
Attachment of Fibrinogen on Ion Beam Treated Polyurethane

Abstract
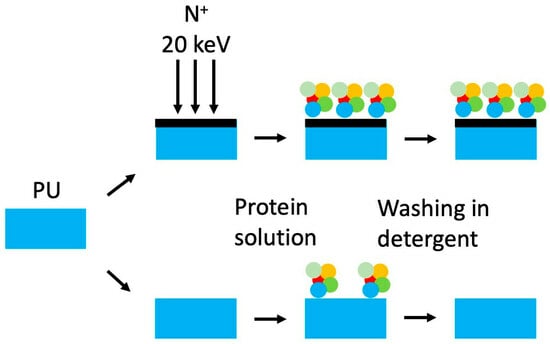
:
1. Introduction
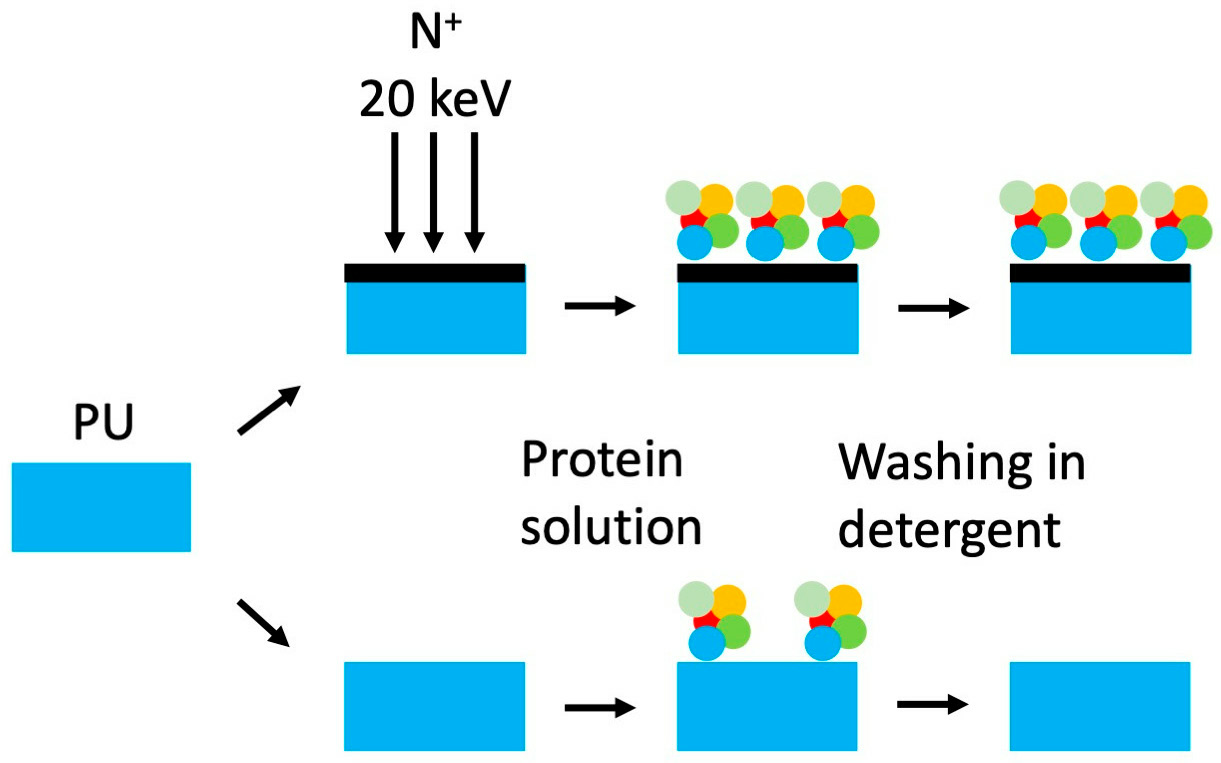
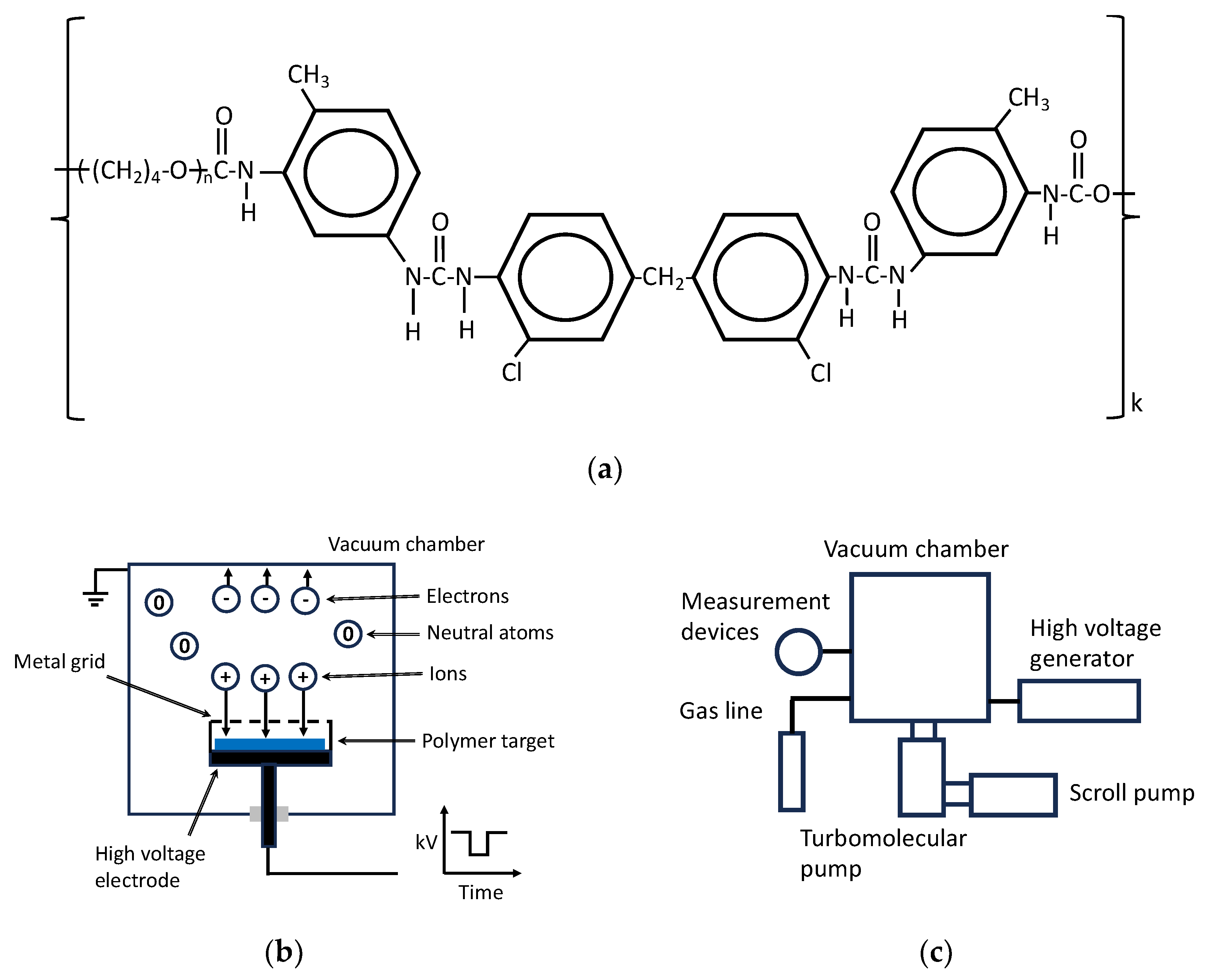
2. Materials and Methods
3. Results
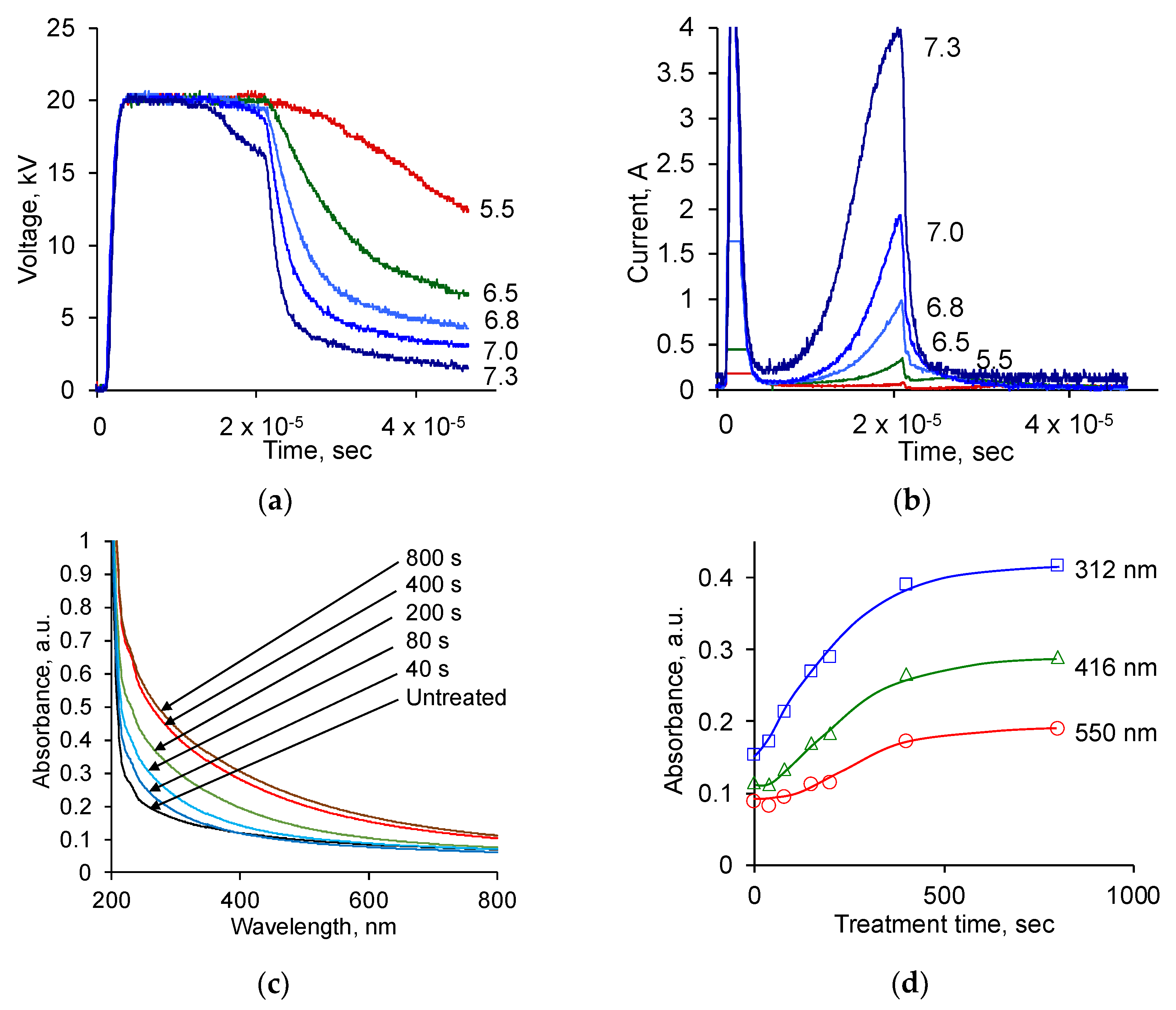
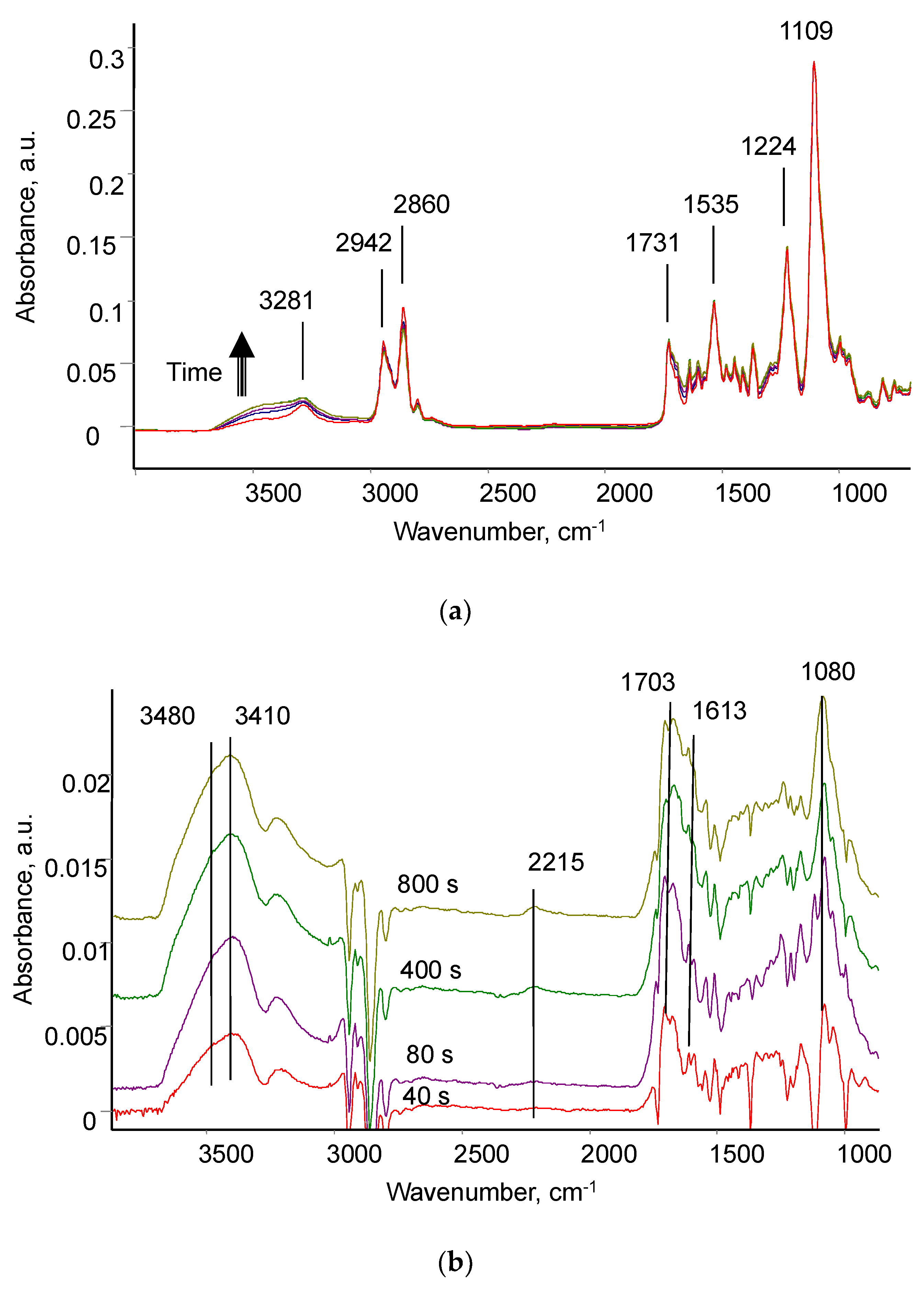
3.1. Evaluation of Treatment Parameters
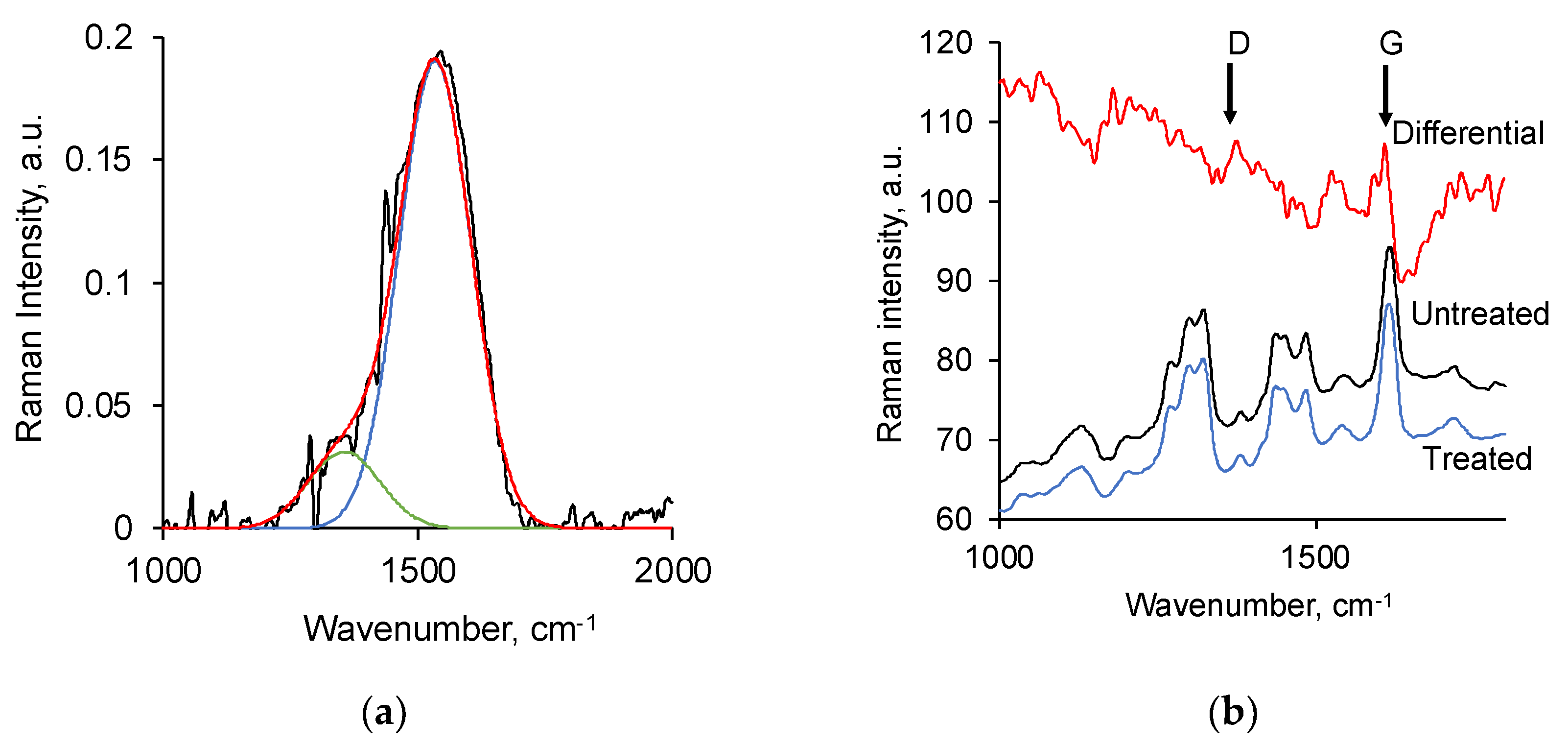
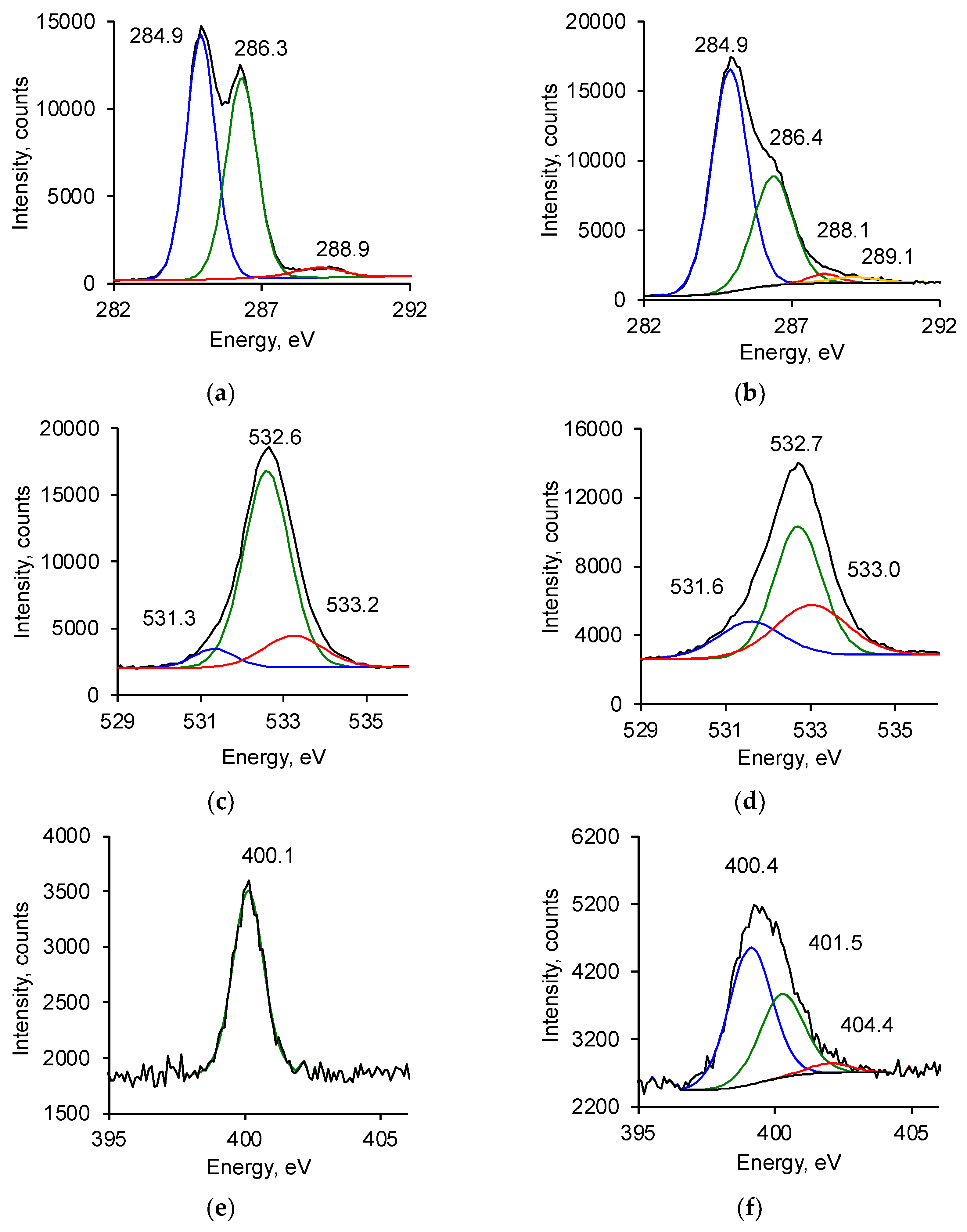
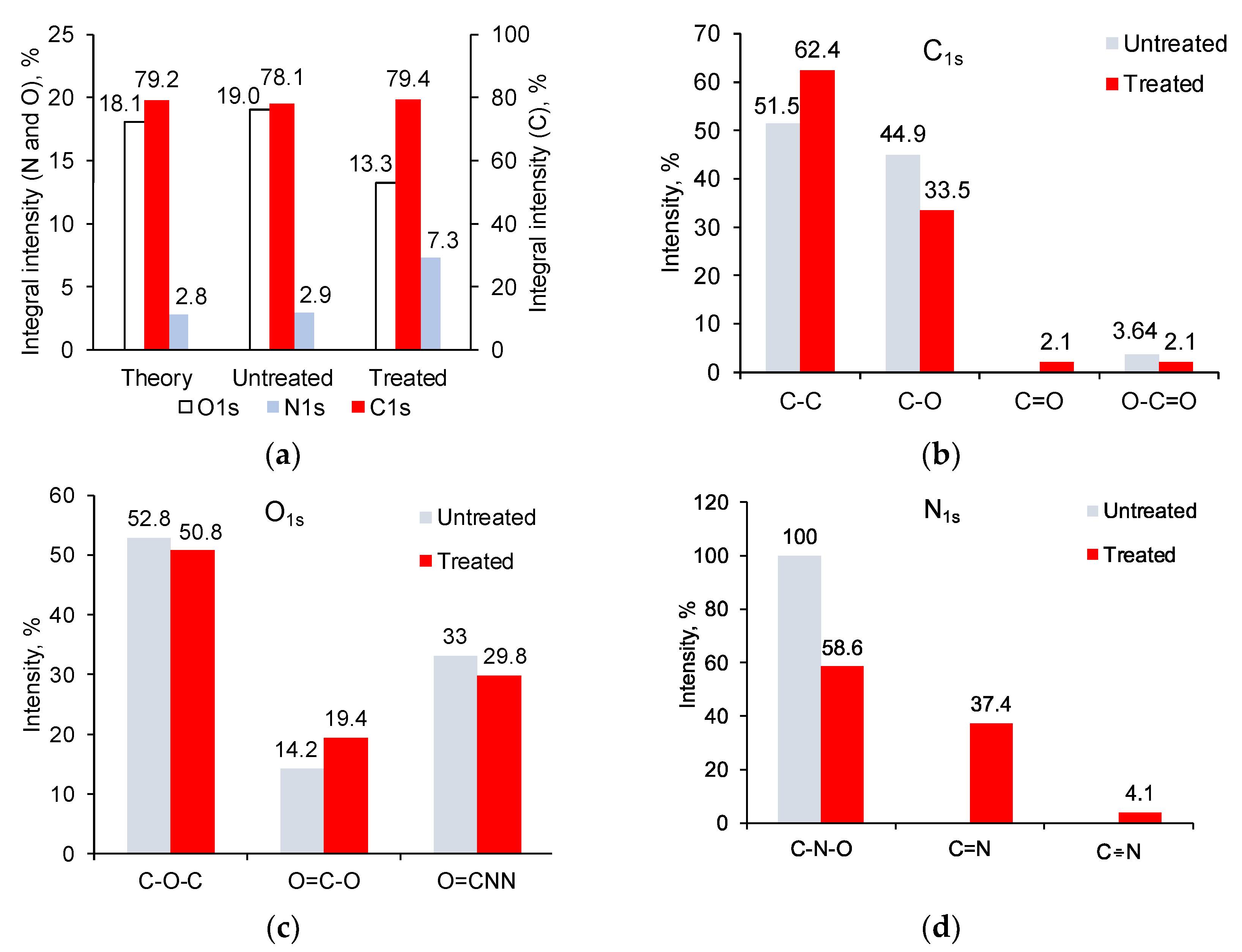
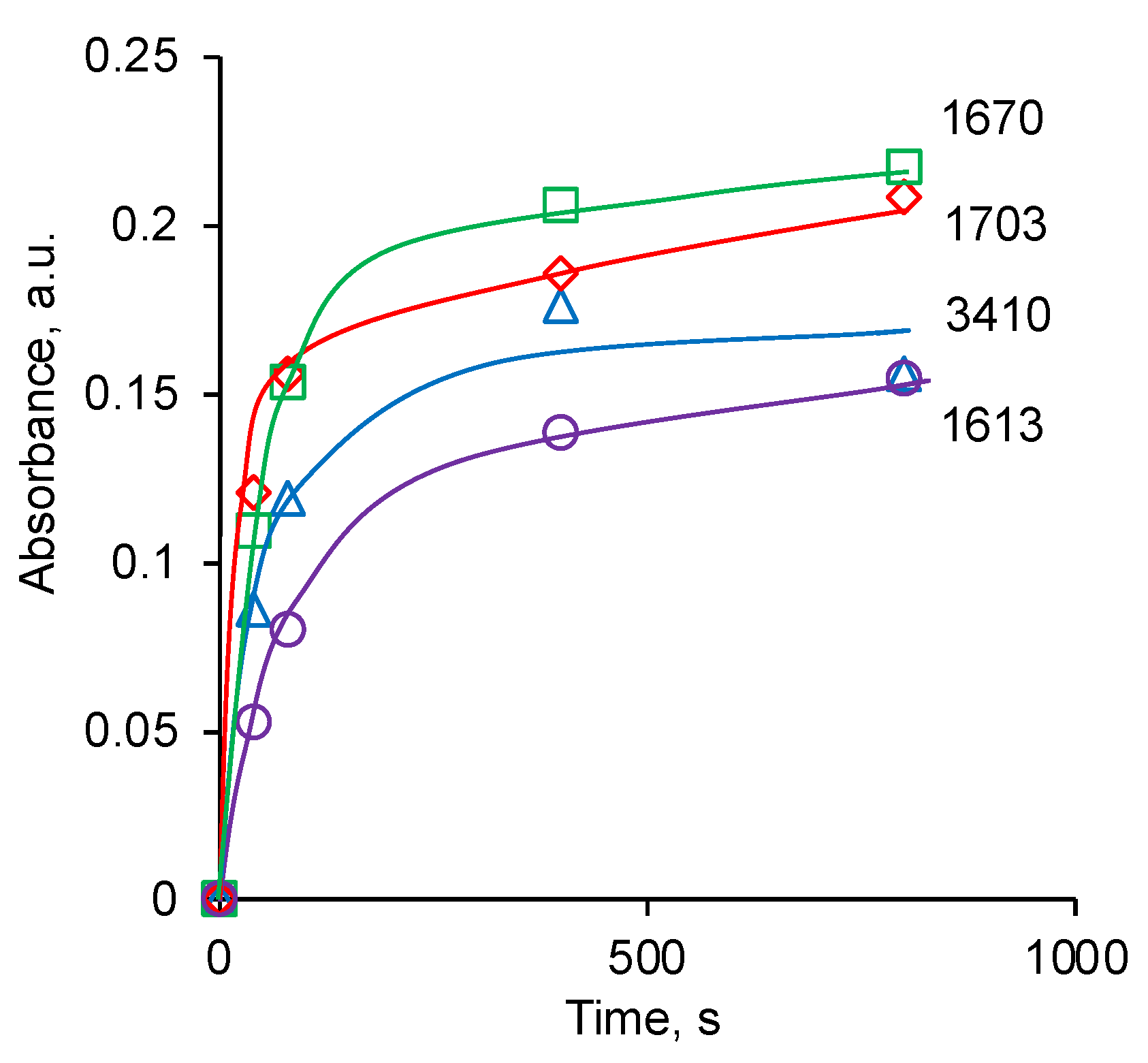
3.2. Ion Beam Treatment of Polyurethane
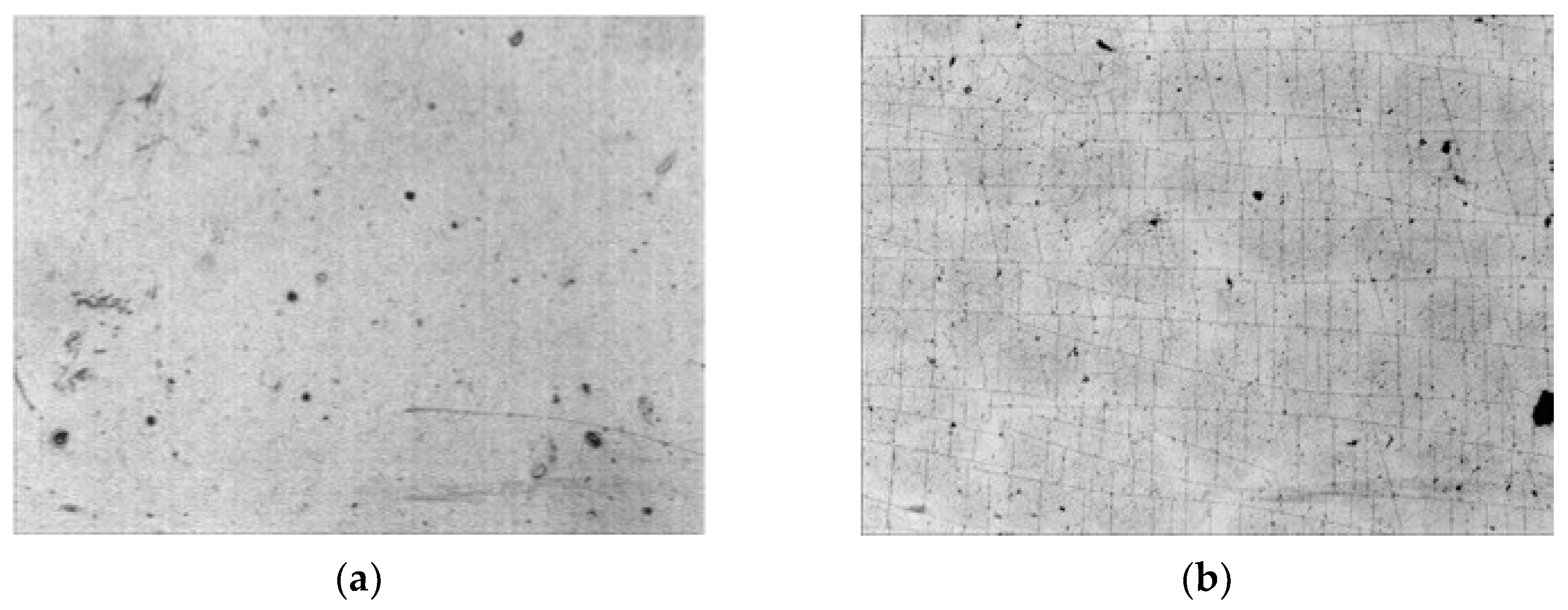
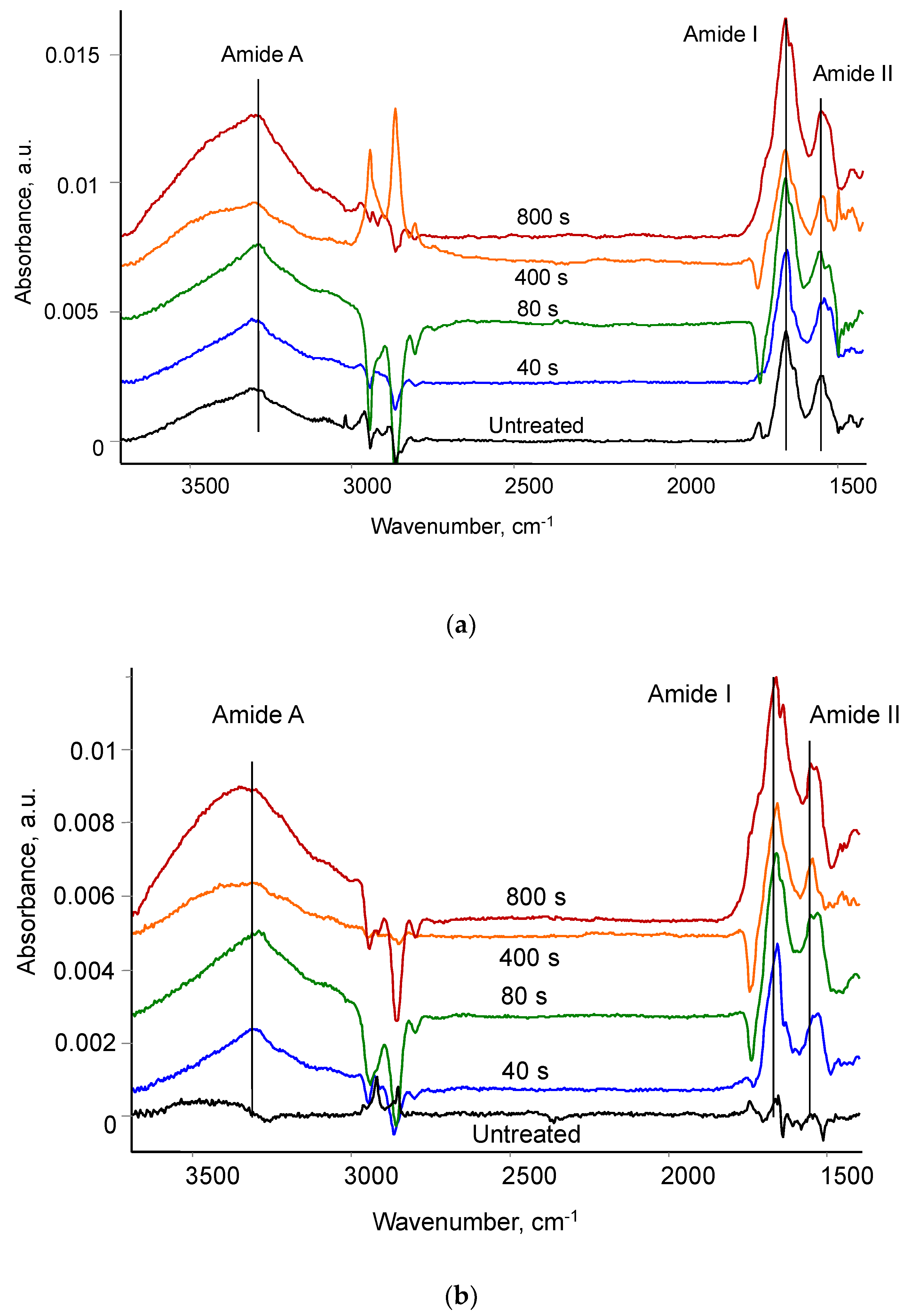
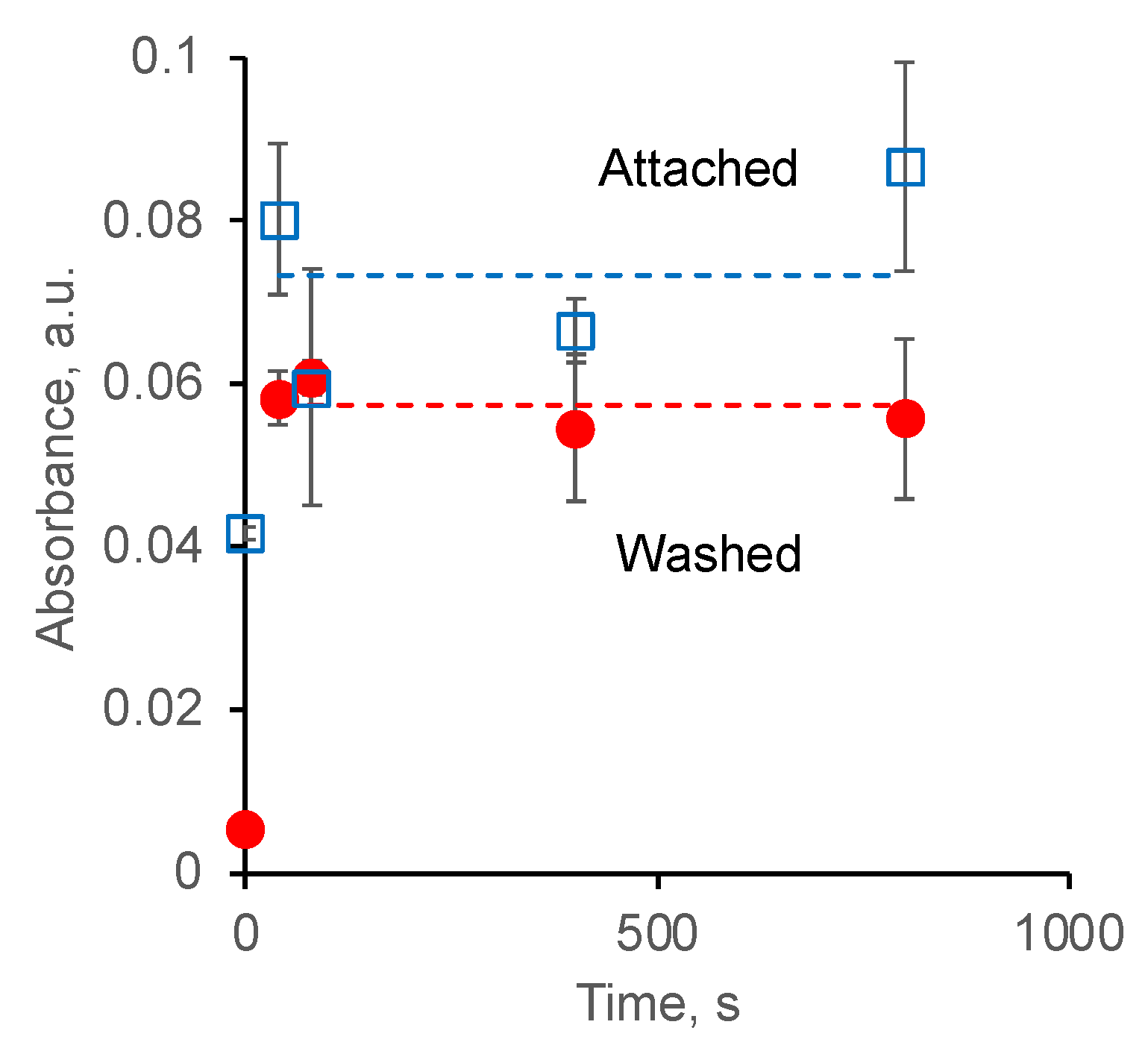
3.3. Attachment of Fibrinogen
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mariani, E.; Lisignoli, G.; Borzì, R.M.; Pulsatelli, L. Biomaterials: Foreign Bodies or Tuners for the Immune Response? Int. J. Mol. Sci. 2019, 20, 636. [Google Scholar] [CrossRef] [PubMed]
- Ward, W.K. A Review of the Foreign-body Response to Subcutaneously-implanted Devices: The Role of Macrophages and Cytokines in Biofouling and Fibrosis. J. Diabetes Sci. Technol. 2008, 2, 768–777. [Google Scholar] [CrossRef]
- Kizhakkedathu, J.N.; Conway, E.M. Biomaterial and cellular implants: Foreign surfaces where immunity and coagulation meet. Blood 2022, 139, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Eaton, J. Natural Responses to Unnatural Materials: A Molecular Mechanism for Foreign Body Reactions. Mol. Med. 1999, 5, 351–358. [Google Scholar] [CrossRef]
- Schmid, A.; Dordick, J.S.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt, B. Industrial biocatalysis today and tomorrow. Nature 2001, 409, 258–268. [Google Scholar] [CrossRef]
- Hu, W.-J.; Eaton, J.W.; Tang, L. Molecular basis of biomaterial-mediated foreign body reactions. Blood 2001, 98, 1231–1238. [Google Scholar] [CrossRef]
- Castner, D.G.; Ratner, B.D. Biomedical surface science: Foundations to frontiers. Surf. Sci. 2002, 500, 28–60. [Google Scholar] [CrossRef]
- Ottenbrite, R.M. (Ed.) Frontiers in Biomedical Polymer Applications; Technomic Publishing Company: Lancaster, UK, 1998. [Google Scholar]
