1. Introduction
The energy transition relies on the development of technologies that make it possible to produce energy in a sustainable manner from resources such as wind, sun, potential energy, etc. The energy produced as part of the energy transition is often intermittent, and it is, therefore, necessary to be able to store and restore it reversibly. Electric mobility is also a major contributor to reducing the impacts of human activity on the environment and the climate since it contributes to reducing greenhouse gas emissions. Lithium-ion batteries (LiBs) are at the heart of energy storage for stationary applications and for electric mobility (electric vehicles, EVs) [
1,
2]. They are now widely used in phones, laptops, portable tools, etc., and their increasing use in EVs is indisputable (about 3 million new electric cars were registered in 2020 including 1.4 million new registrations in Europe despite the pandemic [
3]). It is expected that this market (and the associated LiBs market) will continue to grow in the next decades under the impulsion of the energy transition and since EV prices will reach parity with fossil fuel-powered autos in 2025. Lithium is mainly extracted from primary resources such as ores (spodumene, lepidolite, petalite and amblyogonite) and brines [
4,
5,
6]. Although mining is essential to meet the raw material demand for LiBs production, recycling can contribute to face up the future demand in lithium, cobalt, nickel, manganese and graphite arising from the huge increase in electric vehicle production in the next decade (for LiBs application, the cobalt, nickel and lithium demand is forecasted to increase by 180%, 900% and 1000% between 2019 and 2030, respectively [
3]).
Currently, the recycling of lithium-ion batteries is carried out by pyrometallurgy [
7] but more and more hydrometallurgical processes are developed [
8,
9]. The pyrometallurgical processes consist in putting lithium-ion batteries usually without preliminary dismantling in furnace at high temperature (around 1400 °C), leading to the production of a slag containing lithium and aluminum, and an alloy containing copper, iron, nickel, manganese and cobalt, which can be processed by hydrometallurgy. The hydrometallurgical route relies on deep-discharge, dismantling, grinding, leaching and selective extraction of metals by means of ion-exchange resins, solvent extraction and/or precipitation. The pyrometallurgy route is energy-intensive, responsible for loss of valuable metals in slags, loss of graphite as CO
2 and is unable to separate metals directly whereas hydrometallurgical technologies are selective, less energy-consuming and allow reducing of production of effluents [
5]. Many researches focused on the investigation of alternative unit operations to improve the recycling process efficiency while reducing the process cost and the environmental impact. For instance, the development of unit operations based on the use of microorganisms—ultrawave, deep-eutectic solvents to leach cathodic materials as well as ionic liquids—deep-eutectic solvents or supercritical fluids to extract and separate valuable metals from cathodic materials, are under investigation [
10,
11,
12,
13]. Electrodialysis (ED) is another relevant technology which is widely used in desalination processes [
14,
15]. This technology may advantageously reduce the environmental impact, the capital expenditure and operation expenditure of hydrometallurgical processes. However, few studies concern its use in hydrometallurgical processes, and even less address the use of ED in lithium-ion battery recycling processes [
13,
16,
17,
18]
Lizuka et al. [
16] showed that the use of Selemion CMV cation exchange membrane and BP-1E bipolar membrane in electrodializer allowed in recovering selectively 99% lithium(I) from a leaching solution containing 138.82 mg L
−1 lithium(I) and 1.179 g L
−1 cobalt(II) produced by digesting LiCoO
2 in 1 mol L
−1 nitric acid in the presence of 0.8 (vol) % of H
2O
2, providing that cobalt(II) was previously complexed by ethylenediaminetetraacetic acid (EDTA) at pH = 4. Likewise, Afifah et al. [
17] studied the lithium(I)-cobalt(II) separation in nitric acid by electrodialysis by using five pairs of monovalent selective ions exchange membranes (PC Cell membranes PC-MVK/PC-MVA). The separation efficiency was about 99% and a purity of 95.7% was achieved by applying 3 V/cell and a flowrate of 15 L h
−1. Song and Zhao (2018) [
16] combined precipitation and electrodialysis operations to produce lithium carbonate from acidic sulfate solution at pH 4.1 containing 3.25 g L
−1 Li, 0.25 g L
−1 Co, 0.28 g L
−1 Mn, 0.25 g L
−1 Ni, 90 g L
−1 SO
42−, 0.23 g L
−1 Al, 0.46 g L
−1 Ca, 0.68 g L
−1 Cu, 0.57 g L
−1 Fe, 0.79 g L
−1 Mg, 0.78 g L
−1 Zn, 17.5 g L
−1 Cl and 34 g L
−1 Na. Firstly, NaOH was used to precipitate Ca, Mg, Mn, Ni, Co, Zn, Fe, Al and Cu at pH = 12. Lithium(I) was then precipitated as lithium phosphate by using sodium phosphate. Afterwards, lithium phosphate was dissolved in sulfuric acid. ED with a Nafion 117 cation exchange membrane was used to separate lithium(I) and phosphorus from the resulting solution. Sodium carbonate was added into the resulting solution containing lithium(I) to precipitate lithium carbonate (purity = 99.3 %). Chan et al. (2022) [
13] studied lithium(I)-cobalt(II)-nickel(II)-manganese(II) separation from the leach solution containing 69.41 mg L
−1 Li(I), 195.43 mg L
−1 Ni(II), 182.91 mg L
−1 Mn(II) and 196.24 mg L
−1 Co(II), produced by digesting LiNi
1/3Mn
1/3Co
1/3O
2(NMC111) in 1 mol L
−1 H
2SO
4 in the presence of 0.62% (wt) H
2O
2. Around 99% nickel was recovered by ED after complexation with EDTA at pH = 2. Likewise, 87.3% cobalt was recovered at pH = 3 by ED in a second step, and ED was performed to separate lithium(I) from manganese(II) by means of the monovalent selective cation exchange membrane Neosepta
® CMS (separation yield = 99%).
