heat
Cph. The blue and green lines show the difference between the experimental values of the specific heat capacity and the lattice contribution. This difference corresponds to the magnetic contribution at magnetic fields 0 and 9 T. For clarity, the magnetic contribution to the specific heat Cm is shown in
Figure 7b. The magnetic contribution to the heat capacity demonstrates two maxima, at 4 K and 36 K. The positions of the maxima of the magnetic contribution do not change in the external magnetic field of 9 T. The observation of a peak at 36 K in the magnetic specific heat capacity is consistent with the temperature dependence of average hyperfine magnetic field for one of the magnetic subsystems. This temperature almost coincides with the phase transition temperature
TG = 32.5 K obtained from the discrepancy of the temperature dependence of the magnetic susceptibility measured in ZFC and FC regimes in [
13]. The authors of [
13] found that below 700 K an extrapolated Curie temperature is negative and is equal to 28 K. The negative value of the Curie temperature indicates the antiferromagnetic nature of the exchange interactions between the spins.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}