- Ryan, B.J.; Carolan, N.; Fagain, C. Horseradish and soybean peroxidases: Comparable tools for alternative niches? Trends Biotechnol. 2006, 24, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Boecking, T.; James, M.; Coster, H.G.L.; Chilcott, T.C.; Barrow, K.D. Structural Characterization of Organic Multilayers on Silicon (111) formed by Immobilization of Molecular Films on Functionalized Si-C Linked Monolayers. Langmuir 2004, 20, 9227–9235. [Google Scholar] [CrossRef]
- Williams, R.A.; Blanch, H.W. Covalent Immobilization of Protein Monolayers for Biosensor Applications. Biosens. Bioelectron. 1994, 9, 159–167. [Google Scholar] [CrossRef]
- Peterman, J.H.; Tarcha, P.J.; Chu, V.P.; Butler, J.E. The immunochemistry of sandwich-ELISAs IV. The Antigen Capture Capacity of Antibody Covalently Attached to Bromoacetyl Surface-Functionalized Polystyrene. J. Immunol. Methods 1988, 111, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Cunha, A.G.; Fernández-Lorente, G.; Bevilaqua, J.V.; Destain, J.; Paiva, L.M.C.; Freire, D.M.G.; Fernández-Lafuente, R.; Guisán, J.M. Immobilization of Yarrowia lipolytica lipase--a comparison of stability of physical adsorption and covalent attachment techniques. Appl. Biochem. Biotechnol. 2008, 146, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.Y.; Tye, G.J.; Chew, A.L.; Ng, W.K.; Lai, N.S. Linker-mediated oriented antibody immobilisation strategies for a more efficient immunosensor and diagnostic applications: A review. Biosens. Bioelectron. 2023, 14, 100379. [Google Scholar] [CrossRef]
- Abbas, M.; Torati, S.R.; Kim, C.G. Multifunctional Fe3O4/Au core/satellite nanocubes: An efficient chemical synthesis, characterization and functionalization of streptavidin protein. Dalton Trans. 2017, 46, 2303–2309. [Google Scholar] [CrossRef]
- Baldini, F.; Carloni, A.; Giannetti, A.; Porro, G.; Trono, C. An optical PMMA biochip based on fluorescence anisotropy: Application to C-reactive protein assay. Sens. Actuators 2009, 139, 64–68. [Google Scholar] [CrossRef]