Therefore, only several papers were focused on the use of electrodialysis to separate lithium(I), cobalt(II), nickel(II) and manganese(II) in acidic sulfate media without the use of additives such as chelating agents, which avoid precipitation phenomena during electrodialysis [
13]. The present paper investigates the separation of lithium(I) from manganese(II), nickel(II) and cobalt(II) in acid sulfate media even at relatively low pH by means of electrodialysis using two commercial membranes, i.e., Neosepta
® monovalent-selective cation exchange membrane and CSO cationic exchange membrane. Based on these results, a hybrid flowsheet relying on the use of electrodialysis, solvent extraction and precipitation stages was suggested to concentrate and refine the lithium streams from spent LiCo
1/3Ni
1/3Mn
1/3O
2 (NMC111) cathodes of lithium-ion batteries.
2. Materials and Methods
2.1. Reagents
The aqueous solutions and the synthetic leach solution were prepared by dissolving appropriate amounts of reagents into deionized water (resistivity = 18 Mohms), i.e., Li2SO4 (Aldrich, Saint Louis, MO, USA, purity ≥ 98.5%), CoSO4.7H2O (Aldrich, purity ≥ 99%), NiSO4·6H2O (Aldrich, purity ≥ 98%), MnSO4·H2O (Aldrich, purity ≥ 99%).
The solution of 0.1 mol L−1 sulfuric acid was prepared by diluting appropriate amount of pure H2SO4 (Aldrich, purity = 97%) in deionized water (resistivity = 18 Mohms).
The composition of the synthetic solution was representative of leach solutions of NMC111 cathodic materials, i.e., 2.60 g L
−1 lithium(I), 7.88 g L
−1 cobalt(II), 8.01 g L
−1 nickel(II), 4.40 g L
−1 manganese(II) and 51.45 g L
−1 sulfate [
19]. The pH of the aqueous solutions containing the metal salts was adjusted at pH = 2.83 by adding 0.1 mol L
−1 sulfuric acid to find the best compromise between the faradic yield and the lithium extraction efficiency.
2.2. Setup
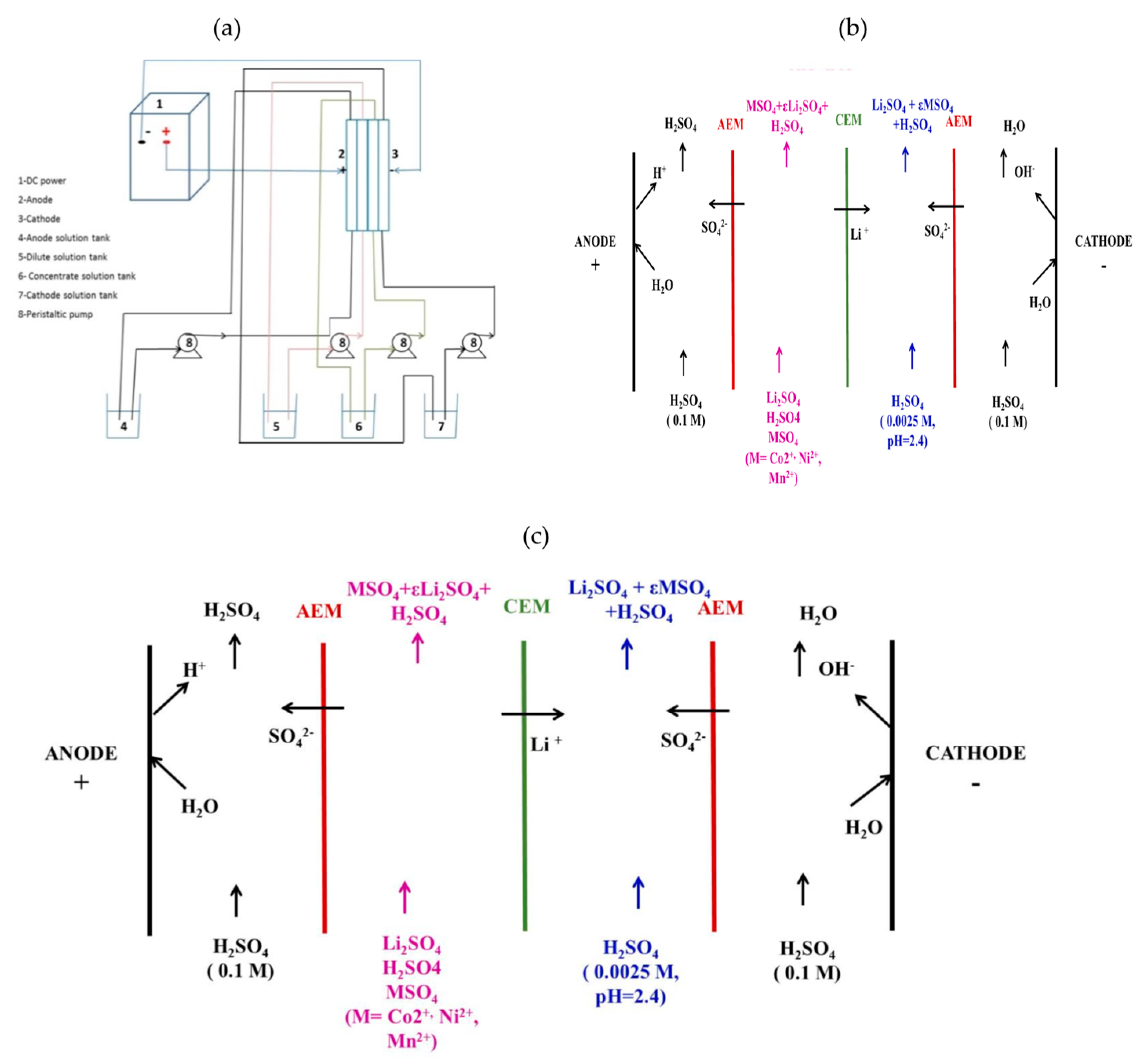
The ED setup reported in
Figure 1a was composed of a DC power (Microlab, Parsippany, NJ, USA), a 2 L-anode solution reservoir, a 2 L-dilute reservoir fed by the leach solution, a 2 L-concentrate reservoir, a 2 L-cathode solution reservoir and four peristaltic pumps (Masterflex L/S model 77250-62, assembled by Parmer Instrument Company, Vernon Hills, IL, USA). The flow rate was fixed at 100 mL min
−1 for all experiments. Neosepta
® AMX (Astom Company, Tokyo, Japan) and Selemion CSO (Asahi Glass Company, Tokyo, Japan) were used as anionic exchange membrane (AEM) and cationic exchange membrane (CEM), respectively. The surface area of these membranes was equal to 10 or 40 cm
2. A Neosepta
