
One-Step Purification of Recombinant Cutinase from an E. coli Extract Using a Stabilizing Triazine-Scaffolded Synthetic Affinity Ligand



Abstract

1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Production and Conventional Purification Process of Cutinase
2.3. Screening of Biomimetic Affinity Ligands via Affinity Chromatographic Assays
2.4. Solid-Phase Synthesis of Ligand 11/3′
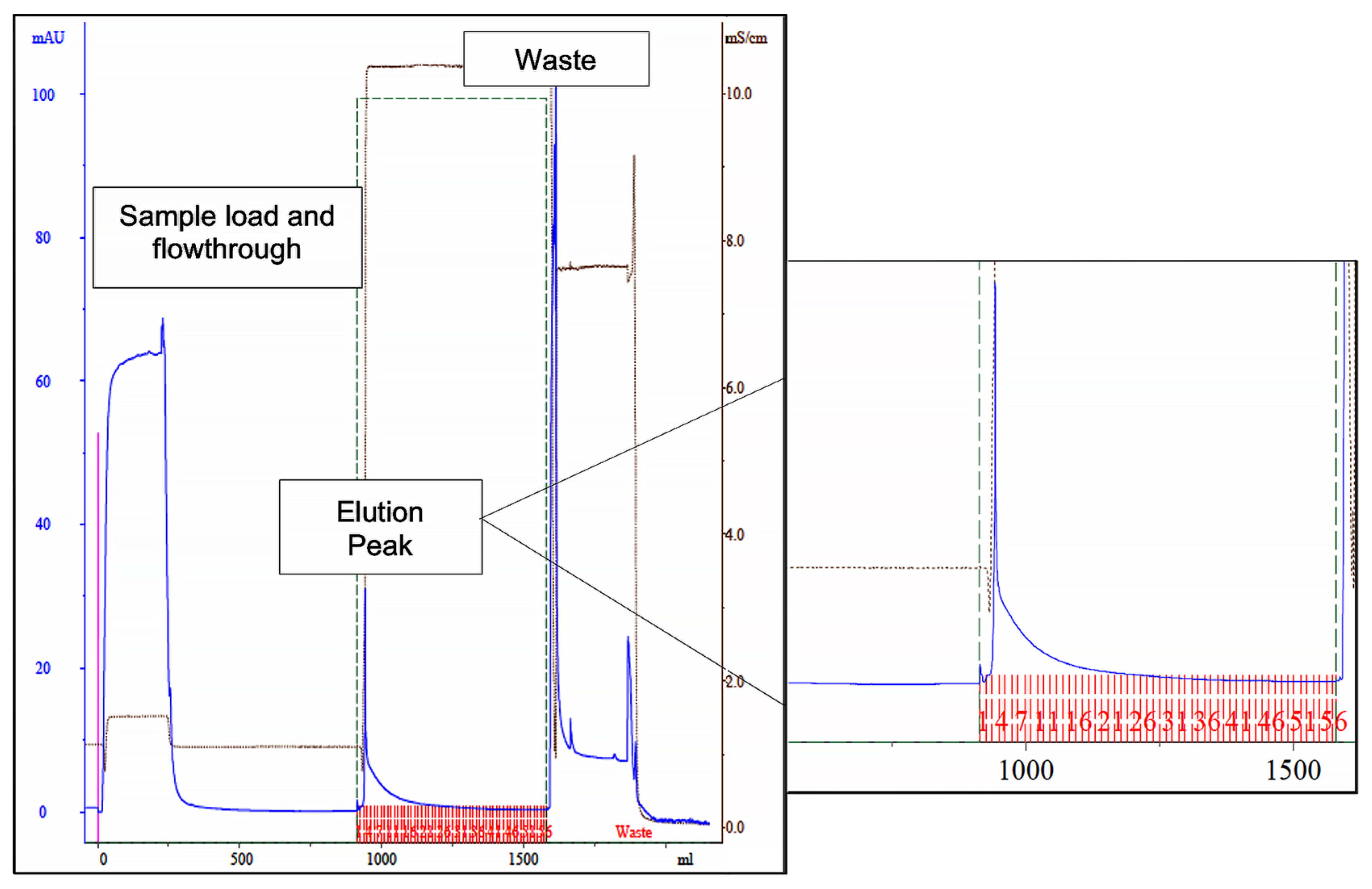
2.4.1. Purification of Cutinase by using Affinity Chromatography with Lead Ligand 11/3′
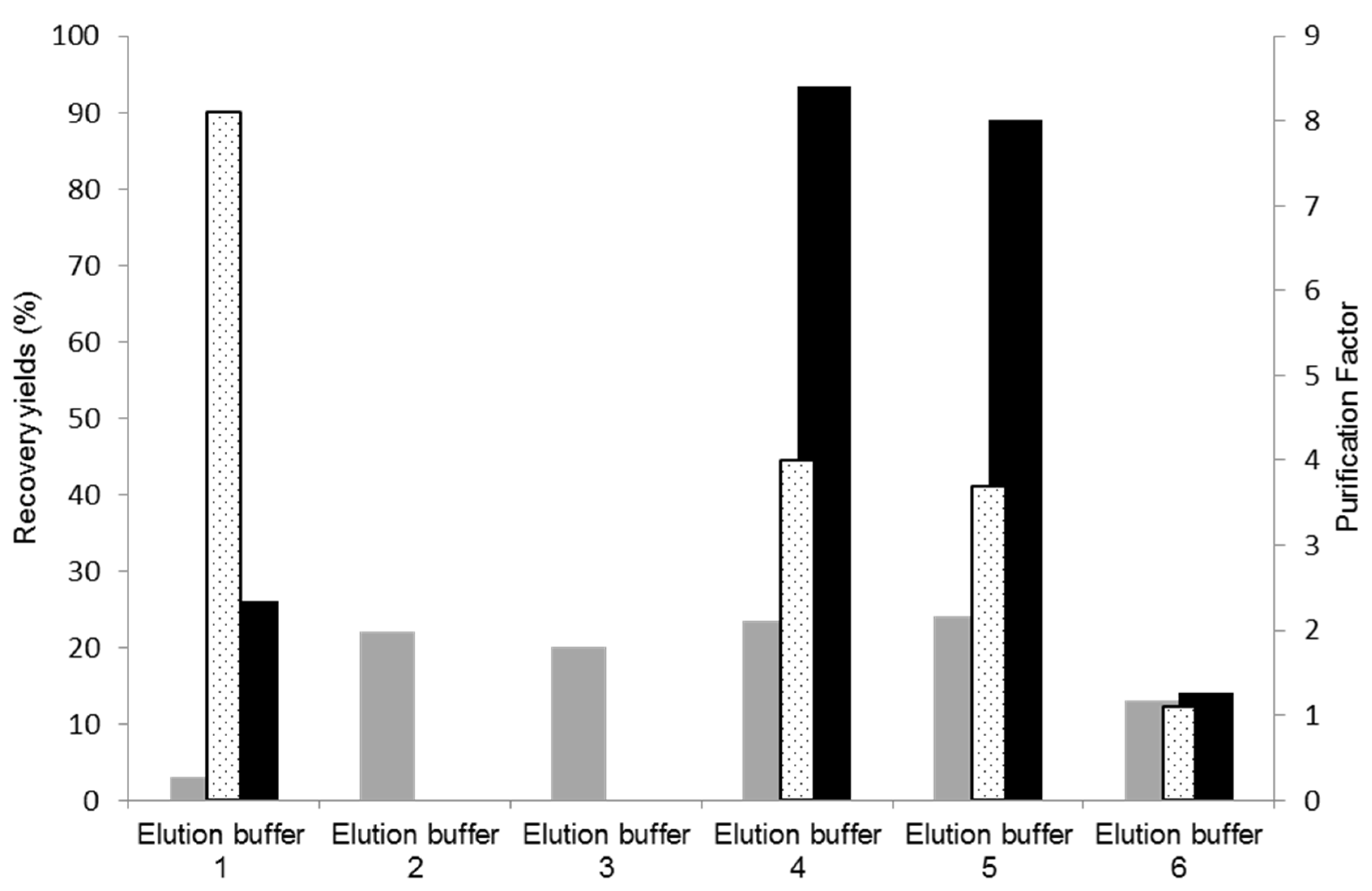
2.4.2. Elution Assays for Optimizing Cutinase Purification
2.5. Protein Quantification Assays
2.6. Enzyme Activity Assay
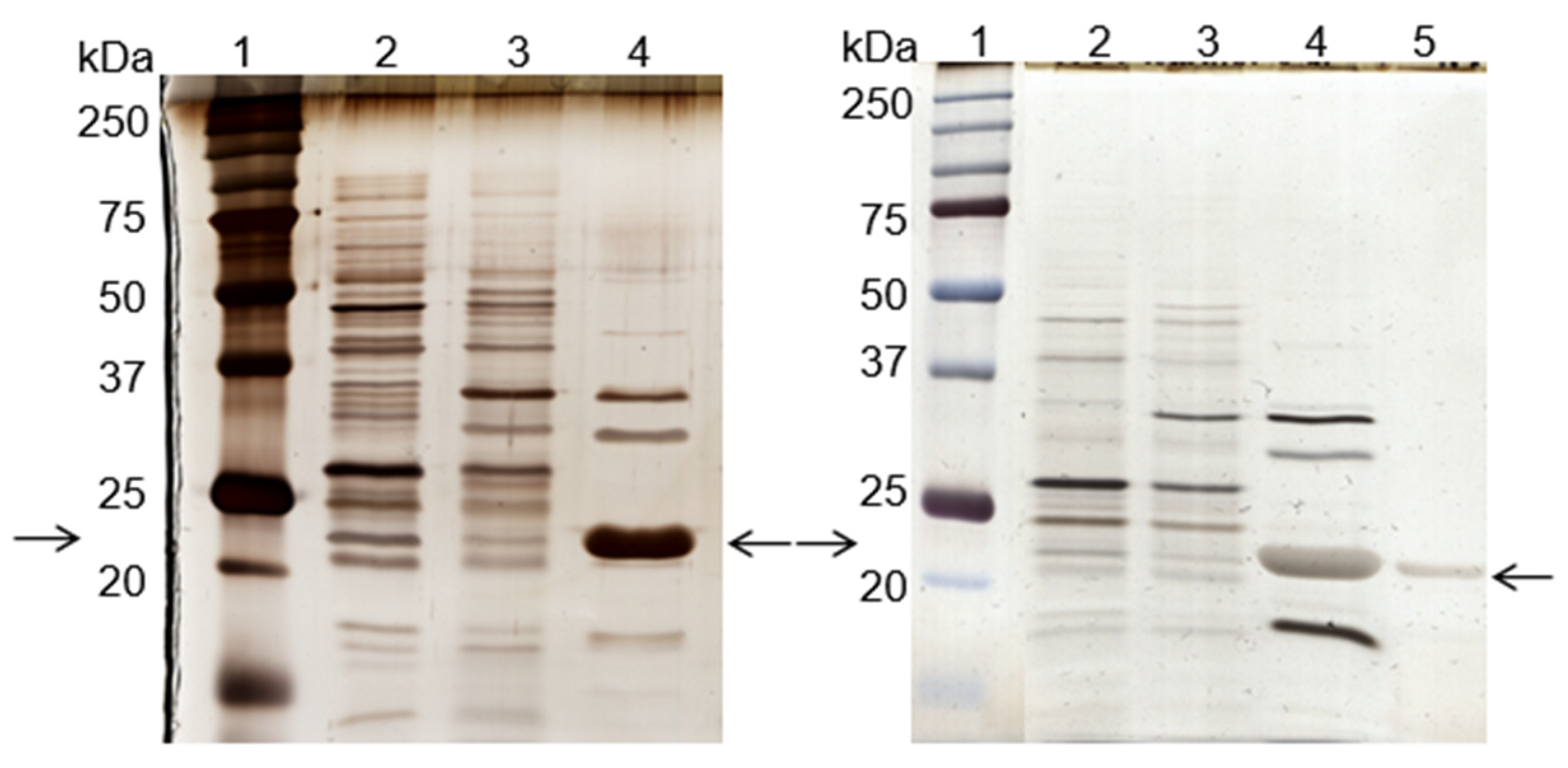
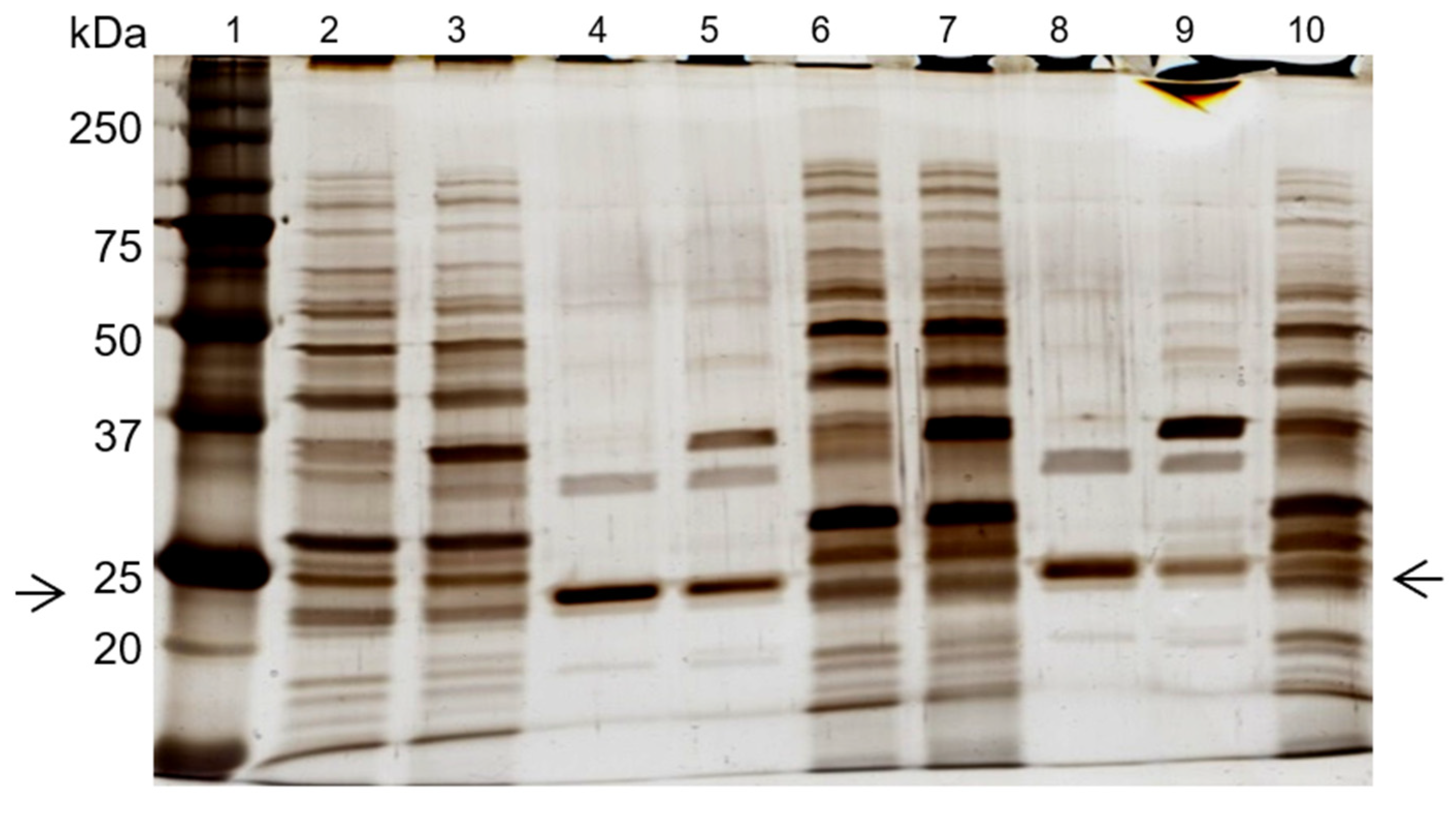
2.7. SDS-PAGE Gel Electrophoresis
3. Results and Discussion
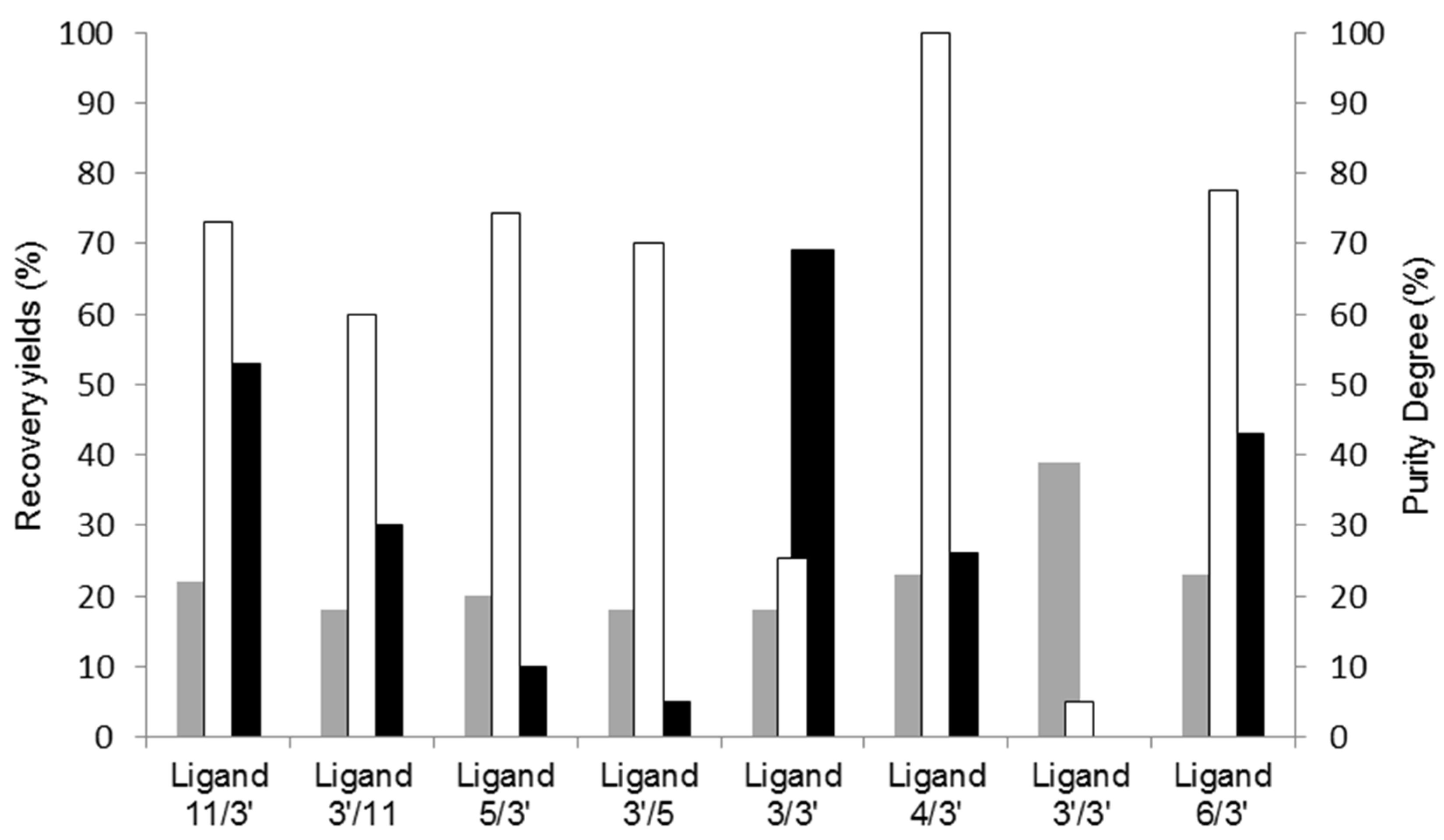
3.1. Screening of Affinity Ligands from a Solid-Phase Combinatorial Library
3.2. Cutinase Purification Using Affinity Chromatography with Ligand 11/3′
3.3. Assessment of Different Elution Buffers to Enhance Cutinase Purification
