
Biologic Treatments for Asthma and Chronic Obstructive Pulmonary Disorder



Abstract
:
1. Introduction
1.1. Asthma Characteristics
1.2. Asthma Pathogenesis Factors: Age, Viruses, Bacteria, Genes and Lifestyle
2. Overlap with Chronic Obstructive Pulmonary Disorder (COPD)
2.1. COPD Pathogenesis
2.2. Asthma-COPD Overlap
2.2.1. Overlap in Symptoms and Phenotypes
2.2.2. Overlap Statistics
2.2.3. Similarities in ACO and Asthma Treatment
2.2.4. Unique Considerations for ACO Patients
3. Diagnosis and Treatment
3.1. General Diagnosis and Treatment for Asthma
3.1.1. Diagnostics
3.1.2. Treatments
3.1.3. Biologic Treatments for Asthma and Asthma-COPD-Overlap
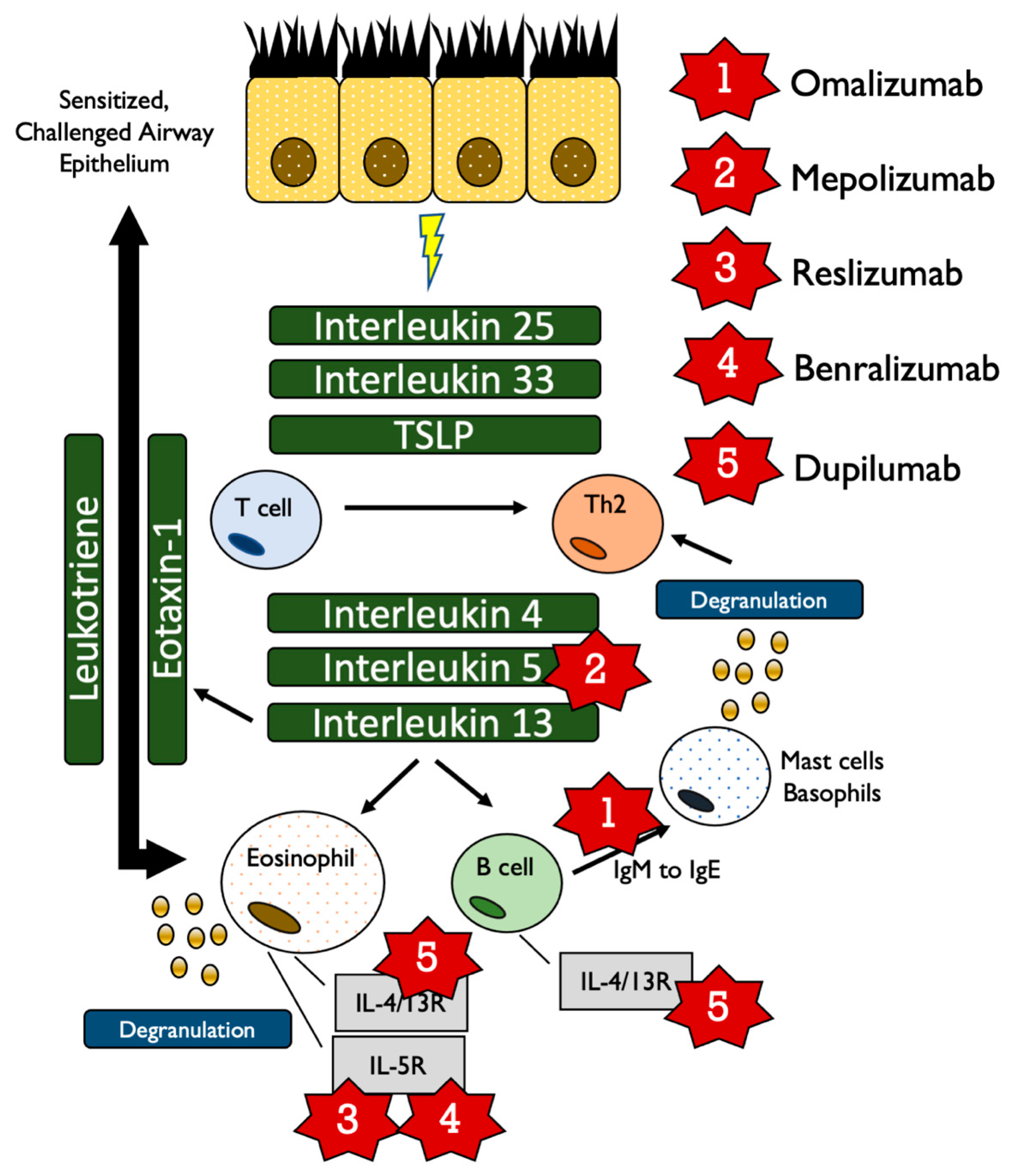
3.2. Compound 1: Omalizumab (Xolair, Approved June, 2003)
3.2.1. Mechanism
3.2.2. Asthma Efficacy
3.2.3. ACO Efficacy
3.3. Compound 2: Mepolizumab (Nucala, Approved November, 2015)
3.3.1. Mechanism
3.3.2. Asthma Efficacy
3.3.3. Efficacy and ACO
3.4. Compound 3: Reslizumab (Cinqair, Approved March, 2016)
3.4.1. Mechanism
3.4.2. Asthma Efficacy
3.4.3. Efficacy and ACO
3.5. Compound 4: Benralizumab (Fasenra, Approved November, 2017)
3.5.1. Mechanism
3.5.2. Asthma Efficacy
3.5.3. Efficacy and ACO
3.6. Compound 5: Dupilumab (Dupixent, Approved October, 2018)
3.6.1. Mechanism
3.6.2. Asthma Efficacy
3.6.3. Efficacy and ACO
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ministry of Health. Labour and Welfare Report: Patient Numbers by Age and Disease; Ministry of Health: Tokyo, Japan, 2017.
- Yamauchi, Y.I. Epidemiology of Asthma: The Present and Near Future. Nihon Naika Gakkai Zasshi 2018, 107, 2059–2066. [Google Scholar] [CrossRef] [Green Version]
- Bush, A. Pathophysiological Mechanisms of Asthma. Front. Pediatr. 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Therapeutic approaches to asthma–chronic obstructive pulmonary disease overlap syndromes. J. Allergy Clin. Immunol. 2015, 136, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Akdis, C.A. Precision medicine and phenotypes, endotypes, genotypes, regiotypes, and theratypes of allergic diseases. J. Clin. Investig. 2019, 129, 1493–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munekata, T. Adult Asthma: From Phenotype to Endotype (Japanese). Allergy 2017, 66, 9–13. [Google Scholar]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, K.; Ogasawara, M. The Role of Histamine in the Pathophysiology of Asthma and the Clinical Efficacy of Antihistamines in Asthma Therapy. Int. J. Mol. Sci. 2019, 20, 1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinose, M.; Sugiura, H.; Nagase, H.; Yamaguchi, M.; Inoue, H.; Sagara, H.; Tamaoki, J.; Tohda, Y.; Munakata, M.; Yamauchi, K.; et al. Japanese guidelines for adult asthma 2017. Allergol. Int. 2017, 66, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, M.E.; Lee, F.E.-H.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.F. Targeting the interleukin pathway in the treatment of asthma. Lancet 2015, 386, 1086–1096. [Google Scholar] [CrossRef]