® monovalent-selective cationic exchange membrane (provided by Eurodia Industrie, Pertuis, France) was also used (surface area = 10 or 40 cm
2). Prior to experiments, the cationic exchange membranes and the anionic exchange membranes were immersed into an aqueous solution containing 0.1 mol L
−1 Li
2SO
4 and deionized water for 24 h, respectively. However anionic exchange membranes were immersed in deionized water for 24 h.
The ED performances were studied by calculating the faradic efficiency (R
f), the permselectivity index for Li(I) toward M (P(Li/M)) and the cations flux (
Ji), defined as follows:
where Δn (mol) denotes the moles number of cations transported through the membrane and F the Faraday constant (96,487 C mol
−1); z is the ion valence, I (A) is the applied current and Δt (s) is the operation time of electrodialysis.
where [Li]
c and [M]
c are the lithium and metal concentrations in the recovery compartment at time t. [Li]
0 and [M]
0 are the initial lithium and metal concentrations in the central compartment, respectively.
where Δ
ni is the mole number of cation
i transferred from central to recovery compartment.
A (m
2) is the active area of membrane and
N is the number of membranes’ pairs in electrodialysis cell (
N = 1 for all our experiments).
2.3. Elemental Analysis
Lithium, cobalt, nickel and manganese concentrations were determined using induced coupled plasma-optical emission spectrometry (ICP-OES, ICAP 6000 Series). Determination of metal concentrations in the aqueous phases were performed at the following wavelengths: 610.362 nm and 670.784 nm for lithium, 238.892 nm, 237.862 nm and 231.160 nm for cobalt, 221.647 nm, 216.556 nm and 230.300 nm for nickel, and 259.373 nm, 294.920 nm and 293.930 nm for manganese. Seven standards containing 5, 10, 20, 25, 50, 80 and 100 mg L−1 lithium, nickel, cobalt and manganese diluted in 0.1 mol L−1 H2SO4 or 0.0025 mol L−1 H2SO4 were prepared by using a multi-elemental standard solution containing 1 g L−1 lithium, 1 g L−1 nickel, 1 g L−1 cobalt and 1 g L−1 manganese dissolved in 5% nitric acid (SCP science).
3. Results and Discussion
3.1. Effect of Current Density on Membrane Selectivity
The ED configuration displayed in
Figure 1b was used to investigate the selectivity of the CSO membrane and the Neosepta
® monovalent-selective cationic exchange membrane for lithium(I) toward cobalt(II), nickel(II) and manganese(II).
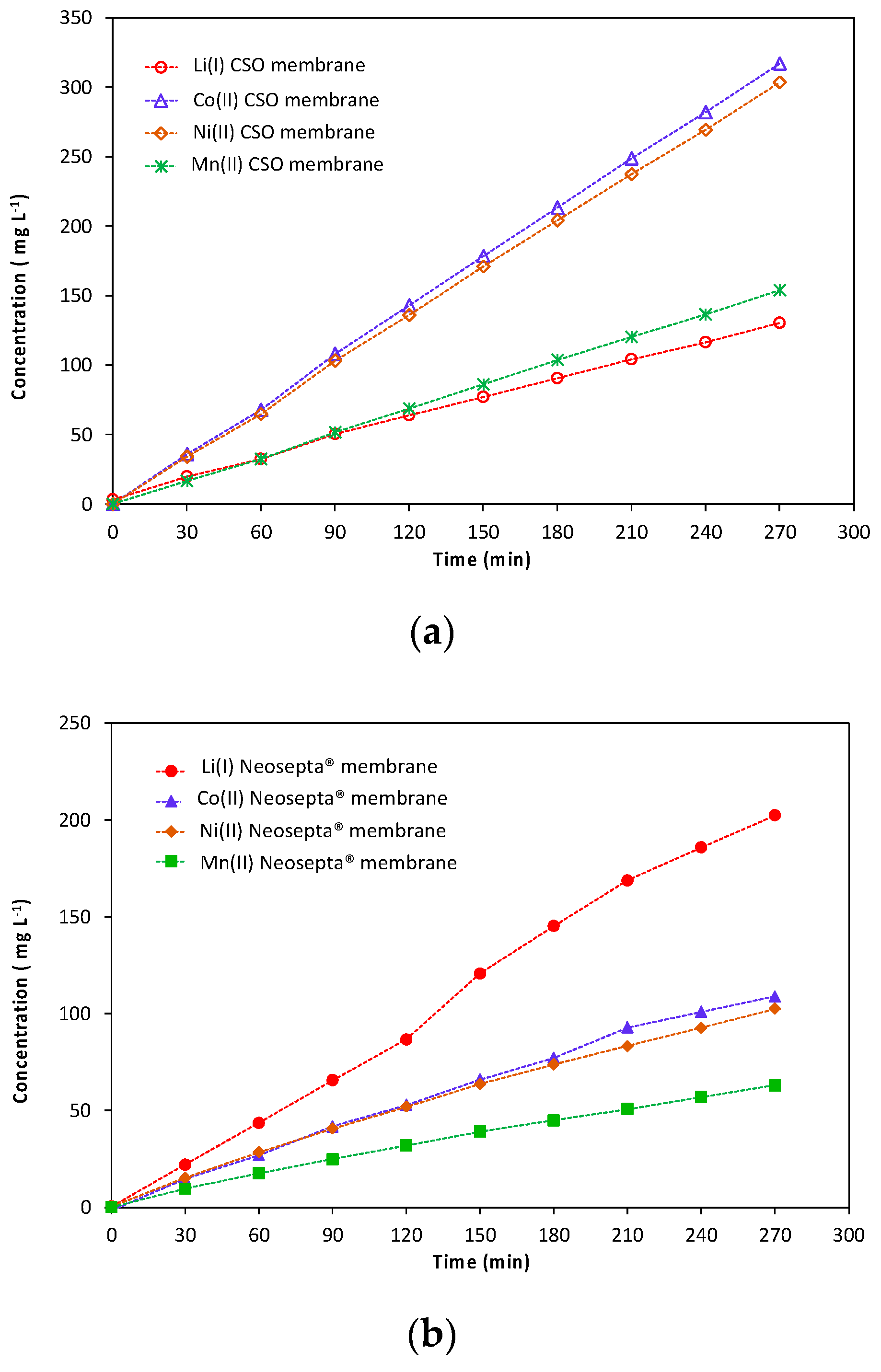
Figure 2 shows the variation of lithium(I), cobalt(II), nickel(II) and manganese(II) concentrations in the recovery compartment as a function of time.