- Bhardwaj, H.; Sumana, G.; Marquette, C.A. A label-free ultrasensitive microfluidic surface Plasmon resonance biosensor for Aflatoxin B1 detection using nanoparticles integrated gold chip. Food Chem. 2020, 307, 125530. [Google Scholar] [CrossRef] [PubMed]
- Ricci, S.; Casalini, S.; Parkula, V.; Selvaraj, M.; Saygin, G.D.; Greco, P.; Biscarini, F.; Mas-Torrent, M. Label-free immunodetection of α-synuclein by using a microfluidics coplanar electrolyte-gated organic field-effect transistor. Biosens. Bioelectron. 2020, 167, 112433. [Google Scholar] [CrossRef] [PubMed]
- Merker, D.; Bertinetti, D.; Merz, R.; Kopnarski, M.; Herberg, F.W.; Popov, C. Enhanced protein immobilization efficacy by nanostructuring of ultrananocrystalline diamond surface. Diam. Relat. Mater. 2023, 136, 109898. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Han, N.; Wu, Y.; Wei, D. Oriented covalent immobilization of recombinant protein A on the glutaraldehyde activated agarose support. Int. J. Biol. Macromol. 2018, 120, 100–108. [Google Scholar] [CrossRef]
- Chen, W.-C.; Chen, Y.-S.; Chang, K.-C.; Chen, C.-H.; Li, D.-J. An in vitro assessment and comparative effectiveness of silanized-glutaraldehyde functionalized titanium surfaces with phosphatidylcholine and type I collagen grafts. Dent. Mater. 2020, 36, 320–328. [Google Scholar] [CrossRef]
- de Andrades, D.; Graebin, N.G.; Kadowaki, M.K.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Immobilization and stabilization of different β-glucosidases using the glutaraldehyde chemistry: Optimal protocol depends on the enzyme. Int. J. Biol. Macromol. 2019, 129, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, M.; Slota, A.J.; Eravuchira, P.J.; Alba, M.; Formentin, P.; Pallarès, J.; Ferré-Borrull, J.; Marsal, L.F. Protein attachment to silane-functionalized porous silicon: A comparison of electrostatic and covalent attachment. J. Colloid Interface Sci. 2015, 452, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Stachelek, S.J.; Finley, M.J.; Alferiev, I.S.; Wang, F.; Tsai, R.K.; Eckells, E.C.; Tomczyk, N.; Connolly, J.M.; Discher, D.E.; Eckmann, D.M.; et al. The effect of CD47 modified polymer surfaces on inflammatory cell attachment and activation. Biomaterials 2011, 32, 4317–4326. [Google Scholar] [CrossRef]