- Bakakos, A.; Loukides, S. Severe Eosinophilic Asthma. J. Clin. Med. 2019, 8, 1375. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef] [Green Version]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Fahy, J.V. Type 2 inflammation in asthma—present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- De Benedictis, F.M.; Attanasi, M. Asthma in childhood. Eur. Respir. Rev. 2016, 25, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasinghe, H.; Kopsaftis, Z.; Carson, K. Asthma Bronchiale and Exercise-Induced Bronchoconstriction. Respiration 2015, 89, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Webley, W.C.; Hahn, D.L. Infection-mediated asthma: Etiology, mechanisms and treatment options, with focus on Chlamydia pneumoniae and macrolides. Respir. Res. 2017, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ekurai, D.; Esaraya, T.; Eishii, H.; Etakizawa, H. Virus-induced exacerbations in asthma and COPD. Front. Microbiol. 2013, 4, 293. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.-Y.; Xu, Y.-J.; Guan, W.-J.; Lin, L.-F. Regional, age and respiratory-secretion-specific prevalence of respiratory viruses associated with asthma exacerbation: A literature review. Arch. Virol. 2018, 163, 845–853. [Google Scholar] [CrossRef]
- Gut, G.; Domany, K.A.; Sadot, E.; Soferman, R.; Fireman, E.; Sivan, Y. Eosinophil cell count in bronchoalveolar lavage fluid in early childhood wheezing: Is it predictive of future asthma? J. Asthma 2019, 57, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Ege, M.J.; Mayer, M.; Normand, A.-C.; Genuneit, J.; Cookson, W.O.; Braun-Fahrländer, C.; Heederik, D.; Piarroux, R.; Von Mutius, E. Exposure to Environmental Microorganisms and Childhood Asthma. N. Engl. J. Med. 2011, 364, 701–709. [Google Scholar] [CrossRef] [PubMed]
- von Mutius, E. The microbial environment and its influence on asthma prevention in early life. J. Allergy Clin. Immunol. 2016, 137, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, N.G.; Hernandez-Leyva, A.; Kau, A.L. The ABCs of wheeze: Asthma and bacterial communities. PLoS Pathog. 2019, 15, e1007645. [Google Scholar] [CrossRef]
- Denning, D.W.; Pashley, C.; Hartl, D.; Wardlaw, A.; Godet, C.; Del Giacco, S.; Delhaes, L.; Sergejeva, S. Fungal allergy in asthma–state of the art and research needs. Clin. Transl. Allergy 2014, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.J.; Sykes, A.; Mallia, P.; Johnston, S.L. Asthma exacerbations: Origin, effect, and prevention. J. Allergy Clin. Immunol. 2011, 128, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Oksel, C.; Custovic, A. Development of allergic sensitization and its relevance to paediatric asthma. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 109–116. [Google Scholar] [CrossRef]
- Murrison, L.B.; Brandt, E.B.; Myers, J.B.; Hershey, G.K.K. Environmental exposures and mechanisms in allergy and asthma development. J. Clin. Investig. 2019, 129, 1504–1515. [Google Scholar] [CrossRef] [Green Version]
- Scherzer, R.; Grayson, M.H. Heterogeneity and the origins of asthma. Ann. AllergyAsthma Immunol. 2018, 121, 400–405. [Google Scholar] [CrossRef]
- De Nijs, S.B.; Venekamp, L.N.; Bel, E.H. Adult-onset asthma: Is it really different? Eur. Respir. Rev. 2013, 22, 44–52. [Google Scholar] [CrossRef]
- Pekkanen, J.; Valkonen, M.; Täubel, M.; Tischer, C.; Leppänen, H.; Kärkkäinen, P.M.; Rintala, H.; Zock, J.-P.; Casas, L.; Probst-Hensch, N.; et al. Indoor bacteria and asthma in adults: A multicentre case–control study within ECRHS II. Eur. Respir. J. 2018, 51, 1701241. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Agustí, A.; Hogg, J.C. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Aldonyte, R.; Bagdonas, E.; Raudoniute, J.; Bruzauskaite, I. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int. J. Chronic Obs. Pulm. Dis. 2015, 10, 995–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosío, B.G.; DaCal, D.; De Llano, L.P. Asthma–COPD overlap: Identification and optimal treatment. Adv. Respir. Dis. 2018, 12, 175346661880566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tho, N.; Park, H.Y.; Nakano, Y. Asthma-COPD overlap syndrome (ACOS): A diagnostic challenge. Respirology 2015, 21, 410–418. [Google Scholar] [CrossRef]
- Ma, A.; Wen, L.; Yin, J.; Hu, Y.; Yue, X.; Li, J.; Dong, X.; Gupta, Y.; Ludwig, R.J.; Krauss-Etschmann, S.; et al. Serum Levels of Autoantibodies Against Extracellular Antigens and Neutrophil Granule Proteins Increase in Patients with COPD Compared to Non-COPD Smokers. Int. J. Chronic Obs. Pulm. Dis. 2020, 15, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, R.L.; Macrea, M.M.; Teodorescu, M. The overlaps of asthma or COPD with OSA: A focused review. Respirology 2017, 22, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Miravitlles, M.; Ribera, A. Understanding the impact of symptoms on the burden of COPD. Respir. Res. 2017, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rogliani, P.; Ora, J.; Puxeddu, E.; Cazzola, M. Airflow obstruction: Is it asthma or is it COPD? Int. J. Chronic Obs. Pulm. Dis. 2016, 11, 3007–3013. [Google Scholar] [CrossRef] [Green Version]
- Llanos, J.-P.; Ortega, H.; Germain, G.; Duh, M.S.; Lafeuille, M.-H.; Tiggelaar, S.; Bell, C.F.; Hahn, B. Health characteristics of patients with asthma, COPD and asthma–COPD overlap in the NHANES database. Int. J. Chronic Obs. Pulm. Dis. 2018, 13, 2859–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, Y.; Yasunaga, H.; Matsui, H.; Hasegawa, W.; Jo, T.; Takami, K.; Fushimi, K.; Nagase, T. Comparison of in-hospital mortality in patients with COPD, asthma and asthma-COPD overlap exacerbations. Respirology 2015, 20, 940–946. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Ichinose, M. Definition and diagnosis of asthma–COPD overlap (ACO). Allergol. Int. 2018, 67, 172–178. [Google Scholar] [CrossRef]
- Uchida, A.; Sakaue, K.; Inoue, H. Epidemiology of asthma-chronic obstructive pulmonary disease overlap (ACO). Allergol. Int. 2018, 67, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Guzman, E.; Khosravi, M.; Mannino, D.M. Asthma, Chronic Obstructive Pulmonary Disease, and Mortality in the U.S. Population. Copd J. Chronic Obs. Pulm. Dis. 2011, 8, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Golpe, R.; Martín-Robles, I.; Sanjuán-López, P.; Pérez-De-Llano, L.; González-Juanatey, C.; López-Campos, J.L.; Arellano-Orden, E. Differences in systemic inflammation between cigarette and biomass smoke-induced COPD. Int. J. Chronic Obs. Pulm. Dis. 2017, 12, 2639–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, M.; Tamaoki, J. Therapeutic approaches of asthma and COPD overlap. Allergol. Int. 2018, 67, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Hayden, L.P.; on behalf of the COPDGene Investigators; Cho, M.H.; Raby, B.A.; Beaty, T.H.; Silverman, E.K.; Hersh, C.P. Childhood asthma is associated with COPD and known asthma variants in COPDGene: A genome-wide association study. Respir. Res. 2018, 19, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, M.J.; Perret, J.L.; Dharmage, S.C.; McDonald, V.M.; McDonald, C.F. Distinguishing adult-onset asthma from COPD: A review and a new approach. Int. J. Chronic Obs. Pulm. Dis. 2014, 9, 945. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; Rhee, C.K.; Kim, K.; Kim, S.H.; Lee, J.Y.; Kim, Y.H.; Yoo, K.H.; Cho, Y.-J.; Jung, K.-S.; Lee, J.H. Heterogeneity of asthma and COPD overlap. Int. J. Chronic Obs. Pulm. Dis. 2018, 13, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, H.; Hamasaki, Y.; Kohno, Y.; Ebisawa, M.; Kondo, N.; Nishima, S.; Nishimuta, T.; Morikawa, A. Japanese guidelines for childhood asthma 2017. Allergol. Int. 2017, 66, 190–204. [Google Scholar] [CrossRef]
- Hernandez-Pacheco, N.; Pino-Yanes, M.; Flores, C. Genomic Predictors of Asthma Phenotypes and Treatment Response. Front. Pediatr. 2019, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Zhang, H.-Y. Genetic Mechanisms of Asthma and the Implications for Drug Repositioning. Genes 2018, 9, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechsler, E.M. Current and Emerging Biologic Therapies for Asthma and COPD. Respir. Care 2018, 63, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H. Severe asthma in Japan. Allergol. Int. 2019, 68, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Pennington, E.; Yaqoob, Z.J.; Al-Kindi, S.G.; Zein, J. Trends in Asthma Mortality in the United States: 1999 to 2015. Am. J. Respir. Crit. Care Med. 2019, 199, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Available online: drugs.com https://www.drugs.com/history-f1.html (accessed on 8 February 2021).
- Tu, X.; Donovan, C.; Kim, R.Y.; Wark, P.A.; Horvat, J.C.; Hansbro, P.M. Asthma-COPD overlap: Current understanding and the utility of experimental models. Eur. Respir. Rev. 2021, 30, 190185. [Google Scholar] [CrossRef]
- Leung, J.M.; Sin, D.D. Asthma-COPD overlap syndrome: Pathogenesis, clinical features, and therapeutic targets. BMJ 2017, 358, j3772. [Google Scholar] [CrossRef] [PubMed]
- Godse, K.; Mehta, A.; Patil, S.; Gautam, M.; Nadkarni, N. Omalizumab-A review. Indian J. Derm. 2015, 60, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Froidure, A.; Mouthuy, J.; Durham, S.R.; Chanez, P.; Sibille, Y.; Pilette, C. Asthma phenotypes and IgE responses. Eur. Respir. J. 2015, 47, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Okayama, Y.; Matsumoto, H.; Odajima, H.; Takahagi, S.; Hide, M.; Okubo, K. Roles of omalizumab in various allergic diseases. Allergol. Int. 2020, 69, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Siergiejko, Z.; Świebocka, E.; Smith, N.; Peckitt, C.; Leo, J.; Peachey, G.; Maykut, R. Oral corticosteroid sparing with omalizumab in severe allergic (IgE-mediated) asthma patients. Curr. Med. Res. Opin. 2011, 27, 2223–2228. [Google Scholar] [CrossRef]
- Garcia, G.; Magnan, A.; Chiron, R.; Contin-Bordes, C.; Berger, P.; Taillé, C.; Devouassoux, G.; De Blay, F.; Couderc, L.-J.; Didier, A.; et al. A Proof-of-Concept, Randomized, Controlled Trial of Omalizumab in Patients with Severe, Difficult-to-Control, Nonatopic Asthma. Chest 2013, 144, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Maltby, S.; Gibson, P.G.; Powell, H.; McDonald, V.M. Omalizumab Treatment Response in a Population with Severe Allergic Asthma and Overlapping COPD. Chest 2017, 151, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Tat, T.S.; Cilli, A. Omalizumab treatment in asthma-COPD overlap syndrome. J. Asthma 2016, 53, 1048–1050. [Google Scholar] [CrossRef]
- Hanania, N.A.; Chipps, B.E.; Griffin, N.M.; Yoo, B.; Iqbal, A.; Casale, T.B. Omalizumab effectiveness in asthma-COPD overlap: Post hoc analysis of PROSPERO. J. Allergy Clin. Immunol. 2019, 143, 1629–1633.e2. [Google Scholar] [CrossRef] [Green Version]
- Kupryś-Lipinska, I.; Pałczyński, C.; Molinska, J.; Kuna, P. Omalizumab therapy in a patient with severe asthma and co-existing chronic obstructive pulmonary disease. Adv. Derm. Allergol. 2019, 36, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Menzella, F.; Lusuardi, M.; Galeone, C.; Facciolongo, N.; Zucchi, L. The clinical profile of benralizumab in the management of severe eosinophilic asthma. Adv. Respir. Dis. 2016, 10, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faverio, P.; Bonaiti, G.; Bini, F.; Vaghi, A.; Pesci, A. Mepolizumab as the first targeted treatment for eosinophilic granulomatosis with polyangiitis: A review of current evidence and potential place in therapy. Clin. Risk Manag. 2018, 14, 2385–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, M.; Mori, F.; Barni, S.; De Martino, M.; Novembre, E. Omalizumab and mepolizumab in the landscape of biological therapy for severe asthma in children: How to choose? Ital. J. Pediatr. 2019, 45, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; Fitzgerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab Treatment in Patients with Severe Eosinophilic Asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Nixon, J.; Newbold, P.; Mustelin, T.; Anderson, G.P.; Kolbeck, R. Monoclonal antibody therapy for the treatment of asthma and chronic obstructive pulmonary disease with eosinophilic inflammation. Pharmacol. Ther. 2017, 169, 57–77. [Google Scholar] [CrossRef] [Green Version]
- Taillé, C.; Chanez, P.; Devouassoux, G.; Didier, A.; Pison, C.; Garcia, G.; Charriot, J.; Bouée, S.; Gruber, A.; Pribil, C.; et al. Mepolizumab in a population with severe eosinophilic asthma and corticosteroid dependence: Results from a French early access programme. Eur. Respir. J. 2020, 55, 1902345. [Google Scholar] [CrossRef]
- Khurana, S.; Brusselle, G.G.; Bel, E.H.; FitzGerald, J.M.; Masoli, M.; Korn, S.; Kato, M.; Albers, F.C.; Bradford, E.S.; Gilson, M.J.; et al. Long-term Safety and Clinical Benefit of Mepolizumab in Patients With the Most Severe Eosinophilic Asthma: The COSMEX Study. Clin. Ther. 2019, 41, 2041–2056.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emma, R.; Morjaria, J.B.; Fuochi, V.; Polosa, R.; Caruso, M. Mepolizumab in the management of severe eosinophilic asthma in adults: Current evidence and practical experience. Adv. Respir. Dis. 2018, 12, 1753466618808490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.; Shackshaft, L.; Green, L.; Roxas, C.; Fernandes, M.; Thompson, L.; D’Ancona, G.; Elstad, M.; Douiri, A.; Nanzer-Kelly, A.; et al. The relationship between fractional exhaled nitric oxide and asthma symptom scores in patients on mepolizumab. In Monitoring Airway Disease; European Respiratory Society (ERS): Lausanne, Switzerland, 2019; Volume 54, p. 2623. [Google Scholar]
- Pavord, I.D.; Chanez, P.; Criner, G.J.; Kerstjens, H.A.; Korn, S.; Lugogo, N.; Martinot, J.-B.; Sagara, H.; Albers, F.C.; Bradford, E.S.; et al. Mepolizumab for Eosinophilic Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2017, 377, 1613–1629. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Kjarsgaard, M.; Capaldi, D.; Radford, K.; Aleman, F.; Parraga, G.; Altman, L.; Wight, T.; O’Byrne, P.M.; Nair, P. Mepolizumab in COPD with eosinophilic bronchitis: A randomized clinical trial. In 5.1 Airway Pharmacology and Treatment; European Respiratory Society (ERS): Lausanne, Switzerland, 2016; Volume 48, p. PA305. [Google Scholar]
- Inserro, A. AJMC.com. Available online: https://www.ajmc.com/view/fda-advisory-committee-votes-against-approving-mepolizumab-for-copd (accessed on 8 February 2021).
- Pelaia, C.; Paoletti, G.; Puggioni, F.; Racca, F.; Pelaia, G.; Canonica, G.W.; Heffler, E. Interleukin-5 in the Pathophysiology of Severe Asthma. Front. Physiol. 2019, 10, 1514. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.K.; Hanania, N.A. Targeting IL-5 in COPD. Int. J. Chronic Obs. Pulm. Dis. 2019, 14, 1045–1051. [Google Scholar] [CrossRef] [Green Version]
- Nair, P.; Bardin, P.; Humbert, M.; Murphy, K.R.; Hickey, L.; Garin, M.; VanLandingham, R.; Chanez, P. Efficacy of Intravenous Reslizumab in Oral Corticosteroid–Dependent Asthma. J. Allergy Clin. Immunol. Pr. 2020, 8, 555–564. [Google Scholar] [CrossRef] [PubMed]
- González, I.D.; Benítez, F.M.; Quirce, S. Benralizumab: A New Approach for the Treatment of Severe Eosinophilic Asthma. J. Investig. Allergol. Clin. Immunol. 2019, 29, 84–93. [Google Scholar] [CrossRef]
- Izumo, T.; Tone, M.; Kuse, N.; Awano, N.; Tanaka, A.; Jo, T.; Yoshimura, H.; Minami, J.; Takada, K.; Inomata, M. Effectiveness and safety of benralizumab for severe asthma in clinical practice (J-BEST): A prospective study. Ann. Transl. Med. 2020, 8, 438. [Google Scholar] [CrossRef] [PubMed]
- Tanosaki, T.; Kabata, H.; Matsusaka, M.; Miyata, J.; Masaki, K.; Mochimaru, T.; Okuzumi, S.; Kuwae, M.; Watanabe, R.; Suzuki, Y.; et al. Clinical characteristics of patients with not well-controlled severe asthma in Japan: Analysis of the Keio Severe Asthma Research Program in Japanese population (KEIO-SARP) registry. Allergol. Int. 2020, 70. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Matsuyama, M.; Arai, N.; Yamada, H.; Hyodo, K.; Nonaka, M.; Kitazawa, H.; Yoshida, K.; Shigemasa, R.; Morishima, Y.; et al. Identification of whole blood gene expressions correlated with responsiveness to benralizumab. J. Allergy Clin. Immunol. 2021, 147, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.W. Biological treatments for severe asthma: A major advance in asthma care. Allergol. Int. 2019, 68, 158–166. [Google Scholar] [CrossRef]
- Bleecker, E.R.; Wechsler, M.E.; Fitzgerald, J.M.; Menzies-Gow, A.; Wu, Y.; Hirsch, I.; Goldman, M.; Newbold, P.; Zangrilli, J.G. Baseline patient factors impact on the clinical efficacy of benralizumab for severe asthma. Eur. Respir. J. 2018, 52, 1800936. [Google Scholar] [CrossRef] [Green Version]
- Maselli, D.J.; Rogers, L.I.; Peters, J. Benralizumab, an add-on treatment for severe eosinophilic asthma: Evaluation of exacerbations, emergency department visits, lung function, and oral corticosteroid use. Clin. Risk Manag. 2018, 14, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busse, W.W.; Bleecker, E.R.; FitzGerald, J.M.; Ferguson, G.T.; Barker, P.; Sproule, S.; Olsson, R.F.; Martin, U.J.; Goldman, M.; Yañez, A.; et al. Long-term safety and efficacy of benralizumab in patients with severe, uncontrolled asthma: 1-year results from the BORA phase 3 extension trial. Lancet Respir. Med. 2019, 7, 46–59. [Google Scholar] [CrossRef]
- Fitzgerald, J.M.; Bleecker, E.R.; Bourdin, A.; Busse, W.W.; Ferguson, G.T.; Brooks, L.; Barker, P.; Martin, U.J. Two-Year Integrated Efficacy And Safety Analysis Of Benralizumab In Severe Asthma. J. Asthma Allergy 2019, 12, 401–413. [Google Scholar] [CrossRef] [Green Version]
- Brightling, C.E.; Bleecker, E.R.; Panettieri, R.A., Jr.; Bafadhel, M.; She, D.; Ward, C.K.; van der Merwe, R. Benralizumab for chronic obstructive pulmonary disease and sputum eosinophilia: A randomised, double-blind, placebo-controlled, phase 2a study. Lancet Respir. Med. 2014, 2, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Criner, G.J.; Celli, B.R.; Singh, D.; Agusti, A.; Papi, A.; Jison, M.; Makulova, N.; Shih, V.H.; Brooks, L.; Barker, P.; et al. Predicting response to benralizumab in chronic obstructive pulmonary disease: Analyses of GALATHEA and TERRANOVA studies. Lancet Respir. Med. 2020, 8, 158–170. [Google Scholar] [CrossRef]
- Barranco, P.; Phillips-Angles, E.; Dominguez-Ortega, J.; Quirce, S. Dupilumab in the management of moderate-to-severe asthma: The data so far. Clin. Risk Manag. 2017, 13, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, K.; Katoh, N.; Fujieda, S.; Izuhara, K.; Oishi, K. Dupilumab: Basic aspects and applications to allergic diseases. Allergol. Int. 2020, 69, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Ford, L.; Pearlman, D.; Spector, S.; Sher, L.; Skobieranda, F.; Wang, L.; Kirkesseli, S.; Rocklin, R.; Bock, B.; et al. Dupilumab in Persistent Asthma with Elevated Eosinophil Levels. N. Engl. J. Med. 2013, 368, 2455–2466. [Google Scholar] [CrossRef] [PubMed]
