

Review of the Brain’s Behaviour after Injury and Disease for Its Application in an Agent-Based Model (ABM)

 , , ,
, , ,  and
and {kind=link}
{kind=link}
Abstract
:1. Introduction
Scope and Methodology
2. Mapping the Brain: The Connectome
3. Brain Damage
3.1. Acquired Brain Injury
3.1.1. Traumatic Brain Injury (TBI)
3.1.2. Non-Traumatic Brain Injury (NTBI)
3.2. Neurological Disorders
3.2.1. Epilepsy
3.2.2. Neurodevelopmental Disorders and Disabilities
3.2.3. Neurodegenerative Diseases
3.3. Psychiatric Disorders
4. Modelling Approaches
4.1. Electrophysiological/Haemodynamical
4.2. Biomechanical
4.3. Mathematical
4.4. Application to a Model of the Boolean Logic Behaviour of Neuronal Self-Organised Communities
5. Conclusions and Future Research Lines
Author Contributions
Funding

Acknowledgments
Conflicts of Interest
References
- Padamsey, Z.; Rochefort, N.L. Paying the brain’s energy bill. Curr. Opin. Neurobiol. 2023, 78, 102668. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, H.; Spooner, F.; Roser, M. Causes of Death. Our World in Data. 2018. Available online: https://ourworldindata.org/causes-of-death (accessed on 19 October 2023).
- Murphy, S.J.; Werring, D.J. Stroke: Causes and clinical features. Medicine 2023, 51, 602–607. [Google Scholar] [CrossRef]
- McKinney, P.A. Brain tumours: Incidence, survival, and aetiology. J. Neurol. Neurosurg. Psychiatry 2004, 75, ii12–ii17. [Google Scholar] [CrossRef]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA A Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Multiple Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.L.; Anderson, J.R.; Dahal, J.; Garcia, J.C.; Yang, J.H.; Sigulinsky, C.L.; Rapp, K.; Emrich, D.P.; Watt, C.B.; Johnstun, H.A.; et al. A pathoconnectome of early neurodegeneration: Network changes in retinal degeneration. Exp. Eye Res. 2020, 199, 108196. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, X.; Hamalainen, T.; Cong, F. Exploring Oscillatory Dysconnectivity Networks in Major Depression During Resting State Using Coupled Tensor Decomposition. IEEE Trans. Biomed. Eng. 2022, 69, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Rubinov, M.; Bullmore, E. Fledgling pathoconnectomics of psychiatric disorders. Trends Cogn. Sci. 2013, 17, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Chojdak-Łukasiewicz, J.; Dziadkowiak, E.; Zimny, A.; Paradowski, B. Cerebral small vessel disease: A review. Adv. Clin. Exp. Med. 2021, 30, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.K.; Stamenova, V.; Turner, G.; Black, S.E.; Levine, B. Pathoconnectomics of cognitive impairment in small vessel disease: A systematic review. Alzheimer’s Dement. 2016, 12, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Gustavsson, A.; Svensson, M.; Wittchen, H.U.; Jönsson, B. The economic cost of brain disorders in Europe. Eur. J. Neurol. 2011, 19, 155–162. [Google Scholar] [CrossRef] [PubMed]
- DiLuca, M.; Olesen, J. The Cost of Brain Diseases: A Burden or a Challenge? Neuron 2014, 82, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Parés-Badell, O.; Barbaglia, G.; Jerinic, P.; Gustavsson, A.; Salvador-Carulla, L.; Alonso, J. Cost of Disorders of the Brain in Spain. PLoS ONE 2014, 9, e105471. [Google Scholar] [CrossRef] [PubMed]
- Pino, R.D.; Díez-Cirarda, M.; Ustarroz-Aguirre, I.; Gonzalez-Larragan, S.; Caprino, M.; Busnatu, S.; Gand, K.; Schlieter, H.; Gabilondo, I.; Gómez-Esteban, J.C. Costs and effects of telerehabilitation in neurological and cardiological diseases: A systematic review. Front. Med. 2022, 9, 832229. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, S.J.; Shahbaz, N.N.; Nisa, Q.; Umer, S.R.; Ali, S.G.; Aziz, M.Y. Cost of Investigating Neurological Disease: Experience of a Tertiary Care Center in Karachi, Pakistan. Cureus 2020, 12, e9291. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Multiple Collaborators. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [PubMed]
- Falk, E.B.; Hyde, L.W.; Mitchell, C.; Faul, J.; Gonzalez, R.; Heitzeg, M.M.; Keating, D.P.; Langa, K.M.; Martz, M.E.; Maslowsky, J.; et al. What is a representative brain? Neuroscience meets population science. Proc. Natl. Acad. Sci. USA 2013, 110, 17615–17622. [Google Scholar] [CrossRef] [PubMed]
- Dotson, V.M.; Duarte, A. The importance of diversity in cognitive neuroscience. Ann. N. Y. Acad. Sci. 2019, 1464, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Green, K.H.; Groep, I.H.V.D.; Brinke, L.W.T.; van der Cruijsen, R.; van Rossenberg, F.; Marroun, H.E. A perspective on enhancing representative samples in developmental human neuroscience: Connecting science to society. Front. Integr. Neurosci. 2022, 16, 981657. [Google Scholar] [CrossRef] [PubMed]
- Siuly, S.; Zhang, Y. Medical Big Data: Neurological Diseases Diagnosis Through Medical Data Analysis. Data Sci. Eng. 2016, 1, 54–64. [Google Scholar] [CrossRef]
- Alonso, S.G.; de la Torre-Díez, I.; Hamrioui, S.; López-Coronado, M.; Barreno, D.C.; Nozaleda, L.M.; Franco, M. Data Mining Algorithms and Techniques in Mental Health: A Systematic Review. J. Med. Syst. 2018, 42, 161. [Google Scholar] [CrossRef]
- Dipietro, L.; Gonzalez-Mego, P.; Ramos-Estebanez, C.; Zukowski, L.H.; Mikkilineni, R.; Rushmore, R.J.; Wagner, T. The evolution of Big Data in neuroscience and neurology. J. Big Data 2023, 10, 116. [Google Scholar] [CrossRef] [PubMed]
- Li, R. Data Mining and Machine Learning Methods for Dementia Research. In Biomarkers for Alzheimer’s Disease Drug Development; Springer: New York, NY, USA, 2018; pp. 363–370. [Google Scholar] [CrossRef]
- Baniya, B.; Athawale, S.V.; Choudhary, M.L.; Ram, N. Neurodegenerative Alzheimer’s Disease Disorders and Deep Learning Approaches. In Data Analysis for Neurodegenerative Disorders; Springer Nature: Singapore, 2023; pp. 49–66. [Google Scholar] [CrossRef]
- Eschenburg, K.M.; Grabowski, T.J.; Haynor, D.R. Learning Cortical Parcellations Using Graph Neural Networks. Front. Neurosci. 2021, 15, 797500. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Y.; Rekik, I.; Massoud, Y.; Solé-Casals, J. Editorial: Graph learning for brain imaging. Front. Neurosci. 2022, 16, 1001818. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Ma, L.; Jiang, T.; Zhang, Y. Unrevealing Reliable Cortical Parcellation of Individual Brains Using Resting-State Functional Magnetic Resonance Imaging and Masked Graph Convolutions. Front. Neurosci. 2022, 16, 838347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Kuai, H.; Chen, J.; Huang, J.; Liang, P.; Zhong, N. Systematic Fusion of Multi-Source Cognitive Networks With Graph Learning—A Study on Fronto-Parietal Network. Front. Neurosci. 2022, 16, 866734. [Google Scholar] [CrossRef] [PubMed]
- Kurucu, M.C.; Rekik, I. Graph neural network based unsupervised influential sample selection for brain multigraph population fusion. Comput. Med. Imaging Graph. 2023, 108, 102274. [Google Scholar] [CrossRef] [PubMed]
- Bessadok, A.; Mahjoub, M.A.; Rekik, I. Graph Neural Networks in Network Neuroscience. IEEE Trans. Pattern Anal. Mach. Intell. 2023, 45, 5833–5848. [Google Scholar] [CrossRef] [PubMed]
- Najarro, E.; Sudhakaran, S.; Risi, S. Towards Self-Assembling Artificial Neural Networks through Neural Developmental Programs. arXiv 2023, arXiv:2307.08197. [Google Scholar] [CrossRef]
- Makram, H. Seven challenges for neuroscience. Funct. Neurol. 2013, 28, 145–151. [Google Scholar]
- Akil, H.; Martone, M.E.; Van Essen, D.C. Challenges and Opportunities in Mining Neuroscience Data. Science 2011, 331, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Brodmann, K. Vergleichende Lokalisationslehre der Groβhirnrinde: In Ihren Prinzipien Dargestellt Auf Grund des Zellenbaues; Barth: Leipzig, Germany, 1909. [Google Scholar]
- Ferris, C.F.; Cai, X.; Qiao, J.; Switzer, B.; Baun, J.; Morrison, T.; Iriah, S.; Madularu, D.; Sinkevicius, K.W.; Kulkarni, P. Life without a brain: Neuroradiological and behavioral evidence of neuroplasticity necessary to sustain brain function in the face of severe hydrocephalus. Sci. Rep. 2019, 9, 16479. [Google Scholar] [CrossRef] [PubMed]
- Miller, A. The Lobotomy Patient. A Decade Later. Can. Med. Assoc. 1967, 96, 1095–1103. [Google Scholar]
- Dickman, H.; Fletke, K.; Redfern, R.E. Prolonged unassisted survival in an infant with anencephaly. BMJ Case Rep. 2016, 2016, bcr2016215986. [Google Scholar] [CrossRef] [PubMed]
- Nagappan, P.G.; Chen, H.; Wang, D.Y. Neuroregeneration and plasticity: A review of the physiological mechanisms for achieving functional recovery postinjury. Mil. Med. Res. 2020, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Louie, D.R.; Peters, S.; Liu-Ambrose, T.; Boyd, L.A.; Eng, J.J. Brain activity during real-time walking and with walking interventions after stroke: A systematic review. J. NeuroEngineering Rehabil. 2021, 18, 8. [Google Scholar] [CrossRef] [PubMed]
- Bigler, E.D. Structural Image Analysis of the Brain in Neuropsychology Using Magnetic Resonance Imaging (MRI) Techniques. Neuropsychol. Rev. 2015, 25, 224–249. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.A.; See, A.P.; Essayed, W.I.; Golby, A.J.; Tie, Y. Challenges and techniques for presurgical brain mapping with functional MRI. NeuroImage Clin. 2018, 17, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, P.; Dang, A.T.; Do, Q.; Netoff, T.I.; Lim, K.O.; Atluri, G. Evaluation of functional MRI-based human brain parcellation: A review. J. Neurophysiol. 2022, 128, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, P.; Kurant, M.; Gigandet, X.; Thiran, P.; Wedeen, V.J.; Meuli, R.; Thiran, J.P. Mapping Human Whole-Brain Structural Networks with Diffusion MRI. PLoS ONE 2007, 2, e597. [Google Scholar] [CrossRef] [PubMed]
- Sagar, S.; Rick, J.; Chandra, A.; Yagnik, G.; Aghi, M.K. Functional brain mapping: Overview of techniques and their application to neurosurgery. Neurosurg. Rev. 2018, 42, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Alkemade, A.; Bazin, P.L.; Balesar, R.; Pine, K.; Kirilina, E.; Möller, H.E.; Trampel, R.; Kros, J.M.; Keuken, M.C.; Bleys, R.L.A.W.; et al. A unified 3D map of microscopic architecture and MRI of the human brain. Sci. Adv. 2022, 8, eabj7892. [Google Scholar] [CrossRef] [PubMed]
- Amunts, K.; Mohlberg, H.; Bludau, S.; Zilles, K. Julich-Brain: A 3D probabilistic atlas of the human brain’s cytoarchitecture. Science 2020, 369, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Borys, D.; Kijonka, M.; Psiuk-Maksymowicz, K.; Gorczewski, K.; Zarudzki, L.; Sokol, M.; Swierniak, A. Non-parametric MRI Brain Atlas for the Polish Population. Front. Neuroinformatics 2021, 15, 684759. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, T.; Li, Y.; Fan, Z.; Xiong, D.; Wang, X.; Gao, M.; Smith, S.M.; Zhu, H. An atlas of trait associations with resting-state and task-evoked human brain functional organizations in the UK Biobank. Imaging Neurosci. 2023, 1, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Cordes, D.; Haughton, V.M.; Arfanakis, K.; Wendt, G.J.; Turski, P.A.; Moritz, C.H.; Quigley, M.A.; Meyeranda, M.E. Mapping Functionally Related Regions of Brain with Functional Connectivity MR Imaging. Am. J. Neuroradiol. 2000, 21, 1636–1644. [Google Scholar]
- Grasby, P.; Frith, C.D.; Friston, K.J.; Simpson, J.; Fletcher, P.C.; Frackowiak, R.S.J.; Dolan, R.J. A graded task approach to the functional mapping of brain areas implicated in auditory-verbal memory. Brain 1994, 117, 1271–1282. [Google Scholar] [CrossRef]
- Mcintosh, A.R. Mapping Cognition to the Brain Through Neural Interactions. Memory 1999, 7, 523–548. [Google Scholar] [CrossRef]
- Tan, L.H.; Chan, A.H.D.; Kay, P.; Khong, P.L.; Yip, L.K.C.; Luke, K.K. Language affects patterns of brain activation associated with perceptual decision. Proc. Natl. Acad. Sci. USA 2008, 105, 4004–4009. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Fox, P.T.; Paus, T. Functional Coactivation Map of the Human Brain. Cereb. Cortex 2008, 18, 2553–2559. [Google Scholar] [CrossRef] [PubMed]
- Laird, A.R.; Eickhoff, S.B.; Rottschy, C.; Bzdok, D.; Ray, K.L.; Fox, P.T. Networks of task co-activations. Neuroimage 2013, 80, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Watanabe, T.; Kawato, M.; Sasaki, Y. Differential Activation Patterns in the Same Brain Region Led to Opposite Emotional States. PLoS Biol. 2016, 14, e1002546. [Google Scholar] [CrossRef] [PubMed]
