

On the Electrochemical Properties of Carbon-Coated NaCrO2 for Na-Ion Batteries




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Material Synthesis
2.2. Material Characterization
2.3. Battery Fabrication and Evaluation
3. Results and Discussion
3.1. SEM and TEM Characterization
3.2. X-ray Diffraction Analysis
3.3. Raman Spectroscopy Results
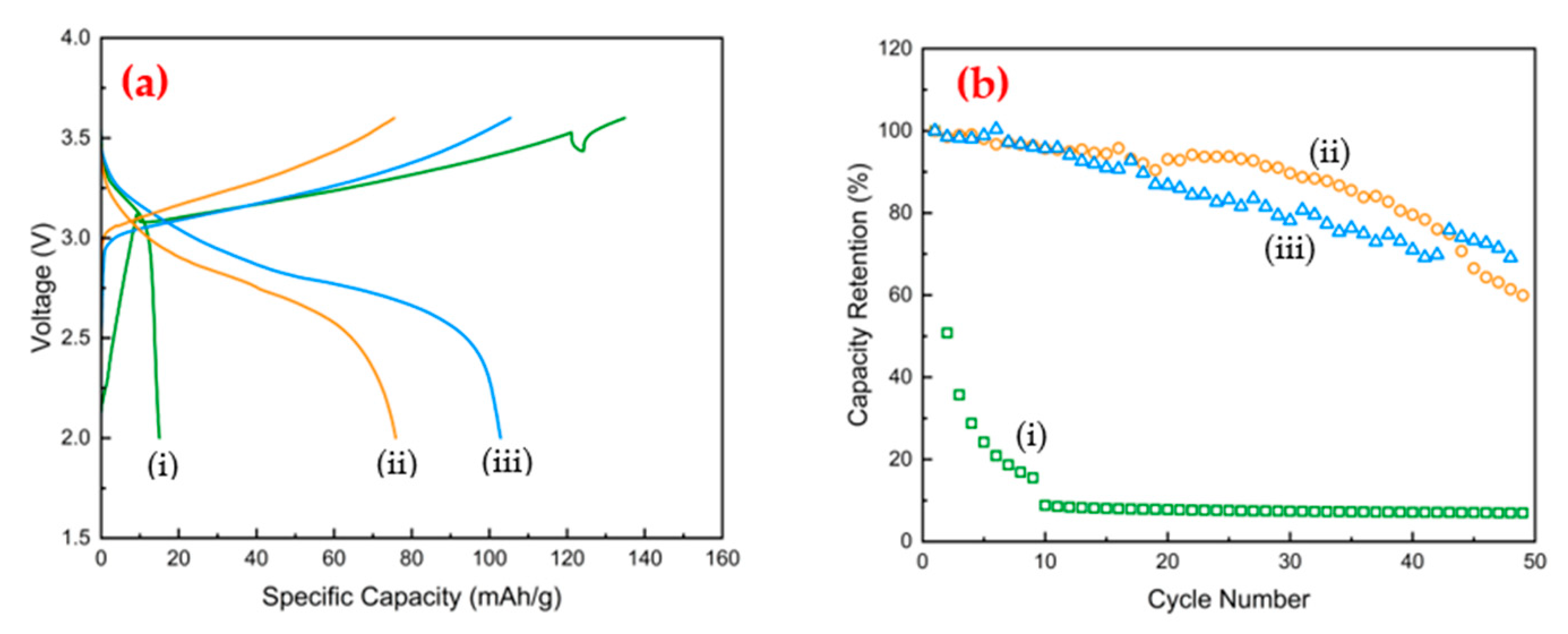
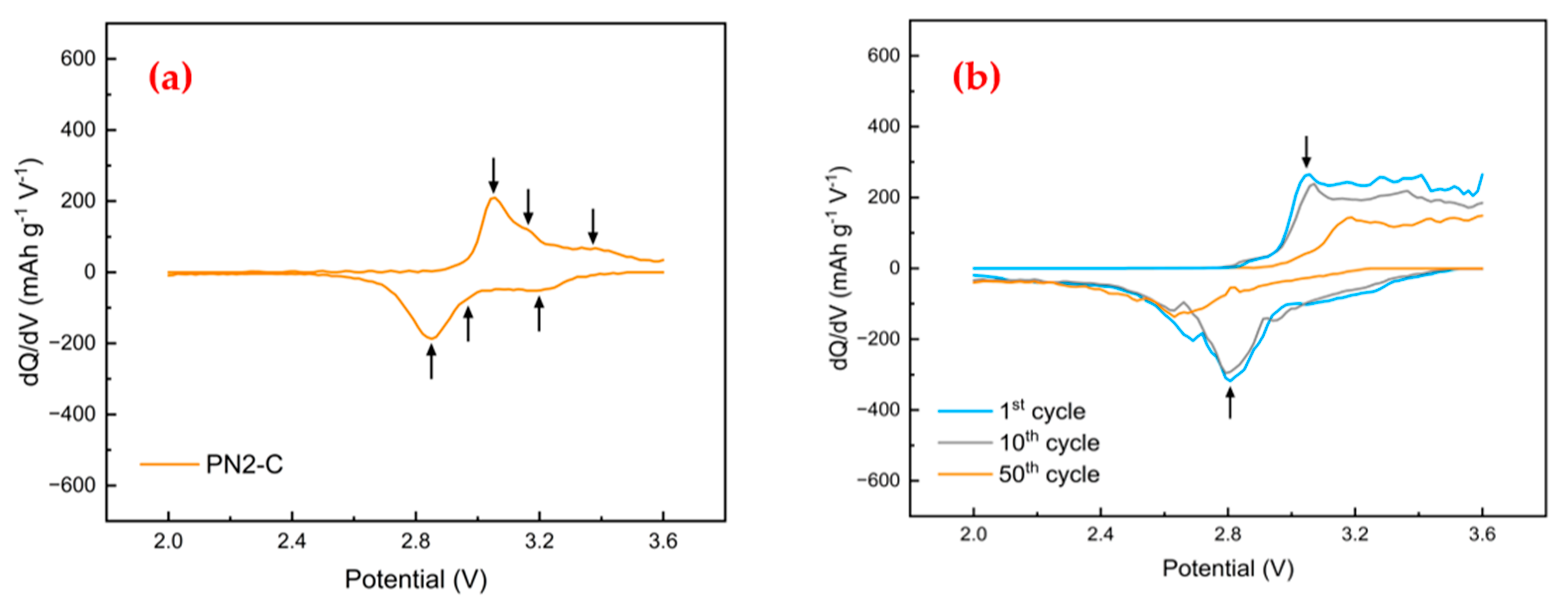
3.4. Battery Cell Evaluation
4. Concluding Remarks
- NaCrO2 reacts with carbon precursors such as PAN at 750 and 900 °C to form carbides. Thus, to form a carbon coating on NaCrO2, the carbonization temperature should be at 650 °C, at which there is no reaction of NaCrO2 with carbon precursors and in-situ formed carbon.
- One-step reaction to synthesize NaCrO2 crystals and carbon coating simultaneously can be done at 900 °C without the formation of carbides because the gas evolution in forming NaCrO2 via the reaction between Na2CO3 and Cr2O3 creates a gaseous envelope and prevents the reactions between the newly formed NaCrO2 with the carbon precursor and in-situ formed carbon. However, such carbon coating does not have high quality because the gas evolution during the formation of NaCrO2 could break the carbon coating on the surface of NaCrO2 and prevent conformal contact between them.
- Ultrafine NaCrO2 particles (diameter < 600 nm) require high CB loading (20–30 wt.% CB) in the electrode to achieve the typical specific capacity of 110 mAh/g.
- Carbon-coated NaCrO2 particles formed via dry powder mixing offer higher specific capacities and better cycle stability than bare NaCrO2, even with the CB loading at 10 wt.%.