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Martínez, A.; Maicas, S. Cutinases: Characteristics and Insights in Industrial Production. Catalysts 2021, 11, 1194. [Google Scholar] [CrossRef]
- Martinez, C.; De Geus, P.; Lauwereys, M.; Matthyssens, G.; Cambillau, C. Fusarium solani cutinase is a lipolytic enzyme with a catalytic serine accessible to solvent. Nature 1992, 356, 615–618. [Google Scholar] [CrossRef]
- Dutta, K.; Sen, S.; Veeranki, V.D. Production, characterization and applications of microbial cutinases. Process Biochem. 2009, 44, 127–134. [Google Scholar] [CrossRef]
- Chen, S.; Su, L.; Chen, J.; Wu, J. Cutinase: Characteristics preparation, and application. Biotechnol. Adv. 2013, 31, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Roussel, A.; Amara, S.; Nyyssölä, A.; Mateos-Diaz, E.; Blangy, S.; Kontkanen, H.; Westerholm-Parvinen, A.; Carrière, F.; Cambillau, C. A cutinase from Trichoderma reesei with a lid-covered active site and kinetic properties of true lipases. J. Mol. Biol. 2014, 426, 3757–3772. [Google Scholar] [CrossRef] [PubMed]
- Longhi, S.; Czjzek, M.; Lamzin, V.; Nicolas, A.; Cambillau, C. Atomic resolution (1.0 A) crystal structure of Fusarium solani cutinase: Stereochemical analysis. J. Mol. Biol. 1997, 268, 779–799. [Google Scholar] [CrossRef] [PubMed]
- Purdy, R.E.; Kolattukudy, P.E. Hydrolysis of plant cuticule by plant pathogens. Properties of cutinase I, cutinase II, and a nonspecific esterase isolated from Fusarium solani pisi. Biochemistry 1975, 14, 2832–2840. [Google Scholar] [CrossRef]
- Egmond, M.R.; de Vlieg, J. Fusarium solani pisi cutinase. Biochimie 2000, 82, 1015–1021. [Google Scholar] [CrossRef]
- Longhi, S.; Cambillau, C. Structure-activity of cutinase, a small lipolytic enzyme. Biochim. Biophys. Acta 1999, 1441, 185–196. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Aires-Barros, M.R.; Cabral, J.M. Cutinase: From molecular level to bioprocess development. Biotechnol. Bioeng. 1999, 66, 17–34. [Google Scholar] [CrossRef]
- Nyyssölä, A. Which properties of cutinases are important for applications? Appl. Microbiol. Biotechnol. 2005, 99, 4931–4942. [Google Scholar] [CrossRef] [PubMed]
- Nikolaivits, E.; Kanelli, M.; Dimarogona, M.; Topakas, E. A Middle-Aged Enzyme Still in Its Prime: Recent Advances in the Field of Cutinases. Catalysis 2018, 8, 612. [Google Scholar] [CrossRef]
- Donelli, I.; Freddi, G.; Nierstrasz, V.A.; Taddei, P. Surface structure and properties of poly-(ethylene terephthalate) hydrolyzed by alkali and cutinase. Polym. Degrad. Chem. 2010, 95, 1542–1550. [Google Scholar] [CrossRef]
- Groß, C.; Hamacher, K.; Schmitz, K.; Jager, S. Cleavage Product Accumulation Decreases the Activity of Cutinase during PET Hydrolysis. J. Chem. Inf. Model. 2017, 57, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Hellesnes, K.N.; Vijayaraj, S.; Fojan, P.; Petersen, E.; Courtad, G. Biochemical Characterization and NMR Study of a PET-Hydrolyzing Cutinase from Fusarium solani pisi. Biochemistry 2023, 62, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- De Barros, D.P.C.; Fernandes, P.; Cabral, J.M.S.; Fonseca, L.P. Synthetic application and activity of cutinase in an aqueous, miniemulsion model system: Hexyl octanoate synthesis. Catal. Today 2011, 173, 95–102. [Google Scholar] [CrossRef]
- De Barros, D.P.C.; Pinto, F.; Pfluck, A.C.D.; Dias, A.S.A.; Fernandes, P.; Fonseca, L.P. Improvement of enzyme stability for alkyl esters synthesis in miniemulsion systems by using media engineering. J. Chem. Technol. Biotechnol. 2018, 93, 1338–1346. [Google Scholar] [CrossRef]
- Sebastião, M.J.; Cabral, J.M.; Aires-Barros, M.R. Synthesis of fatty acid esters by a recombinant cutinase in reversed micelles. Biotechnol. Bioeng. 1993, 42, 326–332. [Google Scholar] [CrossRef]
- Rodriguez, E.L.; Poddar, S.; Iftekhar, S.; Suh, K.; Woolfork, A.G.; Ovbude, S.; Pekarek, S.; Walters, M.; Lott, S.; Hage, D.S. Affinity chromatography: A review of trends and developments over the past 50 years. J. Chromatogr. B 2020, 1157, 122332–122348. [Google Scholar] [CrossRef]
- Chevrel, A.; Candela Innocenti, E.; Golibrzuch, C.; Skudas, R.; Schwämmle, A.; Carrondo, M.J.T.; Kitten, O.; Nissum, M.; Silva, R.J.S. Development of versatile affinity-based system for one step purification process: Case of Group A Streptococcus vaccine. Biotechnol. Bioeng. 2022, 119, 3210–3220. [Google Scholar] [CrossRef]
- Roque, A.C.A.; Silva, C.S.O.; Taipa, M.A. Affinity-based methodologies and ligands for antibody purification: Advances and perspectives. J. Chromatogr. A 2007, 1160, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Topcu̧, A.A.; Kılıc, S.; Özgür, E.; Türkmen, D.; Denizli, A. Inspirations of Biomimetic Affinity Ligands: A Review. ACS Omega 2022, 7, 32897–32907. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-M.; Lin, D.-Q.; Yao, S.-J. Review on biomimetic affinity chromatography with short peptide ligands and its application to protein purification. J. Chromatogr. A 2018, 1571, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Perret, G.; Boschetti, E. Aptamer affinity ligands in protein chromatography. Biochimie 2018, 145, 98–112. [Google Scholar] [CrossRef]
- Lowe, C.R. Combinatorial approaches to affinity chromatography. Curr. Opin. Chem. Biol. 2001, 5, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Lowe, C.R.; Lowe, A.R.; Gupta, G. New developments in affinity chromatography with potential application in the production of biopharmaceuticals. J. Biochem. Biophys. Methods 2001, 49, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.B.; Pina, A.C.; Roque, A.C.A. Rational design of affinity ligands for bioseparation. J. Chromatogr. A 2020, 1619, 460871–460885. [Google Scholar] [CrossRef]
- Sousa, I.T.; Taipa, M.A. Biomimetic Affinity Ligands for Protein Purification. In Protein Downstream Processing: Design, Development and Application of High and Low-Resolution Methods; Labrou, N., Ed.; Springer Science Methods in Molecular Biology Series; Humana: Totowa, NJ, USA, 2021; Volume 2178, pp. 167–199, Chapter 14. [Google Scholar] [CrossRef]
- Ruiu, L.; Roque, A.C.A.; Taipa, M.A.; Lowe, C.R. De novo design, synthesis and screening of a combinatorial library of complementary ligands directed towards the surface of cutinase from Fusarium solani pisi. J. Mol. Recognit. 2006, 19, 372–378. [Google Scholar] [CrossRef]
- Sousa, I.T.; Ruiu, L.; Lowe, C.R.; Taipa, M.A. Synthetic affinity ligands as a novel tool to improve protein stability. J. Mol. Recognit. 2009, 22, 83–90. [Google Scholar] [CrossRef]
- Sousa, I.T.; Lourenço, N.M.T.; Afonso, C.A.M.; Taipa, M.A. Protein Stabilization with a triazine- scaffolded dipeptide-mimic synthetic affinity ligand. J. Mol. Recognit. 2013, 26, 104–112. [Google Scholar] [CrossRef]
- Lauwereys, M.; De Geus, P.; De Meutter, J.; Stanssens, P.; Matthyssens, G. Cloning, Expression and Characterization of Cutinase, a Fungal Lipolytic Enzyme. In Lipases-Structure, Function and Genetic Engineering; Alberghina, L., Schmid, R.D., Verger, R., Eds.; Wiley-VCH: Weinheim, Germany, 1991; pp. 243–251. Available online: http://www.wiley-vch.de/publish/en/company/contact/?sID=v2kgfqie8apmrte0ge7noc68s7 (accessed on 15 November 2023).
- Sebastião, M.J.; Cabral, J.M.S.; Aires-Barros, M.R. Improved purification protocol of a Fusarium solani pisi recombinant cutinase by phase partitioning in aqueous two-phase systems of polyethylene glycol and phosphate. Enzyme Microb. Technol. 1996, 18, 251–260. [Google Scholar] [CrossRef]
- Antoni, G.; Presentini, R.; Neri, P. A simple method for the estimation of amino groups on insoluble matrix beads. Anal. Biochem. 1983, 129, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Calado, C.R.C.; Monteiro, S.M.S.; Cabral, J.M.S.; Fonseca, L.P. Effect of pre-fermentation on the production of cutinase by a recombinant Saccharomyces cerevisiae. J. Biosci. Bioeng. 2002, 93, 354–359. [Google Scholar] [CrossRef]
- Azevedo, A.M.; Gomes, A.C.; Borlido, L.; Santos, I.F.S.; Prazeres, D.M.F.; Aires-Barros, M.R. Capture of human monoclonal antibodies from a clarified cell culture supernatant by phenyl boronate chromatography. J. Mol. Recognit. 2010, 23, 569–576. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Structure of the Biomimetic Adsorbents | Number | Aminated Compound R1 or R2 | Analogue Amino Acids |
|---|---|---|---|
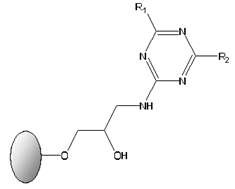 | 3 5 11 3′ 4′ 6 | Tyramine Isoamlylamine 2-Methylbutylamine 4-Aminobenzoic acid 4-Aminophenylacetic acid 4-Aminobutyric acid | Tyrosine Leucine Isoleucine Aspartic acid Glutamic acid Aspartic acid Glutamic acid Glutamic acid |
| Sample | Volume (L) | Protein (mg/L) | Total Protein (mg) | Activity (U/mL) | Total Activity (U) | Specific Activity (U/mg Protein) | Protein Yield (%) | Activity Yield (%) | Purification Factor |
|---|---|---|---|---|---|---|---|---|---|
| First dialysis extract | 0.015 | 255.3 | 3.8 | 49.7 | 745.5 | 195 | 100 | 100 | 1.0 |
| Breakthrough | 0.015 | 100 | 1.5 | 0.3 | 4.5 | 3 | 39 | 1 | 0.0 |
| Washing pool | 0.04 | 27.0 | 1.1 | 0.0 | 0.0 | 0 | 28 | 0 | 0.0 |
| Elution Peak | 0.005 | 45.0 | 0.2 | 56.7 | 283.5 | 1260 | 6 | 38 | 6.5 |
| Elution Pool 1 | 0.030 | 23.5 | 0.7 | 9.7 | 291.0 | 413 | 18 | 39 | 2.1 |
| Total Elution Pool 2 | 0.035 | 26.8 | 0.9 | 21.8 | 763.0 | 813 | 24 | ≅100 | 4.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, L.P.; Taipa, M.Â. One-Step Purification of Recombinant Cutinase from an E. coli Extract Using a Stabilizing Triazine-Scaffolded Synthetic Affinity Ligand. Biomimetics 2024, 9, 57. https://doi.org/10.3390/biomimetics9010057
Fonseca LP, Taipa MÂ. One-Step Purification of Recombinant Cutinase from an E. coli Extract Using a Stabilizing Triazine-Scaffolded Synthetic Affinity Ligand. Biomimetics. 2024; 9(1):57. https://doi.org/10.3390/biomimetics9010057
Chicago/Turabian StyleFonseca, Luís P., and M. Ângela Taipa. 2024. "One-Step Purification of Recombinant Cutinase from an E. coli Extract Using a Stabilizing Triazine-Scaffolded Synthetic Affinity Ligand" Biomimetics 9, no. 1: 57. https://doi.org/10.3390/biomimetics9010057
APA StyleFonseca, L. P., & Taipa, M. Â. (2024). One-Step Purification of Recombinant Cutinase from an E. coli Extract Using a Stabilizing Triazine-Scaffolded Synthetic Affinity Ligand. Biomimetics, 9(1), 57. https://doi.org/10.3390/biomimetics9010057