- Nakuci, J.; Yeon, J.; Kim, J.H.; Kim, S.P.; Rahnev, D. Multiple brain activation patterns for the same task. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Baek, C.Y.; Kim, H.D.; Yoo, D.Y.; Kang, K.Y.; Lee, J.W. Change in activity patterns in the prefrontal cortex in different phases during the dual-task walking in older adults. J. NeuroEngineering Rehabil. 2023, 20, 86. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E. The Brain’s Default Mode Network. Annu. Rev. Neurosci. 2015, 38, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Buckner, R.L.; DiNicola, L.M. The brain’s default network: Updated anatomy, physiology and evolving insights. Nat. Rev. Neurosci. 2019, 20, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, J.; Bernhardt, B.C.; Leech, R.; Bzdok, D.; Jefferies, E.; Margulies, D.S. The default mode network in cognition: A topographical perspective. Nat. Rev. Neurosci. 2021, 22, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Buckner, R.L. The brain’s default network: Origins and implications for the study of psychosis. Dialogues Clin. Neurosci. 2013, 15, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.H.; Kording, K.P.; Parvizi, J.; Fox, M.D. Causal mapping of human brain function. Nat. Rev. Neurosci. 2022, 23, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Sporns, O.; Tononi, G.; Kötter, R. The Human Connectome: A Structural Description of the Human Brain. PLoS Comput. Biol. 2005, 1, e42. [Google Scholar] [CrossRef] [PubMed]
- Crossley, N.A. Connectome analysis and psychiatric disorders. In Connectome Analysis; Elsevier: Amsterdam, The Netherlands, 2023; pp. 423–432. [Google Scholar] [CrossRef]
- Van Essen, D.C.; Barch, D.M. The human connectome in health and psychopathology. World Psychiatry 2015, 14, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Finger, S.; Almli, C. Brain damage and neuroplasticity: Mechanisms of recovery or development? Brain Res. Rev. 1985, 10, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Grafman, J. Conceptualizing functional neuroplasticity. J. Commun. Disord. 2000, 33, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Flügge, G. Adult Neuroplasticity: More Than 40 Years of Research. Neural Plast. 2014, 2014, 541870. [Google Scholar] [CrossRef] [PubMed]
- Kolb, B.; Gibb, R. Brain Plasticity and Behaviour in the Developing Brain. J. Can. Acad. Child Adolesc. Psychiatry 2011, 20, 265. [Google Scholar] [PubMed]
- Park, D.C.; Bischof, G.N. The aging mind: Neuroplasticity in response to cognitive training. Dialogues Clin. Neurosci. 2013, 15, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Viel, T.A.; Toricelli, M.; Pereira, A.R.; Abrao, G.S.; Malerba, H.N.; Maia, J.; Buck, H.S. Mechanisms of neuroplasticity and brain degeneration: Strategies for protection during the aging process. Neural Regen. Res. 2021, 16, 58. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.H.; Kirby, A.J.; Finnerty, G.T. Rewiring the connectome: Evidence and effects. Neurosci. Biobehav. Rev. 2018, 88, 51–62. [Google Scholar] [CrossRef]
- Seven, Y.B.; Mitchell, G.S. Mechanisms of compensatory plasticity for respiratory motor neuron death. Respir. Physiol. Neurobiol. 2019, 265, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.L.; Kolanowski, A.M.; Gill, D.J. Plasticity in Early Alzheimer Disease. Top. Geriatr. Rehabil. 2011, 27, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Obara, K.; Muramatsu, K. Diaschisis. Neurol. Res. 1993, 15, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, Y.; Zhu, Y.; Torigoe, M.; Kita, Y.; Murakami, F. From migration to settlement: The pathways, migration modes and dynamics of neurons in the developing brain. Proc. Jpn. Acad. Ser. B 2016, 92, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaei, H.T.; Lai, C.; Anton, E.S. Neuronal migration in the adult brain: Are we there yet? Nat. Rev. Neurosci. 2007, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, B.; Parnavelas, J.G. Modes of neuronal migration in the developing cerebral cortex. Nat. Rev. Neurosci. 2002, 3, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Irastorza, I.T. Glial Localization of the Cannabinoid CB1 and CB2 Receptors in a Mouse Model of Alzheimer’s Disease. Ph.D. Thesis, Neuroscience Department, Faculty of Medicine and Nursery, Universidad del País Vasco/Euskal Herriko Unibersitatea (UPV/EHU), Leioa, Spain, 2021. [Google Scholar]
- Van Essen, D.C. Cartography and Connectomes. Neuron 2013, 80, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Alemán-Gómez, Y.; Griffa, A.; Houde, J.C.; Najdenovska, E.; Magon, S.; Cuadra, M.B.; Descoteaux, M.; Hagmann, P. A multi-scale probabilistic atlas of the human connectome. Sci. Data 2022, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, H. Human brain mapping: Hemodynamic response and electrophysiology. Clin. Neurophysiol. 2008, 119, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, J.B.; Barger, S.W. Why neurons die: Cell death in the nervous system. Anat. Rec. 1998, 253, 79–90. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Miura, M. Programmed Cell Death in Neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, M.P.; Nikoletopoulou, V.; Barde, Y.A. Death of developing neurons: New insights and implications for connectivity. J. Cell Biol. 2013, 203, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Ham, J. Programmed cell death during neuronal development: The sympathetic neuron model. Cell Death Differ. 2014, 21, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Finger, S. Chapter 51 Recovery of function. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2009; pp. 833–841. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef] [PubMed]
- Pettmann, B.; Henderson, C.E. Neuronal Cell Death. Neuron 1998, 20, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Chang, H.Y.; Sang, T.K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef]
- Zafarlotfi, S.; Quadri, M.; Borodovsky, J. Understanding Brain Damage and Sleep Apnea: A Review. Health Outcomes Res. Med. 2010, 1, e103–e110. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chen, W.; Harnod, T.; Lin, C.L.; Hsu, W.H.; Lin, C.C.; Chang, Y.L.; Wang, I.K.; Kao, C.H. Sleep apnea and risk of traumatic brain injury and associated mortality and healthcare costs: A population-based cohort study. Ann. Transl. Med. 2019, 7, 644. [Google Scholar] [CrossRef] [PubMed]
- Barañano, K.; Burd, I. CNS Malformations in the Newborn. Matern. Heal. Neonatol. Perinatol. 2022, 8, 1. [Google Scholar] [CrossRef]
- Chaudhari, B.P.; Ho, M.L. Congenital Brain Malformations: An Integrated Diagnostic Approach. Semin. Pediatr. Neurol. 2022, 42, 100973. [Google Scholar] [CrossRef] [PubMed]
- Giustini, A.; Pistarini, C.; Pisoni, C. Traumatic and nontraumatic brain injury. In Neurological Rehabilitation; Elsevier: Amsterdam, The Netherlands, 2013; pp. 401–409. [Google Scholar] [CrossRef]
- Hohmann, U.; Dehghani, F.; Hohmann, T. Assessment of Neuronal Damage in Brain Slice Cultures Using Machine Learning Based on Spatial Features. Front. Neurosci. 2021, 15, 740178. [Google Scholar] [CrossRef]
- Mamere, A.; Saraiva, L.; Matos, A.; Carneiro, A.; Santos, A. Evaluation of Delayed Neuronal and Axonal Damage Secondary to Moderate and Severe Traumatic Brain Injury Using Quantitative MR Imaging Techniques. Am. J. Neuroradiol. 2009, 30, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Majdan, M.; Plancikova, D.; Brazinova, A.; Rusnak, M.; Nieboer, D.; Feigin, V.; Maas, A. Epidemiology of traumatic brain injuries in Europe: A cross-sectional analysis. Lancet Public Health 2016, 1, e76–e83. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef]
- Goldman, L.; Siddiqui, E.M.; Khan, A.; Jahan, S.; Rehman, M.U.; Mehan, S.; Sharma, R.; Budkin, S.; Kumar, S.N.; Sahu, A.; et al. Understanding Acquired Brain Injury: A Review. Biomedicines 2022, 10, 2167. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M.; Read, C.A. Psychiatric comorbidity following traumatic brain injury. Brain Inj. 2007, 21, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Hammond, F.M.; Corrigan, J.D.; Ketchum, J.M.; Malec, J.F.; Dams-O’Connor, K.; Hart, T.; Novack, T.A.; Bogner, J.; Dahdah, M.N.; Whiteneck, G.G. Prevalence of Medical and Psychiatric Comorbidities Following Traumatic Brain Injury. J. Head Trauma Rehabil. 2019, 34, E1–E10. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, Y.; Yuryshinetz, I.; Zabenko, Y.; Pivneva, T. Mild traumatic brain injury as a pathological process. Heliyon 2023, 9, e18342. [Google Scholar] [CrossRef] [PubMed]
- Dean, P.J.A.; Sterr, A. Long-term effects of mild traumatic brain injury on cognitive performance. Front. Hum. Neurosci. 2013, 7, 30. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef]
- Danna-Dos-Santos, A.; Mohapatra, S.; Santos, M.; Degani, A.M. Long-term effects of mild traumatic brain injuries to oculomotor tracking performances and reaction times to simple environmental stimuli. Sci. Rep. 2018, 8, 4583. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.S.; Kubilus, C.A.; Stern, R.A. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, S.K.; Sridhar, S.; Char, S.; Pandya, K.; Mehta, K. Prevalence of comorbidities post mild traumatic brain injuries: A traumatic brain injury model systems study. Front. Hum. Neurosci. 2023, 17, 1158483. [Google Scholar] [CrossRef] [PubMed]
- Das, A.S.; Vicenty-Padilla, J.C.; Chua, M.M.; Jeelani, Y.; Snider, S.B.; Regenhardt, R.W.; Al-Mufti, F.; Du, R.; Izzy, S. Cerebrovascular injuries in traumatic brain injury. Clin. Neurol. Neurosurg. 2022, 223, 107479. [Google Scholar] [CrossRef]
- Rashid, B. Mechanical Characterization of Brain Tissue in Compression, Tension and Shear Under Dynamic Conditions. Ph.D. Thesis, School of Mechanical and Materials Engineering, University College Dublin, Dublin, Ireland, 2012. [Google Scholar]
- Traumatic Brain Injury. 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459300/ (accessed on 9 October 2023).
- Mokri, B. The Monro-Kellie hypothesis: Applications in CSF volume depletion. Neurology 2001, 56, 1746–1748. [Google Scholar] [CrossRef]
- Kalisvaart, A.C.J.; Wilkinson, C.M.; Gu, S.; Kung, T.F.C.; Yager, J.; Winship, I.R.; van Landeghem, F.K.H.; Colbourne, F. An update to the Monro-Kellie doctrine to reflect tissue compliance after severe ischemic and hemorrhagic stroke. Sci. Rep. 2020, 10, 22013. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Haces, M.; Tang, J.; Acosta, G.; Fernandez, J.; Shi, R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.S.; Sharp, D.J. Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1221–1233. [Google Scholar] [CrossRef]
- Graham, N.S.N.; Jolly, A.; Zimmerman, K.; Bourke, N.J.; Scott, G.; Cole, J.H.; Schott, J.M.; Sharp, D.J. Diffuse axonal injury predicts neurodegeneration after moderate—Severe traumatic brain injury. Brain 2020, 143, 3685–3698. [Google Scholar] [CrossRef] [PubMed]
- Dodd, W.S.; Panther, E.J.; Pierre, K.; Hernandez, J.S.; Patel, D.; Lucke-Wold, B. Traumatic Brain Injury and Secondary Neurodegenerative Disease. Trauma Care 2022, 2, 510–522. [Google Scholar] [CrossRef]
- Li, X.Y.; Feng, D.F. Diffuse axonal injury: Novel insights into detection and treatment. J. Clin. Neurosci. 2009, 16, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Vo, D.T.; Phan, C.C.; Le, H.G.N.; Vo, T.P.; Mai, U.T.T.; Le, H.K.; Ha, T.B.T. Diffuse axonal injury: A case report and MRI findings. Radiol. Case Rep. 2022, 17, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Markl, M.; Leupold, J. Gradient echo imaging. J. Magn. Reson. Imaging 2012, 35, 1274–1289. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.; Haacke, E.M.; Thurnher, M.M.; Barkhof, F. Susceptibility-weighted Imaging: Technical Essentials and Clinical Neurologic Applications. Radiology 2021, 299, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Skarupa, D.J.; Khan, M.; Hsu, A.; Madbak, F.G.; Ebler, D.J.; Yorkgitis, B.; Rahmathulla, G.; Alcindor, D.; Joseph, B. Trends in civilian penetrating brain injury: A review of 26, 871 patients. Am. J. Surg. 2019, 218, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Günther, M.; Dahlberg, M.; Rostami, A.; Azadali, A.; Arborelius, U.P.; Linder, F.; Rostami, E. Incidence, Demographics, and Outcomes of Penetrating Trauma in Sweden During the Past Decade. Front. Neurol. 2021, 12, 730405. [Google Scholar] [CrossRef] [PubMed]
- Vakil, M.T.; Singh, A.K. A review of penetrating brain trauma: Epidemiology, pathophysiology, imaging assessment, complications, and treatment. Emerg. Radiol. 2017, 24, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Raymont, V.; Salazar, A.M.; Lipsky, R.; Goldman, D.; Tasick, G.; Grafman, J. Correlates of posttraumatic epilepsy 35 years following combat brain injury. Neurology 2010, 75, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Penetrating Head Trauma. 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459254/ (accessed on 11 October 2023).