- Rabe, K.F.; Nair, P.; Brusselle, G.; Maspero, J.F.; Castro, M.; Sher, L.; Zhu, H.; Hamilton, J.D.; Swanson, B.N.; Khan, A.; et al. Efficacy and Safety of Dupilumab in Glucocorticoid-Dependent Severe Asthma. N. Engl. J. Med. 2018, 378, 2475–2485. [Google Scholar] [CrossRef]
- Bassani, C.; Rossi, L.; Siveris, K.; Sferelli, R.L.; Saraiva, L.; Tanno, L.K. Use of dupilumab on the treatment of moderate-to-severe asthma: A systematic review. Rev. Da Assoc. Méd. Bras. 2019, 65, 1223–1228. [Google Scholar] [CrossRef]
- Rathinam, K.K.; Abraham, J.J.; Vijayakumar, T.M. Dupilumab in the Treatment of Moderate to Severe Asthma: An Evidence-Based Review. Curr. Res. 2019, 91, 45–51. [Google Scholar] [CrossRef]
- Caminati, M.; Menzella, F.; Guidolin, L.; Senna, G. Targeting eosinophils: Severe asthma and beyond. Drugs Context 2019, 8, 212587. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
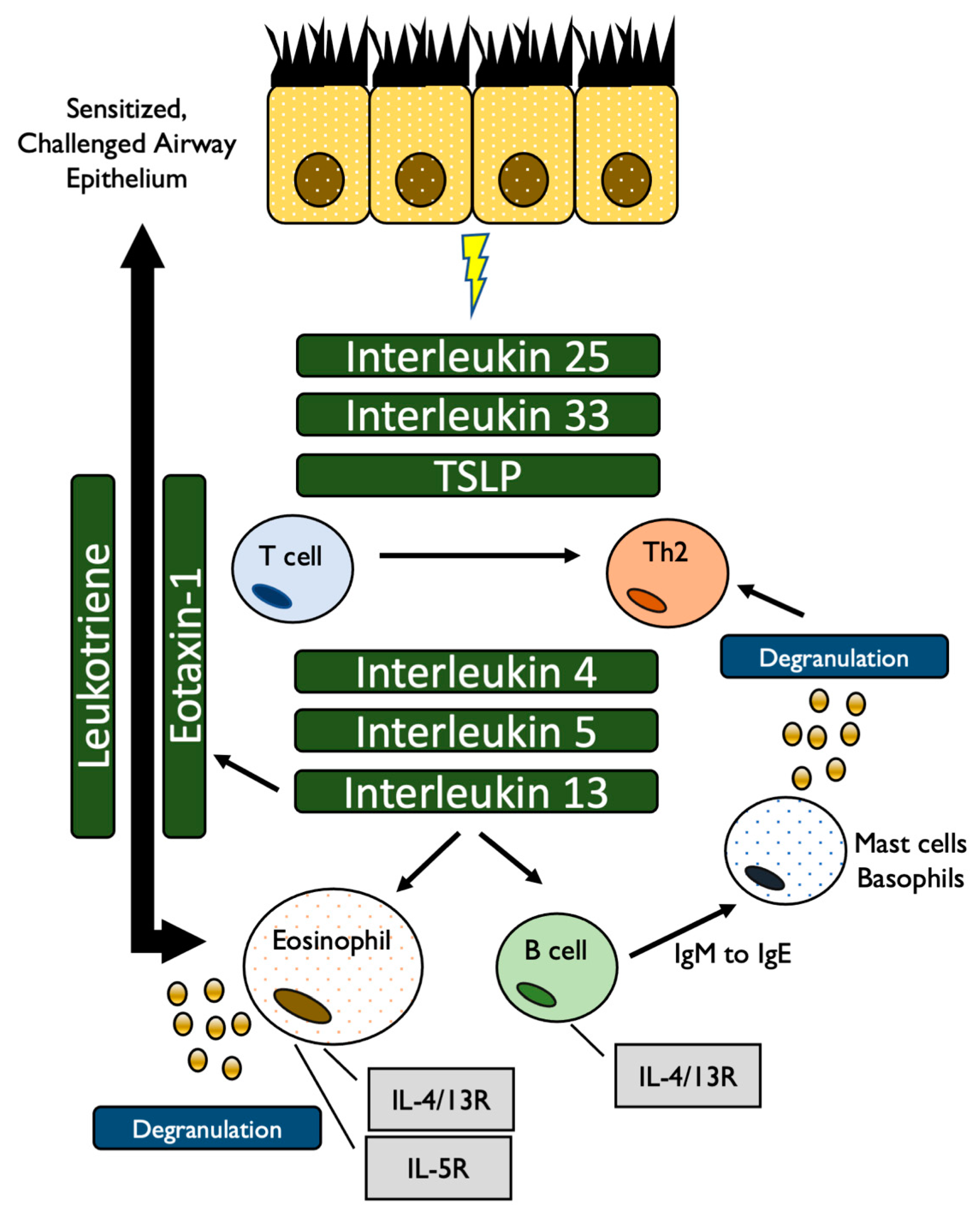
| Cell Type | Secreted Cytokines | Target/Function |
|---|---|---|
| Th2 | IL-4 | Induction of differentiation into Th2 cells Differentiation of B cells Enhance IgG and IgE production |
| IL-5 | Differentiation of B cells Activation of eosinophils Stimulate release of IL-4, IL-13 | |
| IL-13 | Differentiation of B cells IgE production | |
| Epithelium | IL-25, IL-33 | Control secretion of IL-4, IL-5, IL-13 from ILC2 cells |
| TSLP | CD4+ T cells to activate secretion of IL4, IL-5, IL-13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusumoto, M.; Mathis, B.J. Biologic Treatments for Asthma and Chronic Obstructive Pulmonary Disorder. Allergies 2021, 1, 92-107. https://doi.org/10.3390/allergies1020007
Kusumoto M, Mathis BJ. Biologic Treatments for Asthma and Chronic Obstructive Pulmonary Disorder. Allergies. 2021; 1(2):92-107. https://doi.org/10.3390/allergies1020007
Chicago/Turabian StyleKusumoto, Misa, and Bryan J. Mathis. 2021. "Biologic Treatments for Asthma and Chronic Obstructive Pulmonary Disorder" Allergies 1, no. 2: 92-107. https://doi.org/10.3390/allergies1020007
APA StyleKusumoto, M., & Mathis, B. J. (2021). Biologic Treatments for Asthma and Chronic Obstructive Pulmonary Disorder. Allergies, 1(2), 92-107. https://doi.org/10.3390/allergies1020007