- Carbon-coated NaCrO2 particles formed via wet solution mixing provide unprecedented specific capacities at 140 mAh/g, which is substantially higher than the typical specific capacity of 110 mAh/g with the LCV and UCV at 2.0 and 3.6 V vs. Na/Na+, respectively.
- The specific capacity at 140 mAh/g achieved with carbon-coated NaCrO2 unambiguously reveals that the typical specific capacity of 110 mAh/g reported by many researchers is controlled by the kinetic factor, i.e., the electronic conductivity at the electrode/electrolyte interface is the rate-limiting step for Na-ion de/intercalation of NaCrO2.
- Although carbon-coated NaCrO2 particles formed via wet solution mixing provide unprecedented specific capacities, cycle stability is not improved because with the specific capacity at 140 mAh/g the Na deintercalation during charge is more than 50% Na ions per formula unit of NaCrO2, which leads to irreversible redox reactions.
- Based on the present study, one possible direction to improve the cycle stability of NaCrO2 in the future is the integration of carbon coating and doping, which remains to be investigated in the future.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, P.F.; You, Y.; Yin, Y.X.; Guo, Y.G. Layered Oxide Cathodes for Sodium-Ion Batteries: Phase Transition, Air Stability, and Performance. Adv. Energy Mater. 2018, 8, 1701912. [Google Scholar] [CrossRef]
- Han, M.H.; Gonzalo, E.; Singh, G.; Rojo, T. A Comprehensive Review of Sodium Layered Oxides: Powerful Cathodes for Na-Ion Batteries. Energy Environ. Sci. 2015, 8, 81–102. [Google Scholar] [CrossRef]
- Xia, X.; Dahn, J.R. NaCrO2 Is a Fundamentally Safe Positive Electrode Material for Sodium-Ion Batteries with Liquid Electrolytes. Electrochem. Solid State Lett. 2012, 15, 2012–2015. [Google Scholar] [CrossRef]
- Zhou, Y.N.; Ding, J.J.; Nam, K.W.; Yu, X.; Bak, S.M.; Hu, E.; Liu, J.; Bai, J.; Li, H.; Fu, Z.W.; et al. Phase Transition Behavior of NaCrO2 during Sodium Extraction Studied by Synchrotron-Based X-Ray Diffraction and Absorption Spectroscopy. J. Mater. Chem. A 2013, 1, 11130–11134. [Google Scholar] [CrossRef]
- Komaba, S.; Takei, C.; Nakayama, T.; Ogata, A.; Yabuuchi, N. Electrochemical Intercalation Activity of Layered NaCrO2 vs. LiCrO2. Electrochem. Commun. 2010, 12, 355–358. [Google Scholar] [CrossRef]
- Chen, C.Y.; Matsumoto, K.; Nohira, T.; Hagiwara, R.; Fukunaga, A.; Sakai, S.; Nitta, K.; Inazawa, S. Electrochemical and Structural Investigation of NaCrO2 as a Positive Electrode for Sodium Secondary Battery Using Inorganic Ionic Liquid NaFSA-KFSA. J. Power Sources 2013, 237, 52–57. [Google Scholar] [CrossRef]
- Ding, J.J.; Zhou, Y.N.; Sun, Q.; Fu, Z.W. Cycle Performance Improvement of NaCrO2 Cathode by Carbon Coating for Sodium Ion Batteries. Electrochem. Commun. 2012, 22, 85–88. [Google Scholar] [CrossRef]
- Komaba, S.; Nakayama, T.; Ogata, A.; Shimizu, T.; Takei, C.; Takada, S.; Hokura, A.; Nakai, I. Electrochemically Reversible Sodium Intercalation of Layered NaNi0.5Mn0.5O2 and NaCrO2. ECS Trans. 2009, 16, 43–55. [Google Scholar] [CrossRef]
- Kubota, K.; Ikeuchi, I.; Nakayama, T.; Takei, C.; Yabuuchi, N.; Shiiba, H.; Nakayama, M.; Komaba, S. New Insight into Structural Evolution in Layered NaCrO2 during Electrochemical Sodium Extraction. J. Phys. Chem. C 2015, 119, 166–175. [Google Scholar] [CrossRef]
- Luo, M.; Ortiz, A.L.; Guo, F.; Shi, Z.; Li, L.; Ren, Y.; Zhang, X.; Chen, Z.; Shaw, L.; Chen, W. Mechanical Activation Enhanced Solid-State Synthesis of NaCrO2 Cathode Material. Materialia 2019, 5, 100172. [Google Scholar] [CrossRef]
- Sawicki, M.; Ortiz, A.L.; Luo, M.; Shaw, L. Structural-Defect-Controlled Electrochemical Performance of Sodium Ion Batteries with NaCrO2 Cathodes. ChemElectroChem 2017, 4, 3222–3230. [Google Scholar] [CrossRef]
- Luo, M.; Ortiz, A.L.; Shaw, L. Unraveling Processing-Structure-Electrical Conductivity Relationships of NaCrO2 Cathodes for Na-Ion Batteries. J. Electrochem. Soc. 2019, 166, 3546–3553. [Google Scholar] [CrossRef]
- Luo, M.; Ortiz, A.L.; Shaw, L. Enhancing the Electrochemical Performance of NaCrO2 through Structural Defect Control. ACS Appl. Energy Mater. 2020, 3, 7216–7227. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Park, J.-S.; Jung, H.-G.; Chung, K.-Y.; Aurbach, D.; Sun, Y.-K.; Myung, S.-T. NaCrO2 Cathode for High-Rate Sodium-Ion Batteries. Energy Environ. Sci. 2015, 8, 2019–2026. [Google Scholar] [CrossRef]
- Li, W.; Wang, Y.; Hu, G.; Peng, Z.; Cao, Y.; Zeng, Y.; Du, K. Ti-Doped NaCrO2 as Cathode Materials for Sodium-Ion Batteries with Excellent Long Cycle Life. J. Alloys Compd. 2019, 779, 147–155. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, P.; Zhu, W.; Feng, Z.; Vigeant, M.J.; Demers, H.; Guerfi, A.; Zaghib, K. Enhancing the Electrochemical Performance of an O3–NaCrO2 Cathode in Sodium-Ion Batteries by Cation Substitution. J. Power Sources 2019, 435, 226760. [Google Scholar] [CrossRef]
- Zheng, L.; Bennett, J.C.; Obrovac, M.N. Stabilizing NaCrO2 by Sodium Site Doping with Calcium. J. Electrochem. Soc. 2019, 166, A2058–A2064. [Google Scholar] [CrossRef]
- Wang, Y.; Li, W.; Hu, G.; Peng, Z.; Cao, Y.; Gao, H.; Du, K.; Goodenough, J.B. Electrochemical Performance of Large-Grained NaCrO2 Cathode Materials for Na-Ion Batteries Synthesized by Decomposition of Na2Cr2O7·2H2O. Chem. Mater. 2019, 31, 5214–5223. [Google Scholar] [CrossRef]
- Liang, L.; Sun, X.; Denis, D.K.; Zhang, J.; Hou, L.; Liu, Y.; Yuan, C. Ultralong Layered NaCrO2 Nanowires: A Competitive Wide-Temperature-Operating Cathode for Extraordinary High-Rate Sodium-Ion Batteries. ACS Appl. Mater. Interfaces 2019, 11, 4037–4046. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef]
- Wu, J.; Hu, G.; Du, K.; Peng, Z.; Huang, M.; Fan, J.; Gong, Y.; Guan, D.; Shi, Y.; Liu, R.; et al. Inhibiting Electrochemical Phase Transition of NaCrO2 with Long-Cycle Stability by Surface Fluorination Treatment. Electrochim. Acta 2022, 403, 139641. [Google Scholar] [CrossRef]
- Wang, S.; Chen, F.; Zhu, T.Y.; He, X.D.; Liao, J.Y.; Zhang, L.M.; Ding, X.; Hu, Q.; Chen, C.H. In Situ-Formed Cr2O3 Coating on NaCrO2 with Improved Sodium Storage Performance. ACS Appl. Mater. Interfaces 2020, 12, 44671–44678. [Google Scholar] [CrossRef] [PubMed]
- Ikhe, A.B.; Park, W.B.; Manasi, M.; Ahn, D.; Sohn, K.S.; Pyo, M. Unprecedented Cyclability and Moisture Durability of NaCrO2 Sodium-Ion Battery Cathode via Simultaneous Al Doping and Cr2O3 Coating. ACS Appl. Mater. Interfaces 2023, 15, 14958–14969. [Google Scholar] [CrossRef]
- Song, T.; Chen, L.; Gastol, D.; Dong, B.; Marco, J.F.; Berry, F.; Slater, P.; Reed, D.; Kendrick, E. High-Voltage Stabilization of O3-Type Layered Oxide for Sodium-Ion Batteries by Simultaneous Tin Dual Modification. Chem. Mater. 2022, 34, 4153–4165. [Google Scholar] [CrossRef]
- Xie, M.; Luo, R.; Lu, J.; Chen, R.; Wu, F.; Wang, X.; Zhan, C.; Wu, H.; Albishri, H.M.; Al-Bogami, A.S.; et al. Synthesis-Microstructure-Performance Relationship of Layered Transition Metal Oxides as Cathode for Rechargeable Sodium Batteries Prepared by High-Temperature Calcination. ACS Appl. Mater. Interfaces 2014, 6, 17176–17183. [Google Scholar] [CrossRef] [PubMed]
- Darjazi, H.; Bottoni, L.; Moazami, H.R.; Rezvani, S.J.; Balducci, L.; Sbrascini, L.; Staffolani, A.; Tombesi, A.; Nobili, F. From Waste to Resources: Transforming Olive Leaves to Hard Carbon as Sustainable and Versatile Electrode Material for Li/Na-ion Batteries and Supercapacitors. Mater. Today Sustain. 2023, 21, 100313. [Google Scholar] [CrossRef]
- Nagpure, S.C.; Bhushan, B.; Babu, S.S. Raman and NMR Studies of Aged LiFePO4 Cathode. Appl. Surf. Sci. 2012, 259, 49–54. [Google Scholar] [CrossRef]
- Ren, Y.; Xiang, L.; Yin, X.; Xiao, R.; Zuo, P.; Gao, Y.; Yin, G.; Du, C. Ultrathin Si Nanosheets Dispersed in Graphene Matrix Enable Stable Interface and High Rate Capability of Anode for Lithium-ion Batteries. Adv. Funct. Mater. 2022, 32, 2110046. [Google Scholar] [CrossRef]
- Merlen, A.; Buijnsters, J.G.; Pardanaud, C. A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: From Graphene to Amorphous Carbons. Coatings 2017, 7, 153. [Google Scholar] [CrossRef]
- Liu, W.R.; Guo, Z.Z.; Young, W.S.; Shieh, D.T.; Wu, H.C.; Yang, M.H.; Wu, N.L. Effect of Electrode Structure on Performance of Si Anode in Li-ion Batteries: Si Particle Size and Conductive Additive. J. Power Sources 2005, 140, 139–144. [Google Scholar] [CrossRef]
- Wang, C.; Sawicki, M.; Emani, S.; Liu, C.; Shaw, L. Na3MnCO3PO4—A High Capacity, Multi-Electron Transfer Redox Cathode Material for Sodium Ion Batteries. Electrochim. Acta 2015, 161, 322–328. [Google Scholar] [CrossRef]