The metal transfer rate from the central compartment to the cathodic compartment follows the following order (i) Co(II) ≈ Ni(II) > Mn(II) > Li(I) in the presence of the CSO membranes and (ii) Li(I) > Co(II) ≈ Ni(II) > Mn(II) in the presence of the Neosepta
® monovalent-selective cationic exchange membranes. Thus,
Figure 2 showed that both membranes cannot separate nickel(II) and cobalt(II) because these elements exhibit similar physicochemical properties. The Neosepta
® monovalent-selective cationic exchange membrane exhibits higher selectivity for lithium(I) toward divalent cations than the CSO membrane.
Table 1 shows that the lithium(I) molar flux is 1.6 times higher with the Neosepta
® monovalent-selective cationic exchange membrane than with the CSO membrane. Conversely, the total flux of divalent cations is 2.8 times lower.
The high selectivity of the Neosepta
® monovalent-selective cationic exchange membrane results in higher faradic efficiency for lithium(I) and higher permselectivity indexes for lithium(I) with respect to the different divalent cations. The permselectivity index for the Neosepta
® monovalent-selective cationic exchange membrane (P
Li/Co = 5.6) is slightly lower than those reported in the work of Afifah et al. (2018) [
17], i.e., P(Li/Co) = 6.75 at a flowrate of 15 L h
−1 and for a voltage of 3 V/Cell with a pack containing 5 pairs of monovalent selective membranes PC-MVA/PC-MVK (initial lithium(I) and cobalt(II) concentration = 100 mg L
−1 and 300 mg L
−1, respectively) against P(Li/Co) = 5.6 in this study. However, the value reported in our work regarding the permselectivity index for lithium(I) toward manganese(II) is greater than that obtained by Chan et al. [
13] (P(Li/Mn) = 5.4 in the present study against P(Li/Mn) = 2.81 at a flowrate of 45 L h
−1 and for a voltage of 6 V/Cell with a pack containing 2 pairs of standard anionic membranes PCA PC 400D and 2 Neosepta
® CMS cationic membranes, which are selective towards monovalent cations).
The ED configuration reported in
Figure 1c was used under the operating conditions gathered in
Table 2 (cell 2) to study the impact of the current density on the electrodialysis performance by using the Neosepta
® monovalent-selective cationic exchange membrane. This configuration exhibits an additional compartment between the cathodic compartment and the anodic compartment. This compartment was fed with 0.0025 mol L
−1 H
2SO
4 (pH 2.4). This new configuration delays possible metal precipitation phenomena into the membranes that might occur after several hours of experiments.