- Fink, D. Fundamentals of Ion-Irradiated Polymers; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Odzhaev, V.B.; Kozlov, I.P.; Popok, V.N.; Sviridov, D.B. Ion Implantation of Polymers; Belorussian State University: Minsk, Belorussia, 1998. [Google Scholar]
- Bačáková, L.; Švorčík, V.; Rybka, V.; Miček, I.; Hnatowicz, V.; Lisá, V.; Kocourek, F. Adhesion and proliferation of cultured human aortic smooth muscle cells on polystyrene implanted with N+, F+ and Ar+ ions: Correlation with polymer surface polarity and carbonization. Biomaterials 1996, 17, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, L.; Compagnini, G.; Foti, G. Structural modification of polymer film by ion irradiation. Nucl. Instr. Meth. 1992, 62, 413–422. [Google Scholar] [CrossRef]
- Davenas, J.; Thevenard, P.; Boiteux, G.; Fallavier, M.; Lu, X.L. Hydrogenated carbon layers produced by ion beam irradiation of PMMA and polystyrene films. Nucl. Instrum. Meth. Phys. Res. 1990, 46, 317–323. [Google Scholar] [CrossRef]
- Tóth, A.; Bell, T.; Bertóti, I.; Mohai, M.; Zelei, B. Surface modification of polyethylene by low keV ion beams. Nucl. Instrum. Methods Phys. Res. 1999, 148, 1131–1135. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kaibara, M.; Iwaki, M.; Sasabe, H.; Suzuki, Y.; Kusakabe, M. Selective adhesion and proliferation of cells on ion-implanted polymer domains. Biomaterials 1993, 14, 958. [Google Scholar] [CrossRef] [PubMed]
- Melnig, V.; Apetroaei, N.; Dumitrascu, N.; Suzuki, Y.; Tura, V. Improvement of polyurethane surface biocompatibility by plasma and ion beam techniques. J. Optoelectron. Adv. Mater. 2005, 7, 2521–2528. [Google Scholar]
- Marletta, G. Ion-beam modification of polymer surfaces for biological applications. In Materials Science with Ion Beams; Bernas, H., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 345–371. [Google Scholar]