- Enam, S.; Kazim, S.; Shamim, M.; Tahir, M.; Waheed, S. Management of penetrating brain injury. J. Emergencies Trauma Shock 2011, 4, 395. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P. Current topic: Incidence, aetiology, and outcome of non-traumatic coma: A population based study. Arch. Dis. Child. 2001, 84, 193–199. [Google Scholar] [CrossRef]
- Sarrazin, J.L.; Bonneville, F.; Martin-Blondel, G. Brain infections. Diagn. Interv. Imaging 2012, 93, 473–490. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, A.M.; Sarmast, S.T.; Singh, R. Molecular Biology and Epidemiology of Neurotropic Viruses. Cureus 2020, 12, e9674. [Google Scholar] [CrossRef]
- Godkhindi, V.M.; Monappa, V.; Kairanna, N.V.; Sharma, S.; Vasudevan, G.; Hebbar, K.D. Brain infections that mimic malignancy. Diagn. Histopathol. 2022, 28, 456–466. [Google Scholar] [CrossRef]
- Sekino, N.; Selim, M.; Shehadah, A. Sepsis-associated brain injury: Underlying mechanisms and potential therapeutic strategies for acute and long-term cognitive impairments. J. Neuroinflammation 2022, 19, 101. [Google Scholar] [CrossRef]
- Lv, S.; Zhang, Y.; Steinmann, P.; Zhou, X.N.; Utzinger, J. Helminth Infections of the Central Nervous System Occurring in Southeast Asia and the Far East. In Important Helminth Infections in Southeast Asia: Diversity and Potential for Control and Elimination, Part A; Elsevier: Piscataway, NJ, USA, 2010; pp. 351–408. [Google Scholar] [CrossRef]
- Owusu-Agyei, A.K.; Nabarro, L. Helminth infections and differentials of eosinophilia. Medicine 2021, 49, 766–769. [Google Scholar] [CrossRef]
- Tuberculosis Meningitis. 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541015/ (accessed on 19 October 2023).
- Shadmani, G.; Simkins, T.J.; Assadsangabi, R.; Apperson, M.; Hacein-Bey, L.; Raslan, O.; Ivanovic, V. Autoimmune diseases of the brain, imaging and clinical review. Neuroradiol. J. 2021, 35, 152–169. [Google Scholar] [CrossRef] [PubMed]
- McGlasson, S.; Wiseman, S.; Wardlaw, J.; Dhaun, N.; Hunt, D.P.J. Neurological Disease in Lupus: Toward a Personalized Medicine Approach. Front. Immunol. 2018, 9, 372322. [Google Scholar] [CrossRef]
- Kayser, M.S.; Dalmau, J. The Emerging Link Between Autoimmune Disorders and Neuropsychiatric Disease. J. Neuropsychiatry 2011, 23, 90–97. [Google Scholar] [CrossRef]
- Dickens, F. The toxic effects of oxygen on brain metabolism and on tissue enzymes. Biochem. J. 1946, 40, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; McLin, V.A.; Cudalbu, C. Ammonia toxicity to the brain. J. Inherit. Metab. Dis. 2012, 36, 595–612. [Google Scholar] [CrossRef]
- Sharma, P.; Eesa, M.; Scott, J.N. Toxic and Acquired Metabolic Encephalopathies: MRI Appearance. Am. J. Roentgenol. 2009, 193, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Guennec, L.L.; Marois, C.; Demeret, S.; Wijdicks, E.; Weiss, N. Toxic-metabolic encephalopathy in adults: Critical discussion and pragmatical diagnostic approach. Rev. Neurol. 2022, 178, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Wiethoff, S.; Houlden, H. Neurodegeneration with brain iron accumulation. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 157–166. [Google Scholar] [CrossRef]
- Buyukcoban, S.; Arici, M.A.; Koca, U.; Kalkan, S. A Case Report of Toxic Brain Syndrome Caused by Methyl Bromide. Turk. J. Anesth. Reanim. 2015, 43, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Luvisetto, S. Botulinum Neurotoxins in Central Nervous System: An Overview from Animal Models to Human Therapy. Toxins 2021, 13, 751. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Büttner, A. Neurotoxicology and drug-related disorders. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 181–192. [Google Scholar] [CrossRef]
- Moratalla, R.; Khairnar, A.; Simola, N.; Granado, N.; García-Montes, J.R.; Porceddu, P.F.; Tizabi, Y.; Costa, G.; Morelli, M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog. Neurobiol. 2017, 155, 149–170. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Rego, A.C.; Oliveira, C.R. Cellular and molecular mechanisms involved in the neurotoxicity of opioid and psychostimulant drugs. Brain Res. Rev. 2008, 58, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, J.W. Toxic Encephalopathy. Saf. Health Work 2012, 3, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Büttner, A. Nutritional and systemic metabolic disorders. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 167–173. [Google Scholar] [CrossRef]
- Greene-Schloesser, D.; Robbins, M.E.; Peiffer, A.M.; Shaw, E.G.; Wheeler, K.T.; Chan, M.D. Radiation-induced brain injury: A review. Front. Oncol. 2012, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Belka, C.; Budach, W.; Kortmann, R.D.; Bamberg, M. Radiation induced CNS toxicity—Molecular and cellular mechanisms. Br. J. Cancer 2001, 85, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Nonoguchi, N.; Kawabata, S.; Miyatake, S.I.; Kuroiwa, T. Delayed brain radiation necrosis: Pathological review and new molecular targets for treatment. Med. Mol. Morphol. 2015, 48, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Brown, S.L.; Jenrow, K.A.; Ryu, S. Mechanisms of radiation-induced brain toxicity and implications for future clinical trials. J. Neuro-Oncol. 2008, 87, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.S.; Arevalo, O.; Zorofchian, S.; Patrizz, A.; Riascos, R.; Tandon, N.; Blanco, A.; Ballester, L.Y.; Esquenazi, Y. Cerebral Radiation Necrosis: Incidence, Pathogenesis, Diagnostic Challenges, and Future Opportunities. Curr. Oncol. Rep. 2019, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, Y.R.; Li, X.A.; el Naqa, I.; Hahn, C.A.; Marks, L.B.; Merchant, T.E.; Dicker, A.P. Radiation Dose-Volume Effects in the Brain. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Naik, A.; Shaffer, A.; Goel, M.; Krist, D.T.; Liang, E.; Furey, C.G.; Miller, W.K.; Lawton, M.T.; Barnett, D.H.; et al. Differentiating radiation necrosis from tumor recurrence: A systematic review and diagnostic meta-analysis comparing imaging modalities. J. Neuro-Oncol. 2023, 162, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kenney, K.; Amyot, F.; Haber, M.; Pronger, A.; Bogoslovsky, T.; Moore, C.; Diaz-Arrastia, R. Cerebral Vascular Injury in Traumatic Brain Injury. Exp. Neurol. 2016, 275, 353–366. [Google Scholar] [CrossRef]
- Murphy, S.J.; Werring, D.J. Stroke: Causes and clinical features. Medicine 2020, 48, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Grysiewicz, R.A.; Thomas, K.; Pandey, D.K. Epidemiology of Ischemic and Hemorrhagic Stroke: Incidence, Prevalence, Mortality, and Risk Factors. Neurol. Clin. 2008, 26, 871–895. [Google Scholar] [CrossRef] [PubMed]
- Silasi, G.; Murphy, T.H. Stroke and the Connectome: How Connectivity Guides Therapeutic Intervention. Neuron 2014, 83, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, A.; Ramsey, L.E.; Siegel, J.S.; Shulman, G.L.; Corbetta, M. Brain connectivity and neurological disorders after stroke. Curr. Opin. Neurol. 2016, 29, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xu, H.; Yu, C. Brain Connectivity Plasticity in the Motor Network after Ischemic Stroke. Neural Plast. 2013, 2013, 924192. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Y.; Zhu, W.; Chen, X. Changes in brain functional network connectivity after stroke. Neural Regen. Res. 2014, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Desowska, A.; Turner, D.L. Dynamics of brain connectivity after stroke. Rev. Neurosci. 2019, 30, 605–623. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.R.; Kaiser, M.; Farr, T.D. Functional Connectivity Change in Response to Stroke Is Comparable Across Species: From Mouse to Man. Stroke 2021, 52, 2961–2963. [Google Scholar] [CrossRef] [PubMed]
- McFaline-Figueroa, J.R.; Lee, E.Q. Brain Tumors. Am. J. Med. 2018, 131, 874–882. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, L.M. Brain Tumors. N. Engl. J. Med. 2001, 344, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Herholz, K.; Langen, K.J.; Schiepers, C.; Mountz, J.M. Brain Tumors. Semin. Nucl. Med. 2012, 42, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.; Simjee, S. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Costa, L.L.; ao, E.C.B.; Segundo, L.M.D.B.M. Atualização em epilepsia. Rev. De Med. 2020, 99, 170–181. [Google Scholar] [CrossRef]
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef]
- Beghi, E. The Epidemiology of Epilepsy. Neuroepidemiology 2019, 54, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Carmant, L. Seizures and Epilepsy: An Overview for Neuroscientists. Cold Spring Harb. Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.N.; Filippi, D.; Hauser, W.A. The descriptive epidemiology of epilepsy-A review. Epilepsy Res. 2009, 85, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Staley, K. Epileptic Neurons Go Wireless. Science 2004, 305, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Sander, J.W.; Sisodiya, S.M.; Walker, M.C. Adult epilepsy. Lancet 2006, 367, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Stefan, H.; da Silva, F.H.L. Epileptic Neuronal Networks: Methods of Identification and Clinical Relevance. Front. Neurol. 2013, 4, 38995. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Brodovskaya, A.; Hudson, J.L.; Kapur, J. Connectivity and Neuronal Synchrony during Seizures. J. Neurosci. 2021, 41, 7623–7635. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.K.; Gallico, R.P. Learning Disabilities. Pediatr. Clin. N. Am. 1993, 40, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Polanczyk, G.; de Lima, M.S.; Horta, B.L.; Biederman, J.; Rohde, L.A. The Worldwide Prevalence of ADHD: A Systematic Review and Metaregression Analysis. Am. J. Psychiatry 2007, 164, 942–948. [Google Scholar] [CrossRef]
- Robertson, M.M.; Eapen, V.; Singer, H.S.; Martino, D.; Scharf, J.M.; Paschou, P.; Roessner, V.; Woods, D.W.; Hariz, M.; Mathews, C.A.; et al. Gilles de la Tourette syndrome. Nat. Rev. Dis. Prim. 2017, 3, 16097. [Google Scholar] [CrossRef] [PubMed]
- Zouki, J.J.; Ellis, E.G.; Morrison-Ham, J.; Thomson, P.; Jesuthasan, A.; Al-Fatly, B.; Joutsa, J.; Silk, T.J.; Corp, D.T. Mapping a network for tics in Tourette syndrome using causal lesions and structural alterations. Brain Commun. 2023, 5, fcad105. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.N.; Gratton, C.; Church, J.A.; Dosenbach, N.U.; Black, K.J.; Petersen, S.E.; Schlaggar, B.L.; Greene, D.J. Atypical Functional Connectivity in Tourette Syndrome Differs Between Children and Adults. Biol. Psychiatry 2020, 87, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Duffley, G.; Anderson, D.N.; Ostrem, J.L.; Welter, M.L.; Baldermann, J.C.; Kuhn, J.; Huys, D.; Visser-Vandewalle, V.; Foltynie, T.; et al. Structural connectivity predicts clinical outcomes of deep brain stimulation for Tourette syndrome. Brain 2020, 143, 2607–2623. [Google Scholar] [CrossRef] [PubMed]
- Swanson, J.; Sergeant, J.; Taylor, E.; Sonuga-Barke, E.; Jensen, P.; Cantwell, D. Attention-deficit hyperactivity disorder and hyperkinetic disorder. Lancet 1998, 351, 429–433. [Google Scholar] [CrossRef]
- Faraone, S.V.; Asherson, P.; Banaschewski, T.; Biederman, J.; Buitelaar, J.K.; Ramos-Quiroga, J.A.; Rohde, L.A.; Sonuga-Barke, E.J.S.; Tannock, R.; Franke, B. Attention-deficit/hyperactivity disorder. Nat. Rev. Dis. Prim. 2015, 1, 15020. [Google Scholar] [CrossRef] [PubMed]
- Ougrin, D.; Chatterton, S.; Banarsee, R. Attention deficit hyperactivity disorder (ADHD): Review for primary care clinicians. Lond. J. Prim. Care 2010, 3, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.H.; Wolf, L.E.; Wasserstein, J. Adults with ADHD: An Overview. Ann. N. Y. Acad. Sci. 2001, 931, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Irastorza Eguskiza, L.J.; Bellón, J.M.; Mora, M. Comorbidity of personality disorders and attention-deficit hyperactivity disorder in adults. Rev. De Psiquiatr. Y Salud Ment. 2018, 11, 151–155. [Google Scholar] [CrossRef]
- Thapar, A.; Cooper, M.; Eyre, O.; Langley, K. Practitioner Review: What have we learnt about the causes of ADHD? J. Child Psychol. Psychiatry 2012, 54, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Konrad, K.; Eickhoff, S.B. Is the ADHD brain wired differently? A review on structural and functional connectivity in attention deficit hyperactivity disorder. Hum. Brain Mapp. 2010, 31, 904–916. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, A.; Xia, S.; Branch, C.; Li, X. A review of attention-deficit/hyperactivity disorder from the perspective of brain networks. Front. Hum. Neurosci. 2013, 7, 192. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.R.; Hoexter, M.Q.; Castellanos, X.F.; Rohde, L.A. Abnormal Brain Connectivity Patterns in Adults with ADHD: A Coherence Study. PLoS ONE 2012, 7, e45671. [Google Scholar] [CrossRef]
- Pereira-Sanchez, V.; Franco, A.R.; Vieira, D.; de Castro-Manglano, P.; Soutullo, C.; Milham, M.P.; Castellanos, F.X. Systematic Review: Medication Effects on Brain Intrinsic Functional Connectivity in Patients With Attention-Deficit/Hyperactivity Disorder. J. Am. Acad. Child Adolesc. Psychiatry 2021, 60, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Lyon, G.R. Learning Disabilities. Future Child. 1996, 6, 54. [Google Scholar] [CrossRef]
- Semrud-Clikeman, M. Neuropsychological Aspects for Evaluating Learning Disabilities. Commun. Disord. Q. 2005, 26, 242–247. [Google Scholar] [CrossRef]
- Finn, E.S.; Shen, X.; Holahan, J.M.; Scheinost, D.; Lacadie, C.; Papademetris, X.; Shaywitz, S.E.; Shaywitz, B.A.; Constable, R.T. Disruption of Functional Networks in Dyslexia: A Whole-Brain, Data-Driven Analysis of Connectivity. Biol. Psychiatry 2014, 76, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Turker, S.; Kuhnke, P.; Jiang, Z.; Hartwigsen, G. Disrupted network interactions serve as a neural marker of dyslexia. Commun. Biol. 2023, 6, 1114. [Google Scholar] [CrossRef] [PubMed]
- Richards, T.L.; Berninger, V.W.; Yagle, K.J.; Abbott, R.D.; Peterson, D.J. Changes in DTI Diffusivity and fMRI Connectivity Cluster Coefficients for Students with and without Specific Learning Disabilities In Written Language: Brain’s Response to Writing Instruction. J. Nat. Sci. 2017, 3, e350. [Google Scholar] [PubMed]
- Žarić, G.; Timmers, I.; Gerretsen, P.; Fraga González, G.; Tijms, J.; van der Molen, M.W.; Blomert, L.; Bonte, M. Atypical White Matter Connectivity in Dyslexic Readers of a Fairly Transparent Orthography. Front. Psychol. 2018, 9, 308630. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.K.; Aboud, K.S.; Nguyen, T.Q.; Cutting, L.E. Applying a network framework to the neurobiology of reading and dyslexia. J. Neurodev. Disord. 2018, 10, 37. [Google Scholar] [CrossRef]
- Schurz, M.; Wimmer, H.; Richlan, F.; Ludersdorfer, P.; Klackl, J.; Kronbichler, M. Resting-State and Task-Based Functional Brain Connectivity in Developmental Dyslexia. Cereb. Cortex 2014, 25, 3502–3514. [Google Scholar] [CrossRef] [PubMed]
- Izadi-Najafabadi, S.; Rinat, S.; Zwicker, J.G. Brain functional connectivity in children with developmental coordination disorder following rehabilitation intervention. Pediatr. Res. 2021, 91, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Jolles, D.; Ashkenazi, S.; Kochalka, J.; Evans, T.; Richardson, J.; Rosenberg-Lee, M.; Zhao, H.; Supekar, K.; Chen, T.; Menon, V. Parietal hyper-connectivity, aberrant brain organization, and circuit-based biomarkers in children with mathematical disabilities. Dev. Sci. 2016, 19, 613–631. [Google Scholar] [CrossRef] [PubMed]
- Banker, S.M.; Ramphal, B.; Pagliaccio, D.; Thomas, L.; Rosen, E.; Sigel, A.N.; Zeffiro, T.; Marsh, R.; Margolis, A.E. Spatial Network Connectivity and Spatial Reasoning Ability in Children with Nonverbal Learning Disability. Sci. Rep. 2020, 10, 561. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Cascella, M.; Marwaha, R. Intellectual Disability. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Baburamani, A.A.; Patkee, P.A.; Arichi, T.; Rutherford, M.A. New approaches to studying early brain development in Down syndrome. Dev. Med. Child Neurol. 2019, 61, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, K.; Constantine, A.; Clift, P.; Condliffe, R.; Moledina, S.; Jansen, K.; Inuzuka, R.; Veldtman, G.R.; Cua, C.L.; Tay, E.L.W.; et al. Cardiovascular Complications of Down Syndrome: Scoping Review and Expert Consensus. Circulation 2023, 147, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Asim, A.; Kumar, A.; Muthuswamy, S.; Jain, S.; Agarwal, S. Down syndrome: An insight of the disease. J. Biomed. Sci. 2015, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Bull, M.J. Down Syndrome. N. Engl. J. Med. 2020, 382, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Saini, F.; Dell’ Acqua, F.; Strydom, A. Structural Connectivity in Down Syndrome and Alzheimer’s Disease. Front. Neurosci. 2022, 16, 908413. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Prim. 2020, 6, 9. [Google Scholar] [CrossRef]
- Csumitta, K.D.; Gotts, S.J.; Clasen, L.S.; Martin, A.; Raitano Lee, N. Youth with Down syndrome display widespread increased functional connectivity during rest. Sci. Rep. 2022, 12, 9836. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Y.; Lu, F.M.; Wang, M.Y.; Hu, Z.S.; Zhang, J.; Chen, Z.Y.; Armada-da Silva, P.A.S.; Yuan, Z. Altered Functional Connectivity in the Motor and Prefrontal Cortex for Children With Down’s Syndrome: An fNIRS Study. Front. Hum. Neurosci. 2020, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Schependom, J.V.; D’haeseleer, M. Advances in Neurodegenerative Diseases. J. Clin. Med. 2023, 12, 1709. [Google Scholar] [CrossRef]
- Davenport, F.; Gallacher, J.; Kourtzi, Z.; Koychev, I.; Matthews, P.M.; Oxtoby, N.P.; Parkes, L.M.; Priesemann, V.; Rowe, J.B.; Smye, S.W.; et al. Neurodegenerative disease of the brain: A survey of interdisciplinary approaches. J. R. Soc. Interface 2023, 20, 20220406. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Amor, S.; Puentes, F.; Baker, D.; Valk, P.V.D. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Shah, R.C.; Bennett, D.A. Diagnosis and Management of Dementia: Review. JAMA 2019, 322, 1589. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef] [PubMed]
- Checkoway, H.; Lundin, J.I.; Kelada, S.N. Neurodegenerative diseases. IARC Sci Publ. 2011, 163, 407–419. [Google Scholar]
- Niu, H.; Álvarez Álvarez, I.; Guillén-Grima, F.; Aguinaga-Ontoso, I. Prevalencia e incidencia de la enfermedad de Alzheimer en Europa: Metaanálisis. Neurología 2017, 32, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 2011, 32, 1341–1371. [Google Scholar] [CrossRef] [PubMed]
- Depp, C.; Sun, T.; Sasmita, A.O.; Spieth, L.; Berghoff, S.A.; Nazarenko, T.; Overhoff, K.; Steixner-Kumar, A.A.; Subramanian, S.; Arinrad, S.; et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 2023, 618, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Cain, A.; Taga, M.; McCabe, C.; Green, G.S.; Hekselman, I.; White, C.C.; Lee, D.I.; Gaur, P.; Rozenblatt-Rosen, O.; Zhang, F.; et al. Multicellular communities are perturbed in the aging human brain and Alzheimer’s disease. Nat. Neurosci. 2023, 26, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Stam, C.; Jones, B.; Nolte, G.; Breakspear, M.; Scheltens, P. Small-World Networks and Functional Connectivity in Alzheimer’s Disease. Cereb. Cortex 2006, 17, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Sporns, O.; Saykin, A.J. The human connectome in Alzheimer disease—relationship to biomarkers and genetics. Nat. Rev. Neurol. 2021, 17, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Martensson, G.; Pereira, J.B.; Mecocci, P.; Vellas, B.; Tsolaki, M.; Kłoszewska, I.; Soininen, H.; Lovestone, S.; Simmons, A.; Volpe, G.; et al. Stability of graph theoretical measures in structural brain networks in Alzheimer’s disease. Sci. Rep. 2018, 8, 11592. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.Y.; Mirra, S.S.; Sait, H.B.R.; Sacktor, T.C.; Sigurdsson, E.M. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Aβ and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 2011, 122, 285–292. [Google Scholar] [CrossRef]
- Savioz, A.; Leuba, G.; Vallet, P.G. A framework to understand the variations of PSD-95 expression in brain aging and in Alzheimer’s disease. Ageing Res. Rev. 2014, 18, 86–94. [Google Scholar] [CrossRef]
- Kivisäkk, P.; Carlyle, B.C.; Sweeney, T.; Quinn, J.P.; Ramirez, C.E.; Trombetta, B.A.; Mendes, M.; Brock, M.; Rubel, C.; Czerkowicz, J.; et al. Increased levels of the synaptic proteins PSD-95, SNAP-25, and neurogranin in the cerebrospinal fluid of patients with Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Self, W.K.; Holtzman, D.M. Emerging diagnostics and therapeutics for Alzheimer disease. Nat. Med. 2023, 29, 2187–2199. [Google Scholar] [CrossRef]
- Talarico, G.; Trebbastoni, A.; Bruno, G.; de Lena, C. Modulation of the Cannabinoid System: A New Perspective for the Treatment of the Alzheimer’s Disease. Curr. Neuropharmacol. 2019, 17, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.P.M.; Surendranathan, A.; Bentley, A.; Barker, S.A.H.; Taylor, J.P.; Thomas, A.J.; Allan, L.M.; McNally, R.J.; James, P.W.; McKeith, I.G.; et al. Clinical prevalence of Lewy body dementia. Alzheimer’s Res. Ther. 2018, 10, 19. [Google Scholar] [CrossRef]
- Fahn, S. Classification of movement disorders. Mov. Disord. 2011, 26, 947–957. [Google Scholar] [CrossRef]
- Abdo, W.F.; van de Warrenburg, B.P.C.; Burn, D.J.; Quinn, N.P.; Bloem, B.R. The clinical approach to movement disorders. Nat. Rev. Neurol. 2010, 6, 29–37. [Google Scholar] [CrossRef]
- Heim, B.; Krismer, F.; Marzi, R.D.; Seppi, K. Magnetic resonance imaging for the diagnosis of Parkinson’s disease. J. Neural Transm. 2017, 124, 915–964. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Pietracupa, S.; Martin-Bastida, A.; Piccini, P. Iron metabolism and its detection through MRI in parkinsonian disorders: A systematic review. Neurol. Sci. 2017, 38, 2095–2101. [Google Scholar] [CrossRef]
- Dean, D.C.; Sojkova, J.; Hurley, S.; Kecskemeti, S.; Okonkwo, O.; Bendlin, B.B.; Theisen, F.; Johnson, S.C.; Alexander, A.L.; Gallagher, C.L. Alterations of Myelin Content in Parkinson’s Disease: A Cross-Sectional Neuroimaging Study. PLoS ONE 2016, 11, e0163774. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, C.; Sun, J.; Liang, L.; Lv, H.; Gao, L.; Fang, J.; Ma, T.; Wu, T. Alteration of brain structural connectivity in progression of Parkinson’s disease: A connectome-wide network analysis. NeuroImage Clin. 2021, 31, 102715. [Google Scholar] [CrossRef]
- Boshkovski, T.; Cohen-Adad, J.; Misic, B.; Arnulf, I.; Corvol, J.C.; Vidailhet, M.; Lehéricy, S.; Stikov, N.; Mancini, M. The Myelin-Weighted Connectome in Parkinson’s Disease. Mov. Disord. 2021, 37, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Tinaz, S. Functional Connectome in Parkinson’s Disease and Parkinsonism. Curr. Neurol. Neurosci. Rep. 2021, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Loh, A.; Boutet, A.; Germann, J.; Al-Fatly, B.; Elias, G.J.B.; Neudorfer, C.; Krotz, J.; Wong, E.H.Y.; Parmar, R.; Gramer, R.; et al. A Functional Connectome of Parkinson’s Disease Patients Prior to Deep Brain Stimulation: A Tool for Disease-Specific Connectivity Analyses. Front. Neurosci. 2022, 16, 804125. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Xia, G. Ataxia. CONTINUUM Lifelong Learn. Neurol. 2016, 22, 1208–1226. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H.L. The Spinocerebellar Ataxias. J. Neuro-Ophthalmol. 2009, 29, 227–237. [Google Scholar] [CrossRef]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Odish, O.F.; Caeyenberghs, K.; Hosseini, H.; van den Bogaard, S.J.; Roos, R.A.; Leemans, A. Dynamics of the connectome in Huntington’s disease: A longitudinal diffusion MRI study. NeuroImage Clin. 2015, 9, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Poudel, G.R.; Harding, I.H.; Egan, G.F.; Georgiou-Karistianis, N. Network spread determines severity of degeneration and disconnection in Huntington’s disease. Hum. Brain Mapp. 2019, 40, 4192–4201. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, F.A.; Turner, J.A.; Vergara, V.M.; Miller, R.L.; Mennigen, E.; Liu, J.; Misiura, M.B.; Ciarochi, J.; Johnson, H.J.; Long, J.D.; et al. Whole-Brain Connectivity in a Large Study of Huntington’s Disease Gene Mutation Carriers and Healthy Controls. Brain Connect. 2018, 8, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.T. Prion diseases. Lancet Neurol. 2005, 4, 635–642. [Google Scholar] [CrossRef]
- Knight, R.S.G. PRION DISEASES. J. Neurol. Neurosurg. Psychiatry 2004, 75, i36–i42. [Google Scholar] [CrossRef]
- Ironside, J.W.; Ritchie, D.L.; Head, M.W. Prion diseases. In Neuropathology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 393–403. [Google Scholar] [CrossRef]
- Fornari, S.; Schäfer, A.; Jucker, M.; Goriely, A.; Kuhl, E. Prion-like spreading of Alzheimer’s disease within the brain’s connectome. J. R. Soc. Interface 2019, 16, 20190356. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A. Multiple sclerosis is primarily a neurodegenerative disease. J. Neural Transm. 2013, 120, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Gironi, M.; Arnò, C.; Comi, G.; Penton-Rol, G.; Furlan, R. Multiple Sclerosis and Neurodegenerative Diseases. In Immune Rebalancing; Elsevier: Amsterdam, The Netherlands, 2016; pp. 63–84. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Multiple Sclerosis Review. Pharm. Ther. 2012, 37, 175. [Google Scholar]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2018, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Schoonheim, M.M.; Meijer, K.A.; Geurts, J.J.G. Network Collapse and Cognitive Impairment in Multiple Sclerosis. Front. Neurol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Hogestol, E.A.; Ghezzo, S.; Nygaard, G.O.; Espeseth, T.; Sowa, P.; Beyer, M.K.; Harbo, H.F.; Westlye, L.T.; Hulst, H.E.; Alnaes, D. Functional connectivity in multiple sclerosis modelled as connectome stability: A 5-year follow-up study. Mult. Scler. J. 2021, 28, 532–540. [Google Scholar] [CrossRef]
- Schoonheim, M.M.; Broeders, T.A.; Geurts, J.J. The network collapse in multiple sclerosis: An overview of novel concepts to address disease dynamics. NeuroImage Clin. 2022, 35, 103108. [Google Scholar] [CrossRef] [PubMed]
- Barile, B.; Ashtari, P.; Stamile, C.; Marzullo, A.; Maes, F.; Durand-Dubief, F.; Van Huffel, S.; Sappey-Marinier, D. Classification of multiple sclerosis clinical profiles using machine learning and grey matter connectome. Front. Robot. AI 2022, 9, 926255. [Google Scholar] [CrossRef] [PubMed]
- Manglani, H.R.; Fountain-Zaragoza, S.; Shankar, A.; Nicholas, J.A.; Prakash, R.S. Employing connectome-based models to predict working memory in multiple sclerosis. Brain Connect. 2021, 12, 502–514. [Google Scholar] [CrossRef]
- Tahedl, M.; Levine, S.M.; Greenlee, M.W.; Weissert, R.; Schwarzbach, J.V. Functional Connectivity in Multiple Sclerosis: Recent Findings and Future Directions. Front. Neurol. 2018, 9, 403898. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Cree, B.A. Treatment of Multiple Sclerosis: A Review. Am. J. Med. 2020, 133, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, P.; Rucco, R.; Jacini, F.; Trojsi, F.; Lardone, A.; Baselice, F.; Femiano, C.; Santangelo, G.; Granata, C.; Vettoliere, A.; et al. Brain functional networks become more connected as amyotrophic lateral sclerosis progresses: A source level magnetoencephalographic study. NeuroImage Clin. 2018, 20, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Verstraete, E.; de Reus, M.A.; Veldink, J.H.; van den Berg, L.H.; van den Heuvel, M.P. Correlation between structural and functional connectivity impairment in amyotrophic lateral sclerosis. Hum. Brain Mapp. 2014, 35, 4386–4395. [Google Scholar] [CrossRef] [PubMed]
- Verstraete, E.; van den Heuvel, M.P.; Veldink, J.H.; Blanken, N.; Mandl, R.C.; Hulshoff Pol, H.E.; van den Berg, L.H. Motor Network Degeneration in Amyotrophic Lateral Sclerosis: A Structural and Functional Connectivity Study. PLoS ONE 2010, 5, e13664. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Canu, E.; Valsasina, P.; Riva, N.; Prelle, A.; Comi, G.; Filippi, M. Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; Amminger, G.P.; Aguilar-Gaxiola, S.; Alonso, J.; Lee, S.; Ustun, T.B. Age of onset of mental disorders: A review of recent literature. Curr. Opin. Psychiatry 2007, 20, 359–364. [Google Scholar] [CrossRef]
- Solmi, M.; Radua, J.; Olivola, M.; Croce, E.; Soardo, L.; Salazar de Pablo, G.; Il Shin, J.; Kirkbride, J.B.; Jones, P.; Kim, J.H.; et al. Age at onset of mental disorders worldwide: Large-scale meta-analysis of 192 epidemiological studies. Mol. Psychiatry 2021, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, M.P.; Sporns, O. A cross-disorder connectome landscape of brain dysconnectivity. Nat. Rev. Neurosci. 2019, 20, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Solmi, M.; Seitidis, G.; Mavridis, D.; Correll, C.U.; Dragioti, E.; Guimond, S.; Tuominen, L.; Dargél, A.; Carvalho, A.F.; Fornaro, M.; et al. Incidence, prevalence, and global burden of schizophrenia-data, with critical appraisal, from the Global Burden of Disease (GBD) 2019. Mol. Psychiatry 2023, 28, 5319–5327. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.H.; North, S.W.; Shields, C.G. Schizophrenia: A Review. Am. Fam. Physician 2007, 75, 1821–1829. [Google Scholar] [PubMed]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and Treatment Options. Pharm. Ther. 2014, 39, 638. [Google Scholar]
- Yuan, L.; Ma, X.; Li, D.; Ouyang, L.; Fan, L.; Li, C.; He, Y.; Chen, X. Alteration of a brain network with stable and strong functional connections in subjects with schizophrenia. Schizophrenia 2022, 8, 91. [Google Scholar] [CrossRef]
- Rubinov, M.; Knock, S.A.; Stam, C.J.; Micheloyannis, S.; Harris, A.W.; Williams, L.M.; Breakspear, M. Small-world properties of nonlinear brain activity in schizophrenia. Hum. Brain Mapp. 2009, 30, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Micheloyannis, S.; Pachou, E.; Stam, C.J.; Breakspear, M.; Bitsios, P.; Vourkas, M.; Erimaki, S.; Zervakis, M. Small-world networks and disturbed functional connectivity in schizophrenia. Schizophr. Res. 2006, 87, 60–66. [Google Scholar] [CrossRef]
- Mastrandrea, R.; Piras, F.; Gabrielli, A.; Banaj, N.; Caldarelli, G.; Spalletta, G.; Gili, T. The unbalanced reorganization of weaker functional connections induces the altered brain network topology in schizophrenia. Sci. Rep. 2021, 11, 15400. [Google Scholar] [CrossRef]
- Belmaker, R.; Agam, G. Major Depressive Disorder. N. Engl. J. Med. 2008, 358, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065. [Google Scholar] [CrossRef] [PubMed]
- Scheuer, H.; Alarcón, G.; Demeter, D.V.; Earl, E.; Fair, D.A.; Nagel, B.J. Reduced fronto-amygdalar connectivity in adolescence is associated with increased depression symptoms over time. Psychiatry Res. Neuroimaging 2017, 266, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fan, Y.; Zeng, L.L.; Liu, B.; Ju, Y.; Wang, M.; Dong, Q.; Lu, X.; Sun, J.; Zhang, L.; et al. The neuroprogressive nature of major depressive disorder: Evidence from an intrinsic connectome analysis. Transl. Psychiatry 2021, 11, 102. [Google Scholar] [CrossRef]
- Yang, H.; Chen, X.; Chen, Z.B.; Li, L.; Li, X.Y.; Castellanos, F.X.; Bai, T.J.; Bo, Q.J.; Cao, J.; Chang, Z.K.; et al. Disrupted intrinsic functional brain topology in patients with major depressive disorder. Mol. Psychiatry 2021, 26, 7363–7371. [Google Scholar] [CrossRef] [PubMed]
- Scalabrini, A.; Vai, B.; Poletti, S.; Damiani, S.; Mucci, C.; Colombo, C.; Zanardi, R.; Benedetti, F.; Northoff, G. All roads lead to the default-mode network-global source of DMN abnormalities in major depressive disorder. Neuropsychopharmacology 2020, 45, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Saris, I.M.J.; Penninx, B.W.J.H.; Dinga, R.; van Tol, M.J.; Veltman, D.J.; van der Wee, N.J.A.; Aghajani, M. Default Mode Network Connectivity and Social Dysfunction in Major Depressive Disorder. Sci. Rep. 2020, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.T.; Hadas, I.; Zomorrodi, R.; Voineskos, D.; Farzan, F.; Fitzgerald, P.B.; Blumberger, D.M.; Daskalakis, Z.J. Modulation of functional network properties in major depressive disorder following electroconvulsive therapy (ECT): A resting-state EEG analysis. Sci. Rep. 2020, 10, 17057. [Google Scholar] [CrossRef] [PubMed]
- Korgaonkar, M.S.; Goldstein-Piekarski, A.N.; Fornito, A.; Williams, L.M. Intrinsic connectomes are a predictive biomarker of remission in major depressive disorder. Mol. Psychiatry 2019, 25, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Berwian, I.M.; Wenzel, J.G.; Kuehn, L.; Schnuerer, I.; Kasper, L.; Veer, I.M.; Seifritz, E.; Stephan, K.E.; Walter, H.; Huys, Q.J.M. The relationship between resting-state functional connectivity, antidepressant discontinuation and depression relapse. Sci. Rep. 2020, 10, 22346. [Google Scholar] [CrossRef] [PubMed]
- Repple, J.; Mauritz, M.; Meinert, S.; de Lange, S.C.; Grotegerd, D.; Opel, N.; Redlich, R.; Hahn, T.; Förster, K.; Leehr, E.J.; et al. Severity of current depression and remission status are associated with structural connectome alterations in major depressive disorder. Mol. Psychiatry 2019, 25, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Fava, M.; Kendler, K.S. Major Depressive Disorder. Neuron 2000, 28, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.F.; Firth, J.; Vieta, E. Bipolar Disorder. N. Engl. J. Med. 2020, 383, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Vieta, E.; Berk, M.; Schulze, T.G.; Carvalho, A.F.; Suppes, T.; Calabrese, J.R.; Gao, K.; Miskowiak, K.W.; Grande, I. Bipolar disorders. Nat. Rev. Dis. Prim. 2018, 4, 18008. [Google Scholar] [CrossRef] [PubMed]
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhu, R.; Tian, S.; Wang, H.; Chen, Z.; Wang, X.; Shao, J.; Qin, J.; Shi, J.; Liu, H.; et al. Structural-functional decoupling predicts suicide attempts in bipolar disorder patients with a current major depressive episode. Neuropsychopharmacology 2020, 45, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Sankar, A.; Scheinost, D.; Goldman, D.A.; Drachman, R.; Colic, L.; Villa, L.M.; Kim, J.A.; Gonzalez, Y.; Marcelo, I.; Shinomiya, M.; et al. Graph theory analysis of whole brain functional connectivity to assess disturbances associated with suicide attempts in bipolar disorder. Transl. Psychiatry 2022, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E. Epidemiology of Autistic Disorder and Other Pervasive Developmental Disorders. J. Clin. Psychiatry 2005, 66, 3. [Google Scholar] [PubMed]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism spectrum disorder. Nat. Rev. Dis. Prim. 2020, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Rutter, M. Concepts of Autism: A Review of Research. J. Child Psychol. Psychiatry 1968, 9, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, J.; Fombonne, E.; Scorah, J.; Ibrahim, A.; Durkin, M.S.; Saxena, S.; Yusuf, A.; Shih, A.; Elsabbagh, M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E. The epidemiology of autism: A review. Psychol. Med. 1999, 29, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H. Autism spectrum disorders—A genetics review. Genet. Med. 2011, 13, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Rezazadeh, I.; Frohlich, J.; Loo, S.K.; Jeste, S.S. Brain connectivity in autism spectrum disorder. Curr. Opin. Neurol. 2016, 29, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Berto, S.; Treacher, A.H.; Caglayan, E.; Luo, D.; Haney, J.R.; Gandal, M.J.; Geschwind, D.H.; Montillo, A.A.; Konopka, G. Association between resting-state functional brain connectivity and gene expression is altered in autism spectrum disorder. Nat. Commun. 2022, 13, 3328. [Google Scholar] [CrossRef] [PubMed]
- Kana, R.K.; Uddin, L.Q.; Kenet, T.; Chigani, D.; Müller, R.A. Brain connectivity in autism. Front. Hum. Neurosci. 2014, 8, 349. [Google Scholar] [CrossRef] [PubMed]
- Benkarim, O.; Paquola, C.; Park, B.y.; Hong, S.J.; Royer, J.; Vos de Wael, R.; Lariviere, S.; Valk, S.; Bzdok, D.; Mottron, L.; et al. Connectivity alterations in autism reflect functional idiosyncrasy. Commun. Biol. 2021, 4, 1078. [Google Scholar] [CrossRef]
- Liu, X.; Huang, H. Alterations of functional connectivities associated with autism spectrum disorder symptom severity: A multi-site study using multivariate pattern analysis. Sci. Rep. 2020, 10, 4330. [Google Scholar] [CrossRef] [PubMed]
- Redcay, E.; Moran, J.M.; Mavros, P.L.; Tager-Flusberg, H.; Gabrieli, J.D.E.; Whitfield-Gabrieli, S. Intrinsic functional network organization in high-functioning adolescents with autism spectrum disorder. Front. Hum. Neurosci. 2013, 7, 573. [Google Scholar] [CrossRef] [PubMed]
- Roine, U.; Roine, T.; Salmi, J.; Nieminen-von Wendt, T.; Tani, P.; Leppämäki, S.; Rintahaka, P.; Caeyenberghs, K.; Leemans, A.; Sams, M. Abnormal wiring of the connectome in adults with high-functioning autism spectrum disorder. Mol. Autism 2015, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Vissers, M.E.; X Cohen, M.; Geurts, H.M. Brain connectivity and high functioning autism: A promising path of research that needs refined models, methodological convergence, and stronger behavioral links. Neurosci. Biobehav. Rev. 2012, 36, 604–625. [Google Scholar] [CrossRef] [PubMed]
- Woodbury-Smith, M.R.; Volkmar, F.R. Asperger syndrome. Eur. Child Adolesc. Psychiatry 2008, 18, 2–11. [Google Scholar] [CrossRef]
- Tantam, D. Asperger’s Syndrome. J. Child Psychol. Psychiatry 1988, 29, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Tantam, D. Psychological Disorder in Adolescents and Adults with Asperger Syndrome. Autism 2000, 4, 47–62. [Google Scholar] [CrossRef]
- Javaheripour, N.; Wagner, G.; de la Cruz, F.; Walter, M.; Szycik, G.R.; Tietze, F.A. Altered brain network organization in adults with Asperger’s syndrome: Decreased connectome transitivity and assortativity with increased global efficiency. Front. Psychiatry 2023, 14, 1223147. [Google Scholar] [CrossRef] [PubMed]
- Jenike, M.A. Obsessive-Compulsive Disorder. N. Engl. J. Med. 2004, 350, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.J.; Costa, D.L.C.; Lochner, C.; Miguel, E.C.; Reddy, Y.C.J.; Shavitt, R.G.; van den Heuvel, O.A.; Simpson, H.B. Obsessive-compulsive disorder. Nat. Rev. Dis. Prim. 2019, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Beucke, J.C.; Sepulcre, J.; Talukdar, T.; Linnman, C.; Zschenderlein, K.; Endrass, T.; Kaufmann, C.; Kathmann, N. Abnormally High Degree Connectivity of the Orbitofrontal Cortex in Obsessive-Compulsive Disorder. JAMA Psychiatry 2013, 70, 619. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.J.; Soriano-Mas, C.; Pujol, J.; Ortiz, H.; López-Solà, M.; Hernández-Ribas, R.; Deus, J.; Alonso, P.; Yücel, M.; Pantelis, C.; et al. Altered Corticostriatal Functional Connectivity in Obsessive-compulsive Disorder. Arch. Gen. Psychiatry 2009, 66, 1189. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cao, L.; Li, H.; Gao, Y.; Bu, X.; Liang, K.; Bao, W.; Zhang, S.; Qiu, H.; Li, X.; et al. Abnormal resting-state functional connectivity in patients with obsessive-compulsive disorder: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2022, 135, 104574. [Google Scholar] [CrossRef] [PubMed]
- Thibaut, F. Anxiety disorders: A review of current literature. Dialogues Clin. Neurosci. 2017, 19, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Tibrewal, P.; Looi, J.C.L.; Allison, S.; Bastiampillai, T. Benzodiazepines for the long-term treatment of anxiety disorders? Lancet 2021, 398, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.J. Measures of anxiety: State-Trait Anxiety Inventory (STAI), Beck Anxiety Inventory (BAI), and Hospital Anxiety and Depression Scale-Anxiety (HADS-A). Arthritis Care Res. 2011, 63, S467–S472. [Google Scholar] [CrossRef] [PubMed]
- Perino, M.T.; Myers, M.J.; Wheelock, M.D.; Yu, Q.; Harper, J.C.; Manhart, M.F.; Gordon, E.M.; Eggebrecht, A.T.; Pine, D.S.; Barch, D.M.; et al. Whole-Brain Resting-State Functional Connectivity Patterns Associated with Pediatric Anxiety and Involuntary Attention Capture. Biol. Psychiatry Glob. Open Sci. 2021, 1, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Yu, Y.; Wu, Y.; Kai, Y.; Wang, H.; Chen, Y.; Zu, M.; Pang, X.; Tian, Y. Altered brain entropy and functional connectivity patterns in generalized anxiety disorder patients. J. Affect. Disord. 2023, 332, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Van Dam, N.T.; Feng, C.; Luo, Y.; Ai, H.; Gu, R.; Xu, P. Anxious brain networks: A coordinate-based activation likelihood estimation meta-analysis of resting-state functional connectivity studies in anxiety. Neurosci. Biobehav. Rev. 2019, 96, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Vohryzek, J.; Cabral, J.; Vuust, P.; Deco, G.; Kringelbach, M.L. Understanding brain states across spacetime informed by whole-brain modelling. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2022, 380, 20210247. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Roy, D.; Banerjee, A. Whole-Brain Network Models: From Physics to Bedside. Front. Comput. Neurosci. 2022, 16, 866517. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The human brain in numbers: A linearly scaled-up primate brain. Front. Hum. Neurosci. 2009, 3, 857. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T. Total Number of Synapses in the Adult Human Neocortex. Undergrad. J. Math. Model. ONE + Two 2013, 3, 26. [Google Scholar] [CrossRef]
- Wu, Y.K.; Zenke, F. Nonlinear transient amplification in recurrent neural networks with short-term plasticity. eLife 2021, 10, e71263. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, E.; Jirsa, V. The quest for multiscale brain modeling. Trends Neurosci. 2022, 45, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.; Sheerington, C. On the regulation of the blood-supply of the brain. J. Physiol. 1890, 11, 85. [Google Scholar] [CrossRef]
- Xu, Z.; Xia, M.; Wang, X.; Liao, X.; Zhao, T.; He, Y. Meta-connectomic analysis maps consistent, reproducible, and transcriptionally relevant functional connectome hubs in the human brain. Commun. Biol. 2022, 5, 1056. [Google Scholar] [CrossRef] [PubMed]
- de Lange, S.C.; Scholtens, L.H.; van den Berg, L.H.; Boks, M.P.; Bozzali, M.; Cahn, W.; Dannlowski, U.; Durston, S.; Geuze, E.; van Haren, N.E.M.; et al. Shared vulnerability for connectome alterations across psychiatric and neurological brain disorders. Nat. Hum. Behav. 2019, 3, 988–998. [Google Scholar] [CrossRef]
- Park, B.y.; Kebets, V.; Larivière, S.; Hettwer, M.D.; Paquola, C.; van Rooij, D.; Buitelaar, J.; Franke, B.; Hoogman, M.; Schmaal, L.; et al. Multiscale neural gradients reflect transdiagnostic effects of major psychiatric conditions on cortical morphology. Commun. Biol. 2022, 5, 1024. [Google Scholar] [CrossRef]
- Deco, G.; Tononi, G.; Boly, M.; Kringelbach, M.L. Rethinking segregation and integration: Contributions of whole-brain modelling. Nat. Rev. Neurosci. 2015, 16, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Deco, G.; Van Hartevelt, T.J.; Fernandes, H.M.; Stevner, A.; Kringelbach, M.L. The most relevant human brain regions for functional connectivity: Evidence for a dynamical workspace of binding nodes from whole-brain computational modelling. NeuroImage 2017, 146, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Crofts, J.J.; Forrester, M.; Coombes, S.; O’Dea, R.D. Structure-function clustering in weighted brain networks. Sci. Rep. 2022, 12, 16793. [Google Scholar] [CrossRef]
- Bazinet, V.; Hansen, J.Y.; Misic, B. Towards a biologically annotated brain connectome. Nat. Rev. Neurosci. 2023, 24, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.Y.; Shafiei, G.; Vogel, J.W.; Smart, K.; Bearden, C.E.; Hoogman, M.; Franke, B.; van Rooij, D.; Buitelaar, J.; McDonald, C.R.; et al. Local molecular and global connectomic contributions to cross-disorder cortical abnormalities. Nat. Commun. 2022, 13, 4682. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; Huxley, A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef]
- Robinson, P.A.; Rennie, C.J.; Rowe, D.L.; O’Connor, S.C.; Gordon, E. Multiscale brain modelling. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1043–1050. [Google Scholar] [CrossRef]
- Martín-Loeches, M.; Muñoz Ruata, J.; Martínez-Lebrusant, L.; Gómez-Jarabo, G. Electrophysiology and intelligence: The electrophysiology of intellectual functions in intellectual disability. J. Intellect. Disabil. Res. 2001, 45, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. The recent excitement about neural networks. Nature 1989, 337, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Irastorza-Valera, L.; Benítez, J.M.; Montáns, F.J.; Saucedo-Mora, L. An Agent-Based Model to Reproduce the Boolean Logic Behaviour of Neuronal Self-Organised Communities through Pulse Delay Modulation and Generation of Logic Gates. Biomimetics 2024, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Guiraud, D.; Duffau, H.; Mandonnet, E.; Bonnetblanc, F. Electrophysiological brain mapping: Basics of recording evoked potentials induced by electrical stimulation and its physiological spreading in the human brain. Clin. Neurophysiol. 2017, 128, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Neumann, W.; Degen, K.; Schneider, G.; Kühn, A.A. Toward an electrophysiological “sweet spot” for deep brain stimulation in the subthalamic nucleus. Hum. Brain Mapp. 2017, 38, 3377–3390. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.; Ramdani, S.; Duffau, H.; Dali, M.; Vincent, M.A.; Mandonnet, E.; Guiraud, D.; Bonnetblanc, F. Electrophysiological Mapping During Brain Tumor Surgery: Recording Cortical Potentials Evoked Locally, Subcortically and Remotely by Electrical Stimulation to Assess the Brain Connectivity On-line. Brain Topogr. 2021, 34, 221–233. [Google Scholar] [CrossRef] [PubMed]
- McCreery, D.; Agnew, W.; Yuen, T.; Bullara, L. Charge density and charge per phase as cofactors in neural injury induced by electrical stimulation. IEEE Trans. Biomed. Eng. 1990, 37, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Rossel, O.; Hayashibe, M.; Herbet, G.; Duffau, H.; Guiraud, D.; Bonnetblanc, F. The difference between electrical microstimulation and direct electrical stimulation—Towards new opportunities for innovative functional brain mapping? Rev. Neurosci. 2016, 27, 231–258. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Chen, J.E. Multimodal EEG-fMRI: Advancing insight into large-scale human brain dynamics. Curr. Opin. Biomed. Eng. 2021, 18, 100279. [Google Scholar] [CrossRef] [PubMed]
- van Vugt, M.K.; Sederberg, P.B.; Kahana, M.J. Comparison of spectral analysis methods for characterizing brain oscillations. J. Neurosci. Methods 2007, 162, 49–63. [Google Scholar] [CrossRef]
- Kalatsky, V.A. Fourier Approach for Functional Imaging. In In Vivo Optical Imaging of Brain Function, 2nd ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009. [Google Scholar]
- Mallot, H.A. Fourier Analysis for Neuroscientists. In Springer Series in Bio-/Neuroinformatics; Springer International Publishing: Cham, Switzerland, 2013; pp. 57–81. [Google Scholar] [CrossRef]
- Graham, N. Does the brain perform a Fourier analysis of the visual scene? Trends Neurosci. 1979, 2, 207–208. [Google Scholar] [CrossRef]
- Ochs, A.L. Is Fourier analysis performed by the visual system or by the visual investigator. J. Opt. Soc. Am. 1979, 69, 95. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.X.; Gulbinaite, R. Five methodological challenges in cognitive electrophysiology. NeuroImage 2014, 85, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N.A.; Koch, C.; Harris, K.D.; Carandini, M. Challenges and opportunities for large-scale electrophysiology with Neuropixels probes. Curr. Opin. Neurobiol. 2018, 50, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Destrade, M.; Gilchrist, M.; Murphy, J.; Rashid, B.; Saccomandi, G. Extreme softness of brain matter in simple shear. Int. J. Non-Linear Mech. 2015, 75, 54–58. [Google Scholar] [CrossRef]
- Goriely, A.; Geers, M.G.D.; Holzapfel, G.A.; Jayamohan, J.; Jérusalem, A.; Sivaloganathan, S.; Squier, W.; van Dommelen, J.A.W.; Waters, S.; Kuhl, E. Mechanics of the brain: Perspectives, challenges, and opportunities. Biomech. Model. Mechanobiol. 2015, 14, 931–965. [Google Scholar] [CrossRef]
- Rashid, B.; Destrade, M.; Gilchrist, M.D. Mechanical characterization of brain tissue in simple shear at dynamic strain rates. J. Mech. Behav. Biomed. Mater. 2013, 28, 71–85. [Google Scholar] [CrossRef]
- Gilchrist, M.; Rashid, B.; Murphy, J.; Saccomandi, G. Quasi-static deformations of biological soft tissue. Math. Mech. Solids 2013, 18, 622–633. [Google Scholar] [CrossRef]
- Reiter, N.; Paulsen, F.; Budday, S. Mechanisms of mechanical load transfer through brain tissue. Sci. Rep. 2023, 13, 8703. [Google Scholar] [CrossRef] [PubMed]
- Budday, S.; Ovaert, T.C.; Holzapfel, G.A.; Steinmann, P.; Kuhl, E. Fifty Shades of Brain: A Review on the Mechanical Testing and Modeling of Brain Tissue. Arch. Comput. Methods Eng. 2019, 27, 1187–1230. [Google Scholar] [CrossRef]
- Chavoshnejad, P.; Vallejo, L.; Zhang, S.; Guo, Y.; Dai, W.; Zhang, T.; Razavi, M.J. Mechanical hierarchy in the formation and modulation of cortical folding patterns. Sci. Rep. 2023, 13, 13177. [Google Scholar] [CrossRef] [PubMed]
- Lanir, Y. Mechanistic micro-structural theory of soft tissues growth and remodeling: Tissues with unidirectional fibers. Biomech. Model. Mechanobiol. 2014, 14, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Braeu, F.A.; Seitz, A.; Aydin, R.C.; Cyron, C.J. Homogenized constrained mixture models for anisotropic volumetric growth and remodeling. Biomech. Model. Mechanobiol. 2016, 16, 889–906. [Google Scholar] [CrossRef] [PubMed]
- Walter, C.; Balouchzadeh, R.; Garcia, K.E.; Kroenke, C.D.; Pathak, A.; Bayly, P.V. Multi-scale measurement of stiffness in the developing ferret brain. Sci. Rep. 2023, 13, 20583. [Google Scholar] [CrossRef] [PubMed]
- Antonovaite, N.; Beekmans, S.V.; Hol, E.M.; Wadman, W.J.; Iannuzzi, D. Regional variations in stiffness in live mouse brain tissue determined by depth-controlled indentation mapping. Sci. Rep. 2018, 8, 12517. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.R.; Lynch, J.J.; O’Connor, D.T.; Newport, D.T.; Mulvihill, J.J.E. Mechanical and structural characterisation of the dural venous sinuses. Sci. Rep. 2020, 10, 21763. [Google Scholar] [CrossRef] [PubMed]
- Weickenmeier, J.; de Rooij, R.; Budday, S.; Steinmann, P.; Ovaert, T.; Kuhl, E. Brain stiffness increases with myelin content. Acta Biomater. 2016, 42, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Holtzmann, K.; Gautier, H.O.; Christ, A.F.; Guck, J.; Káradóttir, R.T.; Franze, K. Brain tissue stiffness is a sensitive marker for acidosis. J. Neurosci. Methods 2016, 271, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Rashid, B.; Destrade, M.; Gilchrist, M.D. Inhomogeneous deformation of brain tissue during tension tests. Comput. Mater. Sci. 2012, 64, 295–300. [Google Scholar] [CrossRef]
- Rashid, B.; Destrade, M.; Gilchrist, M.D. Determination of friction coefficient in unconfined compression of brain tissue. J. Mech. Behav. Biomed. Mater. 2012, 14, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Rashid, B.; Destrade, M.; Gilchrist, M.D. Influence of preservation temperature on the measured mechanical properties of brain tissue. J. Biomech. 2013, 46, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiong, H. Brain Tissue Preparation, Sectioning, and Staining. In Current Laboratory Methods in Neuroscience Research; Springer: New York, NY, USA, 2013; pp. 3–30. [Google Scholar] [CrossRef]
- Rashid, B.; Destrade, M.; Gilchrist, M.D. Temperature effects on brain tissue in compression. J. Mech. Behav. Biomed. Mater. 2012, 14, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Cyron, C.J.; Aydin, R.C.; Humphrey, J.D. A homogenized constrained mixture (and mechanical analog) model for growth and remodeling of soft tissue. Biomech. Model. Mechanobiol. 2016, 15, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.M.; Przybyla, L.; Weaver, V.M. Tissue mechanics regulate brain development, homeostasis and disease. J. Cell Sci. 2017, 130, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Cyron, C.J.; Humphrey, J.D. Growth and remodeling of load-bearing biological soft tissues. Meccanica 2016, 52, 645–664. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D. Constrained Mixture Models of Soft Tissue Growth and Remodeling-Twenty Years After. J. Elast. 2021, 145, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Reiter, N.; Roy, B.; Paulsen, F.; Budday, S. Insights into the Microstructural Origin of Brain Viscoelasticity: Prospects for Microstructure-Informed Constitutive Modeling. J. Elast. 2021, 145, 99–116. [Google Scholar] [CrossRef]
- Li, W.; Shepherd, D.E.T.; Espino, D.M. Investigation of the Compressive Viscoelastic Properties of Brain Tissue Under Time and Frequency Dependent Loading Conditions. Ann. Biomed. Eng. 2021, 49, 3737–3747. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Jiang, C.; Jiang, H. A visco-hyperelastic model of brain tissue incorporating both tension/compression asymmetry and volume compressibility. Acta Mech. 2019, 230, 2125–2135. [Google Scholar] [CrossRef]
- Rashid, B.; Destrade, M.; Gilchrist, M.D. Hyperelastic and Viscoelastic Properties of Brain Tissue in Tension. In Proceedings of the Volume 2: Biomedical and Biotechnology. American Society of Mechanical Engineers, IMECE2012, Houston, TX, USA, 9–15 November 2012. [Google Scholar] [CrossRef]
- Zwirner, J.; Scholze, M.; Waddell, J.N.; Ondruschka, B.; Hammer, N. Mechanical Properties of Human Dura Mater in Tension-An Analysis at an Age Range of 2 to 94 Years. Sci. Rep. 2019, 9, 16655. [Google Scholar] [CrossRef] [PubMed]
- Forte, A.E.; Gentleman, S.M.; Dini, D. On the characterization of the heterogeneous mechanical response of human brain tissue. Biomech. Model. Mechanobiol. 2016, 16, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Budday, S.; Sommer, G.; Birkl, C.; Langkammer, C.; Haybaeck, J.; Kohnert, J.; Bauer, M.; Paulsen, F.; Steinmann, P.; Kuhl, E.; et al. Mechanical characterization of human brain tissue. Acta Biomater. 2017, 48, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Holzapfel, G.A.; Fereidoonnezhad, B. Modeling of Damage in Soft Biological Tissues. In Biomechanics of Living Organs; Elsevier: Amsterdam, The Netherlands, 2017; pp. 101–123. [Google Scholar] [CrossRef]
- Saucedo-Mora, L.; García-Bañales, O.; Montáns, F.J.; Benítez, J.M. A two-parameter strain energy function for brain matter: An extension of the Hencky model to incorporate locking. Brain Multiphysics 2021, 2, 100036. [Google Scholar] [CrossRef]
- Vogel, J.W.; Corriveau-Lecavalier, N.; Franzmeier, N.; Pereira, J.B.; Brown, J.A.; Maass, A.; Botha, H.; Seeley, W.W.; Bassett, D.S.; Jones, D.T.; et al. Connectome-based modelling of neurodegenerative diseases: Towards precision medicine and mechanistic insight. Nat. Rev. Neurosci. 2023, 24, 620–639. [Google Scholar] [CrossRef] [PubMed]
- Sarvazyan, A.P.; Urban, M.W.; Greenleaf, J.F. Acoustic Waves in Medical Imaging and Diagnostics. Ultrasound Med. Biol. 2013, 39, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, G.; Qian, L.X.; Liang, S.; Destrade, M.; Cao, Y. Measuring the linear and nonlinear elastic properties of brain tissue with shear waves and inverse analysis. Biomech. Model. Mechanobiol. 2015, 14, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.R.; Rousselet, G.A. An Updated Guide to Robust Statistical Methods in Neuroscience. Curr. Protoc. 2023, 3, e719. [Google Scholar] [CrossRef] [PubMed]
- Bzdok, D. Classical Statistics and Statistical Learning in Imaging Neuroscience. Front. Neurosci. 2017, 11, 273651. [Google Scholar] [CrossRef]
- Garrett, D.D.; Kovacevic, N.; McIntosh, A.R.; Grady, C.L. The Importance of Being Variable. J. Neurosci. 2011, 31, 4496–4503. [Google Scholar] [CrossRef] [PubMed]
- Goris, R.L.T.; Movshon, J.A.; Simoncelli, E.P. Partitioning neuronal variability. Nat. Neurosci. 2014, 17, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, R.; Saiga, R.; Yamamoto, Y.; Uesugi, M.; Takeuchi, A.; Uesugi, K.; Terada, Y.; Suzuki, Y.; De Andrade, V.; De Carlo, F.; et al. Structural diverseness of neurons between brain areas and between cases. Transl. Psychiatry 2021, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Waschke, L.; Kloosterman, N.A.; Obleser, J.; Garrett, D.D. Behavior needs neural variability. Neuron 2021, 109, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.M.; Schwartz, C.; Lin, R.T.; Khan, A.A.; Toga, A.W. Three-Dimensional Statistical Analysis of Sulcal Variability in the Human Brain. J. Neurosci. 1996, 16, 4261–4274. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, F.; Scuro, C.; Pantano, P.; Bilotta, E. Modelling brain dynamics by Boolean networks. Sci. Rep. 2022, 12, 16543. [Google Scholar] [CrossRef] [PubMed]
- Macy, M.W.; Willer, R. From Factors to Actors: Computational Sociology and Agent-Based Modeling. Annu. Rev. Sociol. 2002, 28, 143–166. [Google Scholar] [CrossRef]
- de Marchi, S.; Page, S.E. Agent-Based Models. Annu. Rev. Political Sci. 2014, 17, 1–20. [Google Scholar] [CrossRef]
- DeAngelis, D.L.; Diaz, S.G. Decision-Making in Agent-Based Modeling: A Current Review and Future Prospectus. Front. Ecol. Evol. 2019, 6, 237. [Google Scholar] [CrossRef]
- Miller Neilan, R.; Majetic, G.; Gil-Silva, M.; Adke, A.P.; Carrasquillo, Y.; Kolber, B.J. Agent-based modeling of the central amygdala and pain using cell-type specific physiological parameters. PLoS Comput. Biol. 2021, 17, e1009097. [Google Scholar] [CrossRef] [PubMed]
- Avin, S.; Currie, A.; Montgomery, S.H. An agent-based model clarifies the importance of functional and developmental integration in shaping brain evolution. BMC Biol. 2021, 19, 97. [Google Scholar] [CrossRef]
- Tracy, M.; Cerdá, M.; Keyes, K.M. Agent-Based Modeling in Public Health: Current Applications and Future Directions. Annu. Rev. Public Health 2018, 39, 77–94. [Google Scholar] [CrossRef]
- Benítez, J.M.; García-Mozos, L.; Santos, A.; Montáns, F.J.; Saucedo-Mora, L. A simple agent-based model to simulate 3D tumor-induced angiogenesis considering the evolution of the hypoxic conditions of the cells. Eng. Comput. 2022, 38, 4115–4133. [Google Scholar] [CrossRef]
- Saucedo-Mora, L.; Sanz, M.A.; Montáns, F.J.; Benítez, J.M. A simple agent-based hybrid model to simulate the biophysics of glioblastoma multiforme cells and the concomitant evolution of the oxygen field. Comput. Methods Programs Biomed. 2024, 246, 108046. [Google Scholar] [CrossRef]
- Montijn, J.S.; Seignette, K.; Howlett, M.H.; Cazemier, J.L.; Kamermans, M.; Levelt, C.N.; Heimel, J.A. A parameter-free statistical test for neuronal responsiveness. eLife 2021, 10, e71969. [Google Scholar] [CrossRef] [PubMed]
- Chinesta, F.; Huerta, A.; Rozza, G.; Willcox, K. Model Reduction Methods. In Encyclopedia of Computational Mechanics, 2nd ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef] [PubMed]
- Chinesta, F.; Ladevèze, P.; Cueto, E. A Short Review on Model Order Reduction Based on Proper Generalized Decomposition. Arch. Comput. Methods Eng. 2011, 18, 395–404. [Google Scholar] [CrossRef]
- Chinesta, F.; Ammar, A.; Leygue, A.; Keunings, R. An overview of the proper generalized decomposition with applications in computational rheology. J. Non-Newton. Fluid Mech. 2011, 166, 578–592. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Colonius, T. Guide to Spectral Proper Orthogonal Decomposition. AIAA J. 2020, 58, 1023–1033. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y. Locally linear embedding: A survey. Artif. Intell. Rev. 2011, 36, 29–48. [Google Scholar] [CrossRef]
- Aguado, J.V.; Borzacchiello, D.; Lopez, E.; Abisset-Chavanne, E.; González, D.; Cueto, E.; Chinesta, F. New Trends in Computational Mechanics: Model Order Reduction, Manifold Learning and Data-Driven. In From Microstructure Investigations to Multiscale Modeling: Bridging the Gap; ISTE Ltd.: London, UK, 2017. [Google Scholar] [CrossRef]
- Champaney, V.; Amores, V.J.; Garois, S.; Irastorza-Valera, L.; Ghnatios, C.; Montáns, F.J.; Cueto, E.; Chinesta, F. Modeling systems from partial observations. Front. Mater. 2022, 9, 970970. [Google Scholar] [CrossRef]
- McCulloch, W.S.; Pitts, W. A logical calculus of the ideas immanent in nervous activity. Bull. Math. Biophys. 1943, 5, 115–133. [Google Scholar] [CrossRef]
- Irastorza-Valera, L.; Benítez, J.M.; Montáns, F.J.; Saucedo-Mora, L. Numerical reproduction of the Sherrington-Adrian observations through a community of McCulloch-Pitts neurons with plastic remodelling. bioRxiv 2023. [Google Scholar] [CrossRef]
- McCarthy, J.; Minsky, M.L.; Rochester, N.; Shannon, C.E. A Proposal for the Dartmouth Summer Research Project on Artificial Intelligence. AI Mag. 2006, 27, 12. [Google Scholar]
- Turing, A.M. Computing Machinery and Intelligence. Mind 1950, LIX, 433–460. [Google Scholar] [CrossRef]
- Nagarajan, N.; Stevens, C.F. How does the speed of thought compare for brains and digital computers? Curr. Biol. 2008, 18, R756–R758. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.; Massad, E.; Coutinho, F. Can the human brain do quantum computing? Med. Hypotheses 2004, 63, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Hameroff, S.R. The Brain Is Both Neurocomputer and Quantum Computer. Cogn. Sci. 2007, 31, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Schuman, C.D.; Kulkarni, S.R.; Parsa, M.; Mitchell, J.P.; Date, P.; Kay, B. Opportunities for neuromorphic computing algorithms and applications. Nat. Comput. Sci. 2022, 2, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, B.; Sebastian, A.; Schmuker, M.; Srinivasa, N.; Eleftheriou, E. Low-Power Neuromorphic Hardware for Signal Processing Applications: A Review of Architectural and System-Level Design Approaches. IEEE Signal Process. Mag. 2019, 36, 97–110. [Google Scholar] [CrossRef]
- Marković, D.; Grollier, J. Quantum neuromorphic computing. Appl. Phys. Lett. 2020, 117, 150501. [Google Scholar] [CrossRef]
- Yamashita, R.; Nishio, M.; Do, R.K.G.; Togashi, K. Convolutional neural networks: An overview and application in radiology. Insights Imaging 2018, 9, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J.; Kushibar, K.; Asfaw, D.S.; Valverde, S.; Oliver, A.; Martí, R.; Lladó, X. Deep convolutional neural networks for brain image analysis on magnetic resonance imaging: A review. Artif. Intell. Med. 2019, 95, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Çinar, A.; Yildirim, M. Detection of tumors on brain MRI images using the hybrid convolutional neural network architecture. Med. Hypotheses 2020, 139, 109684. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, A.; Maitra, M. MRI-based brain tumour image detection using CNN based deep learning method. Neurosci. Inform. 2022, 2, 100060. [Google Scholar] [CrossRef]
- Badža, M.M.; Barjaktarović, M.v. Classification of Brain Tumors from MRI Images Using a Convolutional Neural Network. Appl. Sci. 2020, 10, 1999. [Google Scholar] [CrossRef]
- Lin, W.; Tong, T.; Gao, Q.; Guo, D.; Du, X.; Yang, Y.; Guo, G.; Xiao, M.; Du, M.; Qu, X. Convolutional Neural Networks-Based MRI Image Analysis for the Alzheimer’s Disease Prediction From Mild Cognitive Impairment. Front. Neurosci. 2018, 12, 777. [Google Scholar] [CrossRef] [PubMed]
- Vreeken, J. Spiking neural networks, an introduction 2003.
- Ghosh-Dastidar, S.; Adeli, H. SPIKING NEURAL NETWORKS. Int. J. Neural Syst. 2009, 19, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Vo-Ho, V.K.; Bulsara, D.; Le, N. Spiking Neural Networks and Their Applications: A Review. Brain Sci. 2022, 12, 863. [Google Scholar] [CrossRef] [PubMed]
- Kasabov, N.; Capecci, E. Spiking neural network methodology for modelling, classification and understanding of EEG spatio-temporal data measuring cognitive processes. Inf. Sci. 2015, 294, 565–575. [Google Scholar] [CrossRef]
- Wang, J.; Belatreche, A.; Maguire, L.; McGinnity, T.M. An online supervised learning method for spiking neural networks with adaptive structure. Neurocomputing 2014, 144, 526–536. [Google Scholar] [CrossRef]
- Tavanaei, A.; Maida, A. BP-STDP: Approximating backpropagation using spike timing dependent plasticity. Neurocomputing 2019, 330, 39–47. [Google Scholar] [CrossRef]
- Gong, Y.; Chen, T.; Wang, S.; Duan, S.; Wang, L. Lightweight spiking neural network training based on spike timing dependent backpropagation. Neurocomputing 2023, 570, 127059. [Google Scholar] [CrossRef]
- Young, A.R.; Dean, M.E.; Plank, J.S.; Rose, G.S. A Review of Spiking Neuromorphic Hardware Communication Systems. IEEE Access 2019, 7, 135606–135620. [Google Scholar] [CrossRef]
- Huynh, P.K.; Varshika, M.L.; Paul, A.; Isik, M.; Balaji, A.; Das, A. Implementing Spiking Neural Networks on Neuromorphic Architectures: A Review. arXiv 2022, arXiv:2202.08897. [Google Scholar] [CrossRef]
- Crimi, A.; Giancardo, L.; Sambataro, F.; Gozzi, A.; Murino, V.; Sona, D. MultiLink Analysis: Brain Network Comparison via Sparse Connectivity Analysis. Sci. Rep. 2019, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Sotero, R.C.; Canales-Rodríguez, E.J.; Alemán-Gómez, Y.; Melie-García, L. Studying the human brain anatomical network via diffusion-weighted MRI and Graph Theory. NeuroImage 2008, 40, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Fornito, A.; Zalesky, A.; Breakspear, M. Graph analysis of the human connectome: Promise, progress, and pitfalls. NeuroImage 2013, 80, 426–444. [Google Scholar] [CrossRef]
- Farahani, F.V.; Karwowski, W.; Lighthall, N.R. Application of Graph Theory for Identifying Connectivity Patterns in Human Brain Networks: A Systematic Review. Front. Neurosci. 2019, 13, 439505. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.H.; Rowland, C.; Harland, B.; Moslehi, S.; Montgomery, R.D.; Schobert, K.; Watterson, W.J.; Dalrymple-Alford, J.; Taylor, R.P. How neurons exploit fractal geometry to optimize their network connectivity. Sci. Rep. 2021, 11, 2332. [Google Scholar] [CrossRef] [PubMed]
- Montáns, F.J.; Cueto, E.; Bathe, K.J. Machine Learning in Computer Aided Engineering. In Machine Learning in Modeling and Simulation; Springer International Publishing: Cham, Switzerland, 2023; pp. 1–83. [Google Scholar] [CrossRef]
- Gray, H. Anatomy: Descriptive and Surgical, Anatomy of the Human Body; John William Parker: London, UK, 1858. [Google Scholar]
- Chinesta, F.; Cueto, E.; Abisset-Chavanne, E.; Duval, J.L.; Khaldi, F.E. Virtual, Digital and Hybrid Twins: A New Paradigm in Data-Based Engineering and Engineered Data. Arch. Comput. Methods Eng. 2018, 27, 105–134. [Google Scholar] [CrossRef]
- Moya, B.; Badías, A.; Alfaro, I.; Chinesta, F.; Cueto, E. Digital twins that learn and correct themselves. Int. J. Numer. Methods Eng. 2020, 123, 3034–3044. [Google Scholar] [CrossRef]

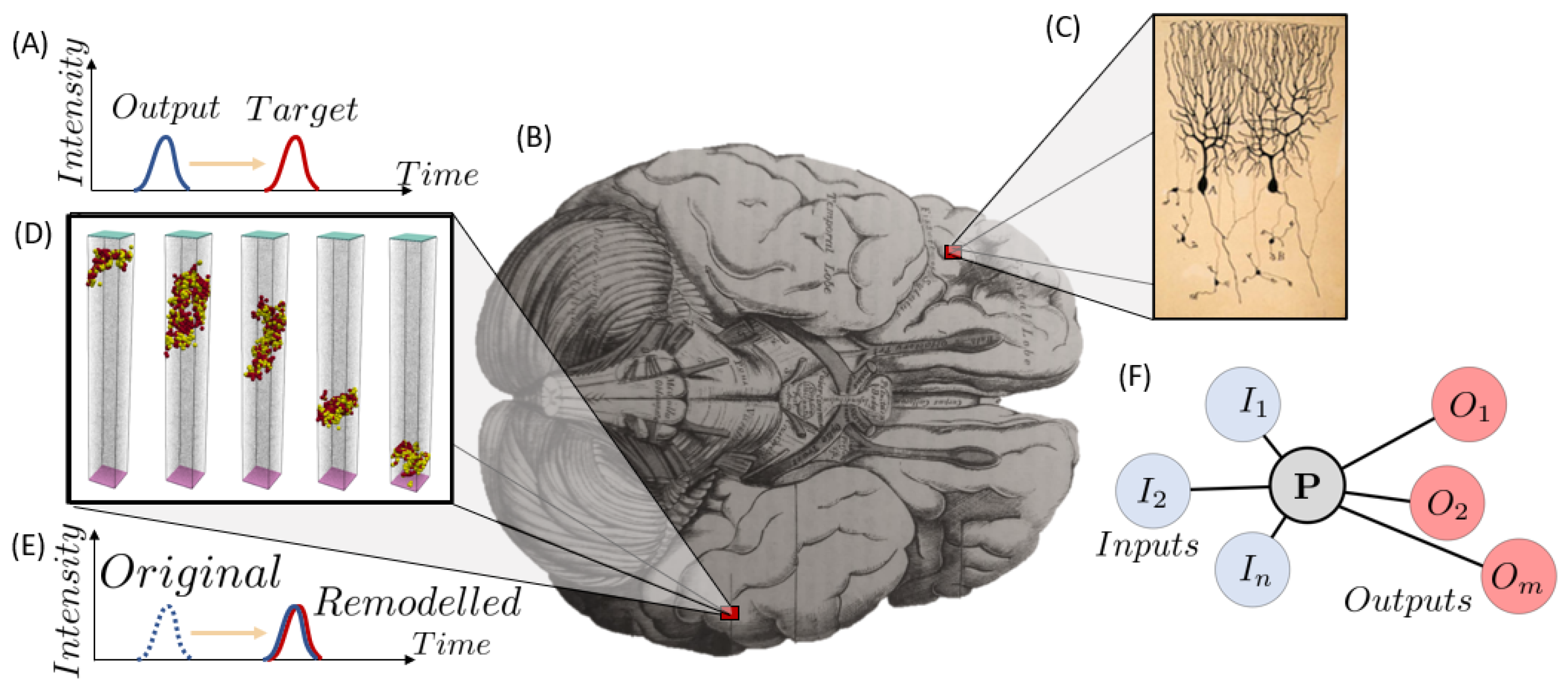
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irastorza-Valera, L.; Soria-Gómez, E.; Benitez, J.M.; Montáns, F.J.; Saucedo-Mora, L. Review of the Brain’s Behaviour after Injury and Disease for Its Application in an Agent-Based Model (ABM). Biomimetics 2024, 9, 362. https://doi.org/10.3390/biomimetics9060362
Irastorza-Valera L, Soria-Gómez E, Benitez JM, Montáns FJ, Saucedo-Mora L. Review of the Brain’s Behaviour after Injury and Disease for Its Application in an Agent-Based Model (ABM). Biomimetics. 2024; 9(6):362. https://doi.org/10.3390/biomimetics9060362
Chicago/Turabian StyleIrastorza-Valera, Luis, Edgar Soria-Gómez, José María Benitez, Francisco J. Montáns, and Luis Saucedo-Mora. 2024. "Review of the Brain’s Behaviour after Injury and Disease for Its Application in an Agent-Based Model (ABM)" Biomimetics 9, no. 6: 362. https://doi.org/10.3390/biomimetics9060362
APA StyleIrastorza-Valera, L., Soria-Gómez, E., Benitez, J. M., Montáns, F. J., & Saucedo-Mora, L. (2024). Review of the Brain’s Behaviour after Injury and Disease for Its Application in an Agent-Based Model (ABM). Biomimetics, 9(6), 362. https://doi.org/10.3390/biomimetics9060362