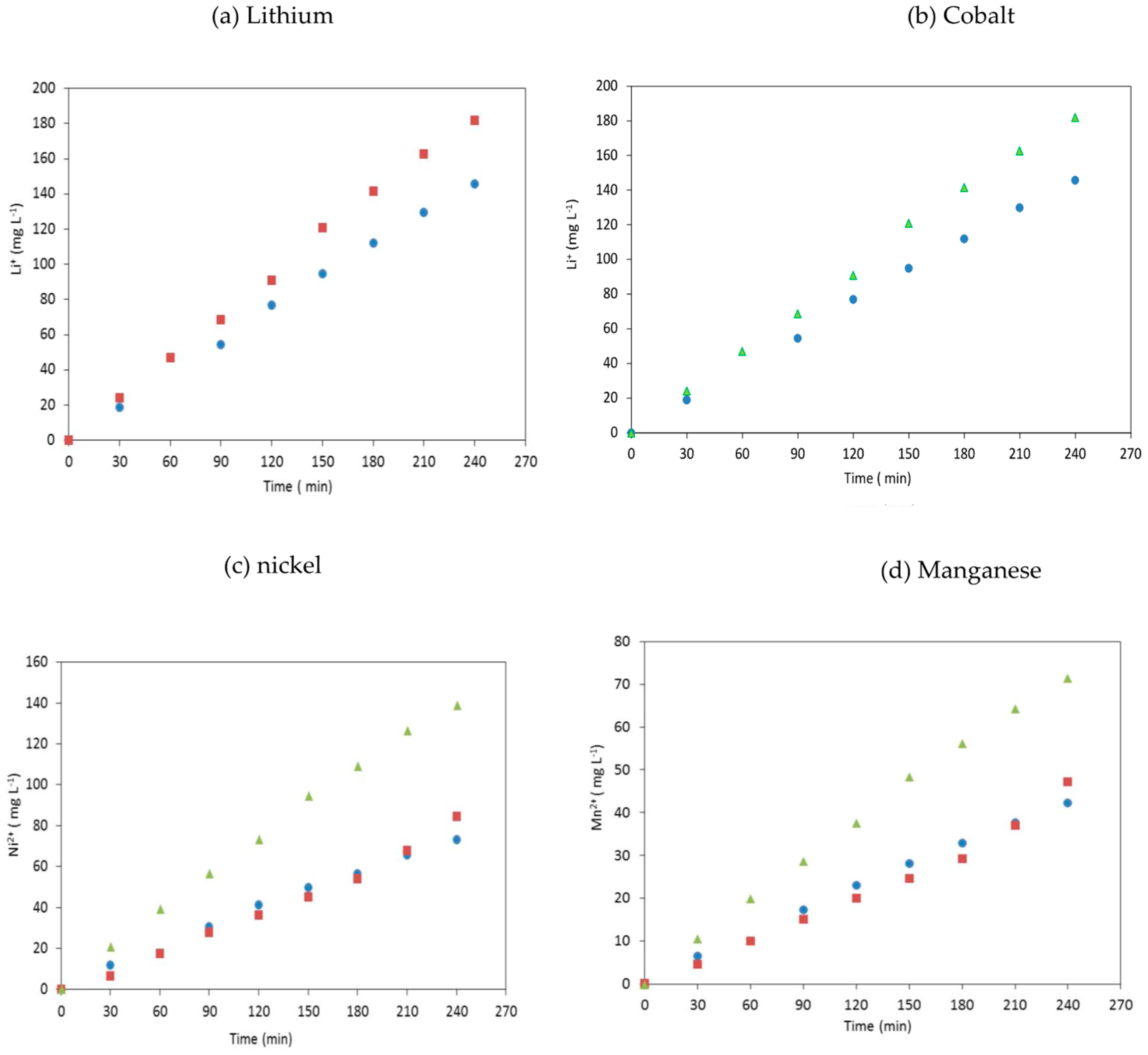
Figure 3 shows the influence of the current density on the metal concentrations in the recovery compartment as a function of time for 4 h. The increase of the current density from 10 mA cm
−2 to 12.5 mA cm
−2 did not influence the transfer of the divalent metals, i.e., cobalt(II), nickel(II) and manganese(II), whereas lithium(I) transfer is slightly affected. The membrane selectivity toward monovalent cations decreased with increasing current as the permselectivity indexes decreased progressively with increasing current density (
Table 2).
The sums of the faradic efficiencies related to metal transport remained almost constant, i.e., 87% at 10 mA cm−2 and 12.5 mA cm−2. Conversely, the faradic efficiency diminished down to 80.3 % when the current density was increased to 15 mA cm−2, likely due to water splitting (water dissociation onto the membrane surface), which was responsible for an increase of the protons transfer.
Given that the limiting current density was equal to j
lim = 18.9 mA cm
−2, a current density of 15 mA cm
−2 corresponding to 80% of the limiting current density was applied [
20]. A decrease of the faradic yield of lithium(I) and an increase of the faradic yield of divalent cations were observed when the current density was close to the limiting current density at the expense of lithium transfer selectivity.
3.2. Metal Precipitation into the Membranes
No precipitation was observed into the Neosepta
® monovalent-selective cationic exchange membrane when the electrodialysis lasted less than 270 min. Conversely, a greenish precipitate was observed inside the Neosepta
® monovalent-selective cationic exchange membrane after performing electrodialysis during 810 min (13.5 h) at 12.5 mA cm
−2 followed by 5 h at 15 mA cm
−2 (configuration 2 in
Figure 1c). Precipitation occurred faster when the concentration ratio between lithium(I) and divalent cations (Ni(II), Co(II), Mn(II)) in the leachate compartment decreased as it was also observed when full lithium-selective membranes were used in ED (divalent cation concentrations remained constant as they were not transferred from the leachate compartment into the recovery compartment, and lithium(I) concentration decreased due to their efficient transfer across the membrane). For instance, a greenish precipitate was observed after 20 min of electrodialysis at 10 mA cm
−2 when the leach solution contained initially 0.5 g L
−1 lithium(I) and 7.88 g L
−1 cobalt(II), 8.09 g L
−1 nickel(II) and 4.40 g L
−1 manganese(II). Precipitation occurred despite the low values of pH into the leachate compartment (pH = 2.8) and the recovery compartment (pH = 2.3). The increase of the local pH and the local concentration of the divalent cations at the membrane surface may be responsible for precipitation. The local increase of pH can be explained by the water splitting at the membrane surface when the current density reaches the limiting value. The high local concentration of divalent cations onto the membrane surface can be explained by the high selectivity of the membrane.
Therefore, the precipitation phenomena could be avoided by mastering the current density so that its value remains lower than the limiting current density.
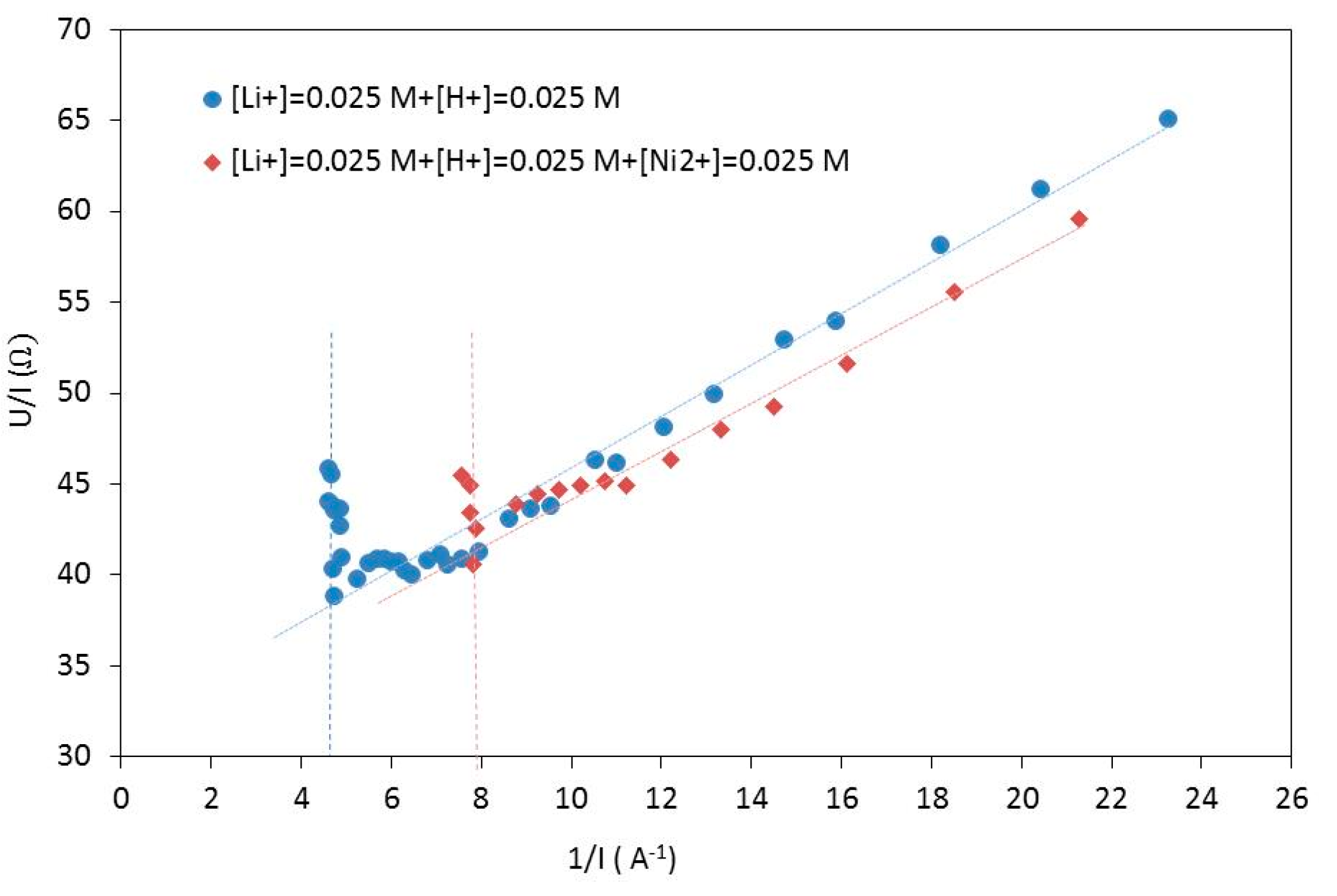
Table 3 gathers the limiting current densities deduced from
Figure 4 by the Cowan and Brown method [
21].
This table shows that there was no influence of the concentration of monovalent cations on the limiting current density. Conversely, the addition of nickel(II) is responsible for a drastic decrease of the limiting current density despite the same high values of ionic conductivity and similar values of pH. Therefore, such a decrease of the limiting current density did not result from a difference in proton transfer between these experiments. The same observation was found by comparing the limiting current densities determined when the electrodialysis was performed with a feed solution containing only lithium(I) and a feed solution containing lithium(I), nickel(II), manganese(II) and cobalt(II).
The limiting current was reached when lithium(I) concentration at the membrane surface was equal to zero. In the case of an ideal lithium(I) selective membrane, the current cannot be transported by the divalent cations, and therefore, only the protons generated by water splitting at the membrane surface can transport the current. Furthermore, water splitting may affect the selectivity of the electro-membrane process as reported by Melnikov et al. [
22]. The control of the current density is particularly important to regulate the fluxes of salt ions and water splitting products (hydrogen and hydroxyl ions). The water splitting led to the formation of hydroxide ions at the membrane surface, which were responsible for an increase of the local pH at the surface membrane. Both the accumulation of divalent cations at the membrane surface and the local increase of pH may lead to metal hydroxide precipitation at the membrane surface.
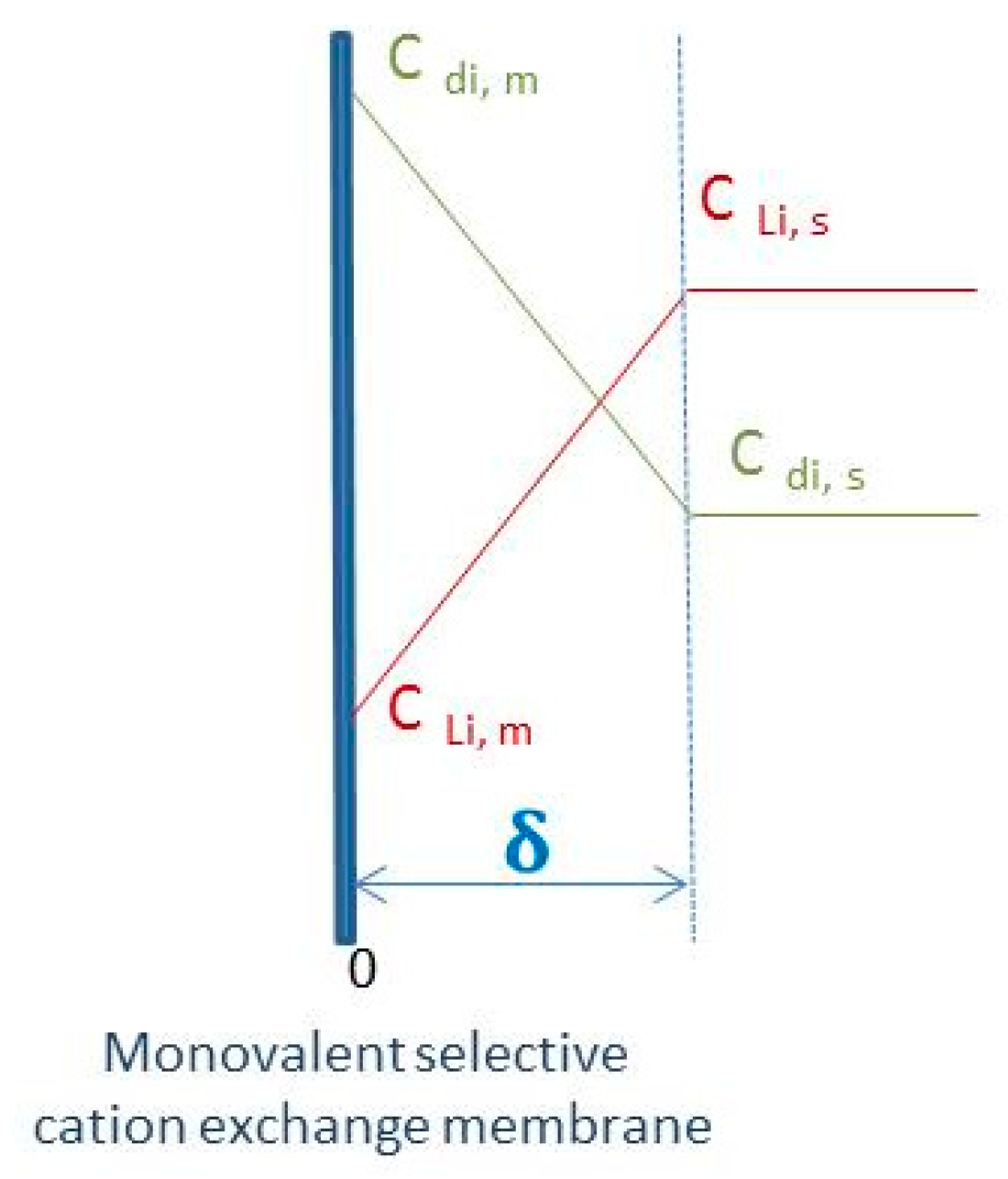
The concentration profiles of lithium(I) and divalent cations in the boundary layer at the vicinity of the membrane depicted in
Figure 5 depends on the difference in transport numbers between the bulk and into the membrane.
In the bulk, the transport number of the species
i corresponds to the fraction of current transported by that species according to the following equation:
where
ui is the electrophoretic mobility in m
2 V
−1 s
−1 of the ion
i of charge
Zi and concentration
Ci.
The transport number of lithium(I) is thus affected by the concentration of the divalent cations in the solution. In the case of an ideal selective membrane toward monovalent cations, the current is carried by the monovalent cations (lithium(I) and protons in the present work) whereas the divalent cations do not contribute to the current:
The transport number of monovalent cations in the membrane was found to be independent on the divalent cation concentration. Thus, the transport numbers of the monovalent cations were not influenced by the divalent cations in the case of an ideal selective membrane. Conversely, the transport numbers of the monovalent cations in solution were influenced by the presence of divalent cations in solution. The difference between the transport numbers of the monovalent cations in the membrane and in the solution increases with the concentration ratio of divalent/monovalent cations in solution. Such an increase contributes to the decrease of the monovalent cation concentration at the vicinity of the membrane, and thus, contributes to the decrease of the limiting current.
The present work evidenced that an increase of the concentration ratio of divalent/monovalent cations in solution was responsible for a decrease of the limiting current density and an increase of precipitation risks into the lithium-selective membrane. Likewise, the decrease of the limiting current density may arise from the slow transport of monovalent cations due to electrostatic repulsion onto the membrane surface caused by the presence of a layer of divalent cations.
Therefore, the use of highly selective membranes in ED requires to determine the value of the limiting current density in operando to check that the current density is always lower than the limiting current density. Monitoring the ionic conductivity in the diluate is then not sufficient and the main challenge is therefore to monitor the limiting current over time [
23,
24,
25].