- Fu, R.K.Y.; Cheung, I.T.L.; Mei, Y.F.; Shek, C.H.; Siu, G.G.; Chu, P.K.; Yang, W.M.; Leng, Y.X.; Huang, Y.X.; Tian, X.B.; et al. Surface modification of polymeric materials by plasma immersion ion implantation. Nucl. Inst. Meth. Phys. Res. 2005, 237, 417–421. [Google Scholar] [CrossRef]
- Kostov, K.G.; Ueda, M.; Tan, I.H.; Leite, N.F.; Beloto, A.F.; Gomes, G.F. Structural effect of nitrogen plasma-based ion implantation on ultra-high molecular weight polyethylene. Surf. Coat. Technol. 2004, 186, 287–290. [Google Scholar] [CrossRef]
- Sahre, K.; Eichhorn, K.-J.; Simon, F.; Pleul, D.; Janke, A.; Gerlach, G. Characterization of ion-beam modified polyimide layers. Surf. Coat. Technol. 2001, 139, 257–264. [Google Scholar] [CrossRef]
- Wong, K.H.; Zinke-Allmang, M.; Wan, W.K.; Zhang, J.Z.; Hu, P. Low energy oxygen ion beam modification of the surface morphology and chemical structure of polyurethane fibers. Nucl. Inst. Meth. Phys. Res. 2006, 243, 63–74. [Google Scholar] [CrossRef]
- Popok, V. Ion implantation of polymers: Formation of nanoparticulate materials. Rev. Adv. Mater. Sci. 2012, 30, 1–26. [Google Scholar]
- Murphy, J.J.; Patel, M.; Powell, S.J.; Smith, P.F. Volatile evolution induced by energetic He++ ions in a poly(ester) based polyurethane. Radiat. Phys. Chem. 2002, 63, 101–108. [Google Scholar] [CrossRef]
- Iwaki, M.; Nakao, A.; Kaibara, M.; Sasabe, H.; Kaneko, S.; Nakajima, H.; Suzuki, Y.; Kusakabe, M.; Fujihana, T. Ion bombardment into inner wall surfaces of tubes and their biomedical applications. Nucl. Inst. Meth. Phys. Res. 1995, 106, 618–623. [Google Scholar] [CrossRef]
- Dejun, L.; Jie, Z.; Hanqing, G.; Mozhu, L.; Fuqing, D.; Qiqing, Z. Surface modification of medical polyurethane by silicon ion bombardment. Nucl. Inst. Meth. Phys. Res. 1993, 82, 57–62. [Google Scholar] [CrossRef]
- Huang, N.; Yang, P.; Leng, Y.X.; Wang, J.; Sun, H.; Chen, J.Y.; Wan, G.J. Surface modification of biomaterials by plasma immersion ion implantation. Surf. Coat. Technol. 2004, 186, 218–226. [Google Scholar] [CrossRef]
- Vyatkin, A.F.; Smirnov, V.V.; Kholopkin, A.I. Development of ion implantation equipment in the USSR. Solid State Technol. 1991, 34, 57. [Google Scholar] [CrossRef]