- Jakobsen, C.L.; Brighi, M.; Andersen, B.P.; Ducrest, G.; Černý, R.; Ravnsbæk, D.B. Expanded Solid-Solution Behavior and Charge-Discharge Asymmetry in NaxCrO2 Na-ion Battery Electrodes. J. Power Sources 2022, 535, 231317. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Takanashi, K.; Nishinobo, T.; Hokura, A.; Yonemura, M.; Matsukawa, T.; Ishigaki, T.; Yamanaka, K.; Ohta, T.; Yabuuchi, N. Layered NaxCrxTi1–xO2 as Bifunctional Electrode Materials for Rechargeable Sodium Batteries. Chem. Mater. 2016, 28, 7006–7016. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Carbon Source | First Treatment | Second Treatment | CB Loading in Electrode | First Discharge Capacity | ||
|---|---|---|---|---|---|---|---|
| Temperature | Time | Temperature | Time | ||||
| B1 | None | 750 °C | 5 h | N/A | N/A | N/A | N/A |
| B2 | None | 900 °C | 5 h | N/A | N/A | N/A | N/A |
| B3 | None | 900 °C | 2 h | N/A | N/A | 10 wt.% 20 wt.% 30 wt.% | 17 mAh/g 78 mAh/g 102 mAh/g |
| PN1-C | PAN | 750 °C | 5 h | N/A | N/A | 10 wt.% | 39 mAh/g |
| PN2-C | PAN | 900 °C | 5 h | N/A | N/A | 10 wt.% | 56 mAh/g |
| PN3-C | PAN | 900 °C | 2 h | 750 °C | 1 h | N/A | N/A |
| PN4-C | PAN | 900 °C | 2 h | 900 °C | 1 h | N/A | N/A |
| ca-C | Citric acid | 900 °C | 2 h | 650 °C | 10 min | N/A | N/A |
| s1-C | Dry sucrose | 900 °C | 2 h | 650 °C | 10 min | 10 wt.% | 60 mAh/g |
| s2-C | Sucrose in solution | 900 °C | 2 h | 650 °C | 1 h | 10 wt.% | 141 mAh/g |
| Sample ID | Peak Position of (003) | d Spacing of (003), Å | Peak Position of (104) | d Spacing of (104), Å | a Parameter (Å) | c Parameter (Å) |
|---|---|---|---|---|---|---|
| B1 | 16.7635° | 5.2843 | 41.7791° | 2.1603 | 2.9754 | 15.8529 |
| B2 | 16.6324° | 5.3256 | 41.6376° | 2.1673 | 2.9793 | 15.9769 |
| B3 | 16.6526° | 5.3192 | 41.6982° | 2.1643 | 2.9750 | 15.9577 |
| Sample ID | PN1-C | ca-C | s1-C |
| ID/IG Ratio | 0.62 | 0.47 | 0.42 |
| D-band FWHM (cm−1) | 124.30396 | 193.6068 | 139.47941 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Z.; Wang, Z.; Shaw, L.L.; Ashuri, M. On the Electrochemical Properties of Carbon-Coated NaCrO2 for Na-Ion Batteries. Batteries 2023, 9, 433. https://doi.org/10.3390/batteries9090433
Shi Z, Wang Z, Shaw LL, Ashuri M. On the Electrochemical Properties of Carbon-Coated NaCrO2 for Na-Ion Batteries. Batteries. 2023; 9(9):433. https://doi.org/10.3390/batteries9090433
Chicago/Turabian StyleShi, Zhepu, Ziyong Wang, Leon L. Shaw, and Maziar Ashuri. 2023. "On the Electrochemical Properties of Carbon-Coated NaCrO2 for Na-Ion Batteries" Batteries 9, no. 9: 433. https://doi.org/10.3390/batteries9090433
APA StyleShi, Z., Wang, Z., Shaw, L. L., & Ashuri, M. (2023). On the Electrochemical Properties of Carbon-Coated NaCrO2 for Na-Ion Batteries. Batteries, 9(9), 433. https://doi.org/10.3390/batteries9090433