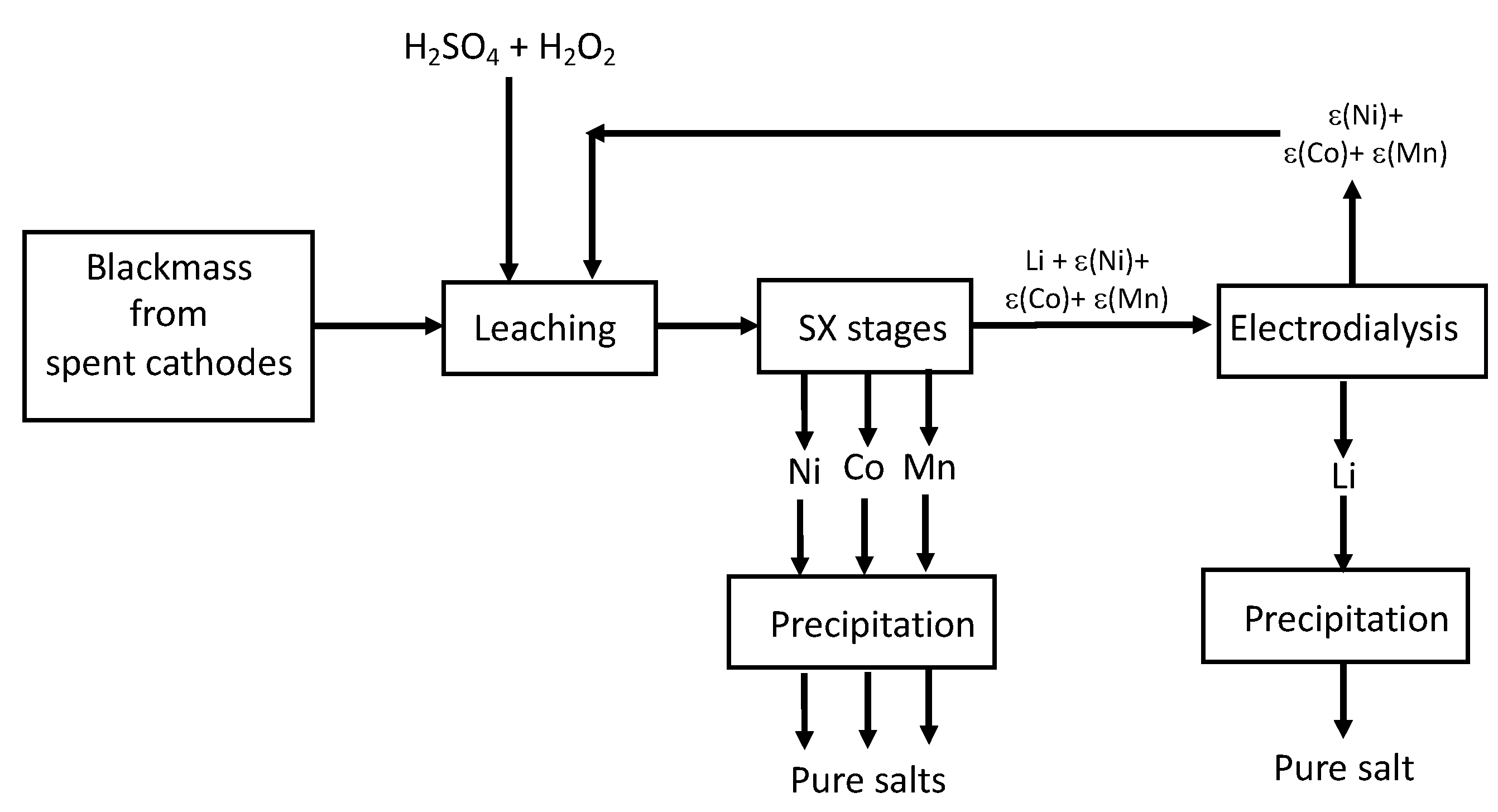
Providing that the current density in the electrodialysis cell could be managed to be lower than the limiting current density, such an electrodialysis technology could be included in a more global flowsheet involving other hydrometallurgical unit operations such as solvent extraction and precipitation as illustrated in
Figure 6. After leaching the blackmass in sulfuric acid in the presence of hydrogen peroxide [
26], cobalt(II), nickel(II) and manganese(II) could be extracted and separated by solvent extraction by using Cyanex
® 272 as extractant [
27]. The remaining stream containing lithium(I) and traces of manganese(II), nickel(II) and cobalt(II) could be afterwards refined by using electrodialysis to produce high-grade lithium salt by precipitation.
4. Conclusions
This paper showed that electrodialysis is an interesting technology to extract selectively and concentrate lithium(I) toward nickel(II), cobalt(II) and manganese(II) from acidic sulfate media. Therefore, this technology could be used in lithium-ion battery recycling processes. More interestingly, electrodialysis could be implemented at the end of the process to remove traces of nickel(II), cobalt(II) and manganese(II) from the lithium solution after cobalt(II), nickel(II) and manganese(II) recovery by liquid-liquid extraction. Thus, electrodialysis could be advantageously used to concentrate the lithium stream and to produce high-grade lithium salts after precipitation with sodium carbonate.
However, there remains a few challenges to solve before implementing electrodialysis technologies in lithium-ion battery recycling, especially when high-selective membranes toward lithium(I) are used. In fact, the difference in transport properties between lithium(I) and divalent cations, i.e., cobalt(II), nickel(II) and manganese(II), through a monovalent ion-selective membrane are responsible for precipitation of the divalent cations into the porosity of the membrane since the limiting current decreases significantly during electrodialysis. The main challenge is therefore to monitor the limiting current over time in order to keep the current value lower than the limiting current.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}