- Begishev, V.; Gavrilov, N.; Mesyats, G.; Klyachkin, Y.; Kondyurina, I.; Osorgina, I.; Kondyurin, A. Modification of polyurethane endoprosthetics surface by pulse ion beam. In Proceedings of the 12th International Conference on High-Power Particle Beams, Haifa, Israel, 7–12 June 1998; Volume 2, pp. 997–1000. [Google Scholar]
- Mesyats, G.; Klyachkin, Y.; Gavrilov, N.; Kondyurin, A. Adhesion of Polytetrafluorethylene modified by an ion beam. Vacuum 1999, 52, 285–289. [Google Scholar] [CrossRef]
- Kondyurin, A.; Maitz, M.F. Surface Modification of ePTFE and Implants Using the Same. U.S. Patent WO2007/022174A3, 15 February 2007. [Google Scholar]
- Kondyurin, A.; Bilek, M. Ion Beam Treatment of Polymers. In Application Aspects from Medicine to Space, 2nd ed.; Elsevier: Oxford, UK, 2014. [Google Scholar]
- Kondyurin, A.V.; Naseri, P.; Tilley, J.M.R.; Nosworthy, N.J.; Bilek, M.M.M.; McKenzie, D.R. Mechanisms for Covalent Immobilization of Horseradish Peroxidase on Ion-Beam-Treated Polyethylene. Scientifica 2012, 2012, 126170. [Google Scholar] [CrossRef] [PubMed]
- Kondyurina, I.; Kondyurin, A. Foreign Body Reaction (Immune Response) for Artificial Implants Can Be Avoided: An Example of Polyurethane in Mice for 1 Week. J. Funct. Biomater. 2023, 14, 432. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.G.; Lamghari, M.; Almeida, C.R.; Oliveira, M.I.; Neves, N.; Ribeiro, A.C.; Barbosa, J.N.; Barros, R.; Maciel, J.; Martins, M.C.L.; et al. Adsorbed fibrinogen leads to improved bone regeneration and correlates with differences in the systemic immune response. Acta Biomater. 2013, 9, 7209–7217. [Google Scholar] [CrossRef] [PubMed]
- Balabiyev, A.; Podolnikova, N.P.; Kilbourne, J.A.; Baluch, D.P.; Lowry, D.; Zare, A.; Ros, R.; Flick, M.J.; Ugarova, T.P. Fibrin polymer on the surface of biomaterial implants drives the foreign body reaction. Biomaterials 2021, 277, 121087. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, Y.; Chen, J.; Yu, P.; Tang, F.; Hu, Z.; Zhou, J.; Liu, L.; Qiu, W.; Ye, Y.; et al. Regulation of biomaterial implantation-induced fibrin deposition to immunological functions of dendritic cells. Mater. Today Bio. 2022, 14, 100224. [Google Scholar] [CrossRef] [PubMed]
- Vilar, R.; Fish, R.J.; Casini, A.; Neerman-Arbez, M. Fibrin(ogen) in human disease: Both friend and foe. Haematologica 2020, 105, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, D.M.; Gonçalves, R.M.; Almeida, C.R.; Pereira, I.O.; Oliveira, M.I.; Neves, N.; Silva, A.M.; Ribeiro, A.C.; Cunha, C.; Almeida, A.R.; et al. Fibrinogen scaffolds with immunomodulatory properties promote in vivo bone regeneration. Biomaterials 2016, 111, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Gavrilov, N.V.; Oks, E.M. High-current pulse sources of broad beams of gas and metal ions for surface treatment. Nucl. Instrum. Methods Phys. Res. 2000, 439, 31–44. [Google Scholar] [CrossRef]
- Anders, A. (Ed.) Handbook of Plasma Immersion Ion Implantation and Deposition; Wiley: New York, NY, USA, 2000. [Google Scholar]
- McKenzie, D.R.; Powles, R. Plasma Immersion Ion Implantation Using Conductive Mesh. US Patent 2007/0268089A1, 22 November 2007. [Google Scholar]
- Osorgina, I.V.; Porozova, S.E.; Plaksin, S.A.; Morozov, I.A. Effect of Long-Term Exposure within the Body on the Condition of Polyurethane Membranes of Breast Prostheses. Biomed. Eng. 2016, 50, 63–66. [Google Scholar] [CrossRef]
- Budnikov, V.I.; Begishev, V.P. Sanitary and chemical evaluation of polyester urethane elastomers. Hyg. Sanit. 1997, 2, 48–49. [Google Scholar]
- Romanova, V.A.; Begishev, V.P. Regulating of properties of polymeric matrix on base of polyurethane at production of composites. Compos. Mater. Constr. 2006, 4, 39–43. [Google Scholar]
- Ziegler, J.F.; Manoyan, J. The stopping of ions in compounds. Nucl. Instr. and Meth. 1998, 35, 215. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Biersack, J.P. The Stopping and Range of Ions in Solid; Pergamon: New York, NY, USA, 1985. [Google Scholar]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. 2000, 61, 14095–14107. [Google Scholar] [CrossRef]
- Piazza, F.; Grambole, D.; Scheider, D.; Casiraghi, C.; Ferrari, A.C.; Robertson, J. Protective diamond-like carbon coatings for future optical storage disks. Diamond Relat. Mater. 2005, 14, 994–999. [Google Scholar] [CrossRef]
- Stagg, B.; Charalampopoulos, T. Refractive index of pyrolitic graphite, amorphous carbon, and flame soot in the temperature range 25 to 600 °C. Combust. Flame 1993, 94, 381–396. [Google Scholar] [CrossRef]
- Gambino, R.J.; Thompson, J.A. Spin resonance spectroscopy of amorphous carbon films. Solid State Commun. 1980, 34, 15–18. [Google Scholar] [CrossRef]
- Fusco, G.; Tagliaferro, A.; Milne, W.I.; Robertson, J. Paramagnetic centres in tetrahedral amorphous carbon. Diam. Relat. Mater. 1997, 6, 783–786. [Google Scholar] [CrossRef]
- Barklie, R.C. Characterisation of defects in amorphous carbon by electron paramagnetic resonance. Diam. Relat. Mater. 2001, 10, 174–181. [Google Scholar] [CrossRef]
- Viana, G.A.; Lacerda, R.G.; Freire, F.L.; Marques, F.C. ESR investigation of graphite-like amorphous carbon films revealing itinerant states as the ones responsible for the signal. J. Non-Cryst. Solids 2008, 354, 2135–2137. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title 1 | Title 2 | Title 3 | |||
|---|---|---|---|---|---|
| Water | CH2I2 | Total | Polar | Dispersic | |
| Untreated | 94 | 55 | 32.6 | 1.2 | 31.4 |
| Treated after 30 min | 37 | 39 | 65.4 | 25.3 | 40.1 |
| Treated, after 1 month | 57 | 50 | 50.6 | 16.4 | 34.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chudinov, V.; Shardakov, I.; Kondyurina, I.; Kondyurin, A. Attachment of Fibrinogen on Ion Beam Treated Polyurethane. Biomimetics 2024, 9, 234. https://doi.org/10.3390/biomimetics9040234
Chudinov V, Shardakov I, Kondyurina I, Kondyurin A. Attachment of Fibrinogen on Ion Beam Treated Polyurethane. Biomimetics. 2024; 9(4):234. https://doi.org/10.3390/biomimetics9040234
Chicago/Turabian StyleChudinov, Vyacheslav, Igor Shardakov, Irina Kondyurina, and Alexey Kondyurin. 2024. "Attachment of Fibrinogen on Ion Beam Treated Polyurethane" Biomimetics 9, no. 4: 234. https://doi.org/10.3390/biomimetics9040234