Omics Approaches for Identifying Physiological Adaptations to Genome Instability in Aging

Abstract
:1. Introduction
2. Adaptive Response to Stress
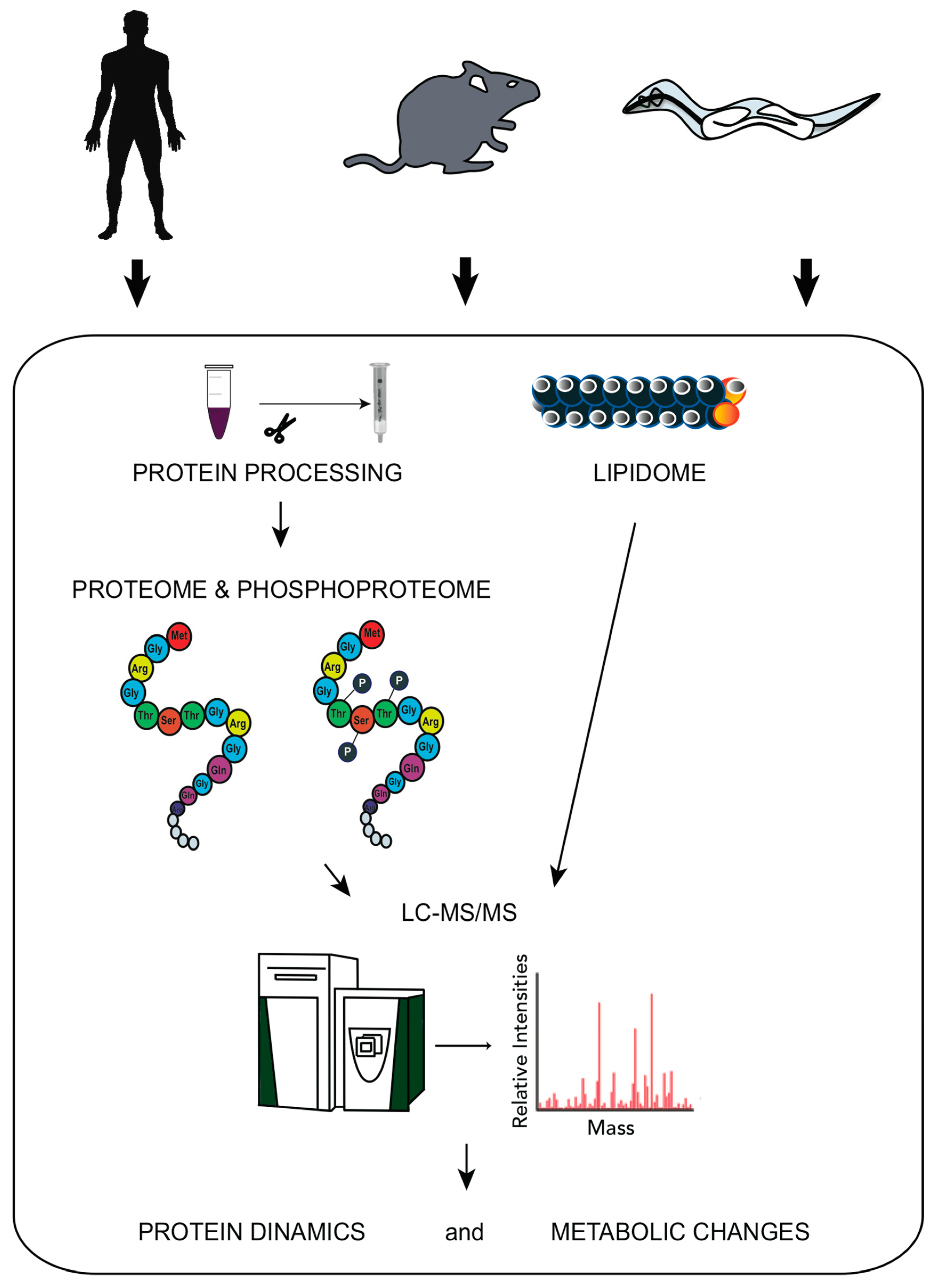
2.1 High-Throughput Approaches as a Tool to Identify Organismal Response Mechanisms upon Stress
2.2 In Vivo Models to Study Adaptations to Nucleotide-Excision Repair (NER) Defects
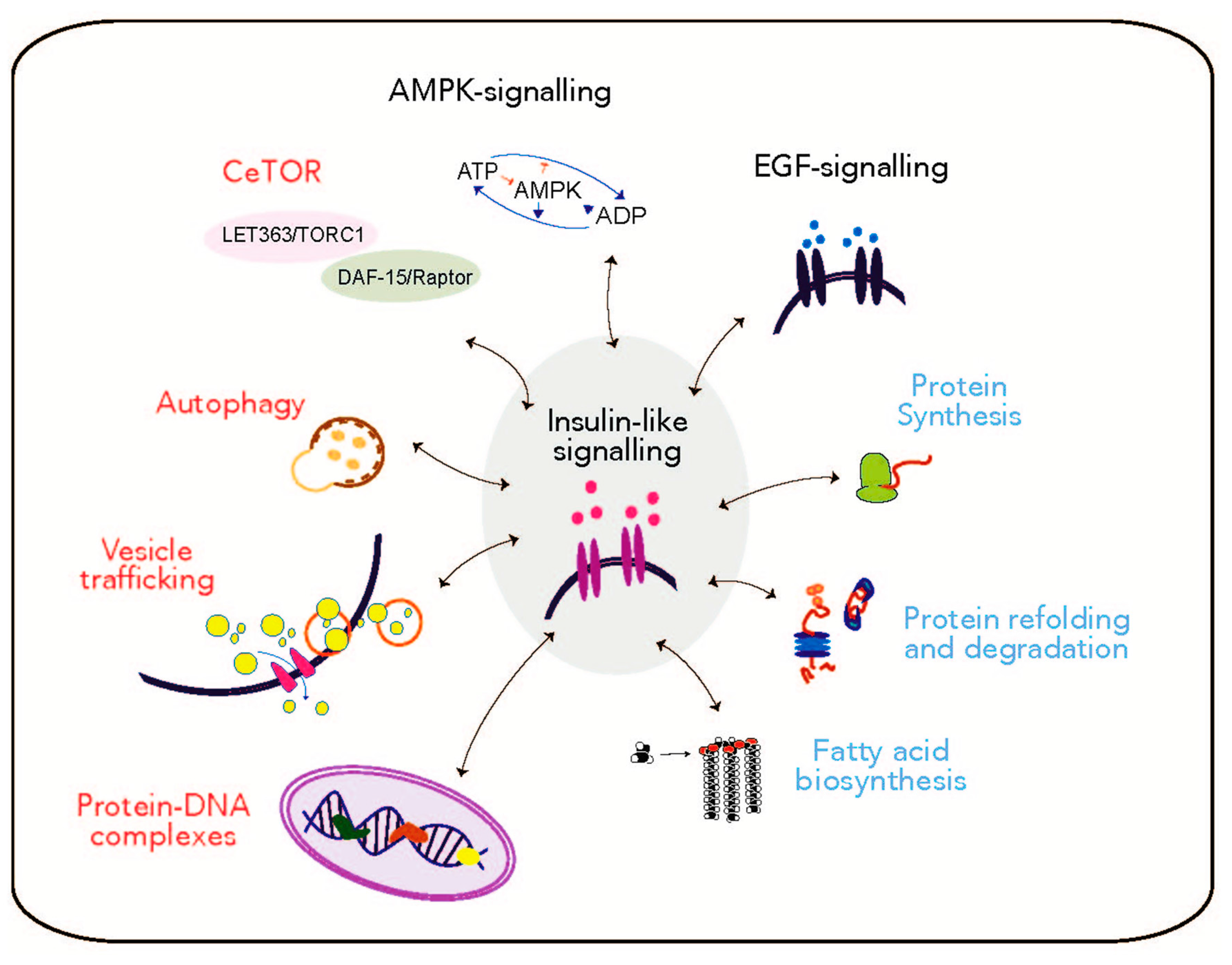
2.3 The Response to Unrepaired DNA Damage upon Nucleotide Excision Repair (NER) Deficiencies Involves Mechanisms that Regulate the Aging Process
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fortini, P.; Pascucci, B.; Parlanti, E.; D’Errico, M.; Simonelli, V.; Dogliotti, E. The base excision repair: Mechanisms and its relevance for cancer susceptibility. Biochimie 2003, 85, 1053–1071. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Hieda, K.; Nikaido, O. Wavelength dependent formation of thymine dimers and (6-4) photoproducts in DNA by monochromatic ultraviolet light ranging from 150 to 365 nm. Photochem. Photobiol. 1991, 54, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Setlow, R.B.; Carrier, W.L. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J. Mol. Biol. 1966, 17, 237–254. [Google Scholar] [CrossRef]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; Suh, Y. Genome Instability and Aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Edifizi, D.; Schumacher, B. Genome Instability in Development and Aging: Insights from Nucleotide Excision Repair in Humans, Mice, and Worms. Biomolecules 2015, 5, 1855–1869. [Google Scholar] [CrossRef] [PubMed]
- Edifizi, D.; Nolte, H.; Babu, V.; Castells-Roca, L.; Mueller, M.M.; Brodesser, S.; Krüger, M.; Schumacher, B. Multilayered Reprogramming in Response to Persistent DNA Damage in C. elegans. Cell Rep. 2017, 20, 2026–2043. [Google Scholar] [CrossRef] [PubMed]
- Garinis, G.A.; Uittenboogaard, L.M.; Stachelscheid, H.; Fousteri, M.; van IJcken, W.; Breit, T.M.; van Steeg, H.; Mullenders, L.H. F.; van der Horst, G.T. J.; Brüning, J.C.; et al. Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nat. Cell Biol. 2009, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Castells-Roca, L.; Babu, V.; Ermolaeva, M.A.; Muller, R.-U.; Frommolt, P.; Williams, A.B.; Greiss, S.; Schneider, J.I.; Benzing, T.; et al. DAF-16/FOXO and EGL-27/GATA promote developmental growth in response to persistent somatic DNA damage. Nat. Cell Biol. 2014, 16, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.; O’Connor, G.; DiStefano, P.S. Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell 2006, 5, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Qiang, L.; Han, W.; Ming, M.; Viollet, B.; He, Y.Y. Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene 2013, 32, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.G.; Chan, R.B.; Bravo, F.V.; Miranda, A.; Silva, R.R.; Zhou, B.; Marques, F.; Pinto, V.; Cerqueira, J.J.; di Paolo, G.; et al. The impact of chronic stress on the rat brain lipidome. Mol. Psychiatry 2015, 21, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, M.; Sakamoto, K. Fatty-acid metabolism is involved in stress-resistance mechanisms of Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2009, 390, 1402–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Ly, T.; Pourkarimi, E.; Murillo, A.B.; Gartner, A.; Lamond, A.I.; Kenyon, C. Deep proteome analysis identifies age-related processes in C. elegans. Cell Syst. 2016, 3, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Liang, V.; Ullrich, M.; Lam, H.; Chew, Y.L.; Banister, S.; Song, X.; Zaw, T.; Kassiou, M.; Götz, J.; Nicholas, H.R. Altered proteostasis in aging and heat shock response in C. elegans revealed by analysis of the global and de novo synthesized proteome. Cell. Mol. Life Sci. 2014, 71, 3339–3361. [Google Scholar] [CrossRef] [PubMed]
- Von Stechow, L.; Olsen, J.V. Proteomics insights into DNA damage response and translating this knowledge to clinical strategies. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, Y.; Niranjan, M. Bridging the gap between transcriptome and proteome measurements identifies post-translationally regulated genes. Bioinformatics 2013, 29, 3060–3066. [Google Scholar] [CrossRef] [PubMed]
- Lackner, D.H.; Schmidt, M.W.; Wu, S.; Wolf, D.A.; Bähler, J. Regulation of transcriptome, translation, and proteome in response to environmental stress in fission yeast. Genome Biol. 2012, 13, R25. [Google Scholar] [CrossRef] [PubMed]
- Derks, K.W.J.; Hoeijmakers, J.H.J.; Pothof, J. The DNA damage response: The omics era and its impact. DNA Repair 2014, 19, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Hirai, M.Y.; Yano, M.; Goodenowe, D.B.; Kanaya, S.; Kimura, T.; Awazuhara, M.; Arita, M.; Fujiwara, T.; Saito, K. Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2004, 101, 10205–10210. [Google Scholar] [CrossRef] [PubMed]
- Montoliu, I.; Scherer, M.; Beguelin, F.; DaSilva, L.; Mari, D.; Salvioli, S.; Martin, F.-P.J.; Capri, M.; Bucci, L.; Ostan, R.; et al. Serum profiling of healthy aging identifies phospho- and sphingolipid species as markers of human longevity. Aging 2014, 6, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.S.; Taubert, S. Function and Regulation of Lipid Biology in Caenorhabditis elegans Aging. Front Physiol. 2012, 3, 143. [Google Scholar] [CrossRef] [PubMed]
- Begley, T.J.; Rosenbach, A.S.; Ideker, T.; Samson, L.D. Damage recovery pathways in Saccharomyces cerevisiae revealed by genomic phenotyping and interactome mapping. Mol. Cancer Res. 2002, 1, 103–112. [Google Scholar] [PubMed]
- Begley, T.J.; Rosenbach, A.S.; Ideker, T.; Samson, L.D. Hot spots for modulating toxicity identified by genomic phenotyping and localization mapping. Mol. Cell 2004, 16, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Said, M.R.; Begley, T.J.; Oppenheim, A.V.; Lauffenburger, D.A.; Samson, L.D. Global network analysis of phenotypic effects: Protein networks and toxicity modulation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2004, 101, 18006–18011. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, S.C.; Teixeira, M.C.; Cabrito, T.R.; Sá-Correia, I. Yeast toxicogenomics: Genome-wide responses to chemical stresses with impact in environmental health, pharmacology, and biotechnology. Front. Genet. 2012, 3, 63. [Google Scholar] [CrossRef] [PubMed]
- Copes, N.; Edwards, C.; Chaput, D.; Saifee, M.; Barjuca, I.; Nelson, D.; Paraggio, A.; Saad, P.; Lipps, D.; Stevens, S.M.; et al. Metabolome and proteome changes with aging in Caenorhabditis elegans. Exp. Gerontol. 2015, 72, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Ippolito, D.L.; Lewis, J.A.; Yu, C.; Leon, L.R.; Stallings, J.D. Alteration in circulating metabolites during and after heat stress in the conscious rat: Potential biomarkers of exposure and organ-specific injury. BMC Physiol. 2014, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- De Fatima Magliarelli, H.; Matondo, M.; Mészáros, G.; Goginashvili, A.; Erbs, E.; Zhang, Z.; Mihlan, M.; Wolfrum, C.; Aebersold, R.; Sumara, I.; et al. Liver ubiquitome uncovers nutrient-stress-mediated trafficking and secretion of complement C3. Cell Death Dis. 2016, 7, e2411. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, S.; Aude-Garcia, C.; Armand, L.; Collin-Faure, V.; Chevallet, M.; Diemer, H.; Gerdil, A.; Proamer, F.; Strub, J.-M.; Habert, A.; et al. Comparative proteomic analysis of the molecular responses of mouse macrophages to titanium dioxide and copper oxide nanoparticles unravels some toxic mechanisms for copper oxide nanoparticles in macrophages. PLoS ONE 2015, 10, e0124496. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, B.; Seshi, B.; Ratanaprayul, W.; Dalal, N.; Lin, L.; Raval, A.; Chakravarti, D.N. Proteome profiling of aging in mouse models: Differential expression of proteins involved in metabolism, transport, and stress response in kidney. Proteomics 2009, 9, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Stauch, K.L.; Purnell, P.R.; Villeneuve, L.M.; Fox, H.S. Data for mitochondrial proteomic alterations in the aging mouse brain. Data Brief 2015, 4, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Chorell, E.; Moritz, T.; Branth, S.; Antti, H.; Svensson, M.B. Predictive metabolomics evaluation of nutrition-modulated metabolic stress responses in human blood serum during the early recovery phase of strenuous physical exercise. J. Proteome Res. 2009, 8, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Glass, D.; Spector, T.D. Omics technologies and the study of human ageing. Nat. Rev. Genet. 2013, 14, 601–607. [Google Scholar] [PubMed]
- Schumacher, B.; Garinis, G.A.; Hoeijmakers, J.H.J. Age to survive: DNA damage and aging. Trends Genet. 2008, 24, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kamileri, I.; Karakasilioti, I.; Sideri, A.; Kosteas, T.; Tatarakis, A.; Talianidis, I.; Garinis, G.A. Defective transcription initiation causes postnatal growth failure in a mouse model of nucleotide excision repair (NER) progeria. Proc. Natl. Acad. Sci. USA. 2012, 109, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, M.; Andressoo, J.O.; Holcomb, V.B.; von Lindern, M.; Jong, W.M. C.; de Zeeuw, C.I.; Suh, Y.; Hasty, P.; Hoeijmakers, J.H.J.; van der Horst, G.T. J.; et al. Adaptive stress response in segmental progeria resembles long-lived dwarfism and calorie restriction in mice. PLoS Genet. 2006, 2, e192. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; van der Pluijm, I.; Moorhouse, M.J.; Kosteas, T.; Robinson, A.R.; Suh, Y.; Breit, T.M.; van Steeg, H.; Niedernhofer, L.J.; van IJcken, W.; et al. Delayed and Accelerated Aging Share Common Longevity Assurance Mechanisms. PLoS Genet. 2008, 4, e1000161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, C.S.; Ramsey, M.M.; Sonntag, W.E. A critical analysis of the role of growth hormone and IGF-1 in aging and lifespan. Trends Genet. 2002, 18, 295–301. [Google Scholar] [CrossRef]
- Van der Pluijm, I.; Garinis, G.A.; Brandt, R.M.C.; Gorgels, T.G.M.F.; Wijnhoven, S.W.; Diderich, K.E.M.; de Wit, J.; Mitchell, J.R.; van Oostrom, C.; Beems, R.; et al. Impaired Genome Maintenance Suppresses the Growth Hormone–Insulin-Like Growth Factor 1 Axis in Mice with Cockayne Syndrome. PLoS Biol. 2006, 5, e2. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Pamenter, R.W.; Rocha, J.S.; Masternak, M.M.; Panici, J.A.; Bartke, A. Long-lived growth hormone receptor knockout mice show a delay in age-related changes of body composition and bone characteristics. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Castells-Roca, L.; Mueller, M.M.; Schumacher, B. Longevity through DNA damage tolerance. Cell Cycle 2015, 14, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Vermeulen, W. Nucleotide Excision Repair in Caenorhabditis elegans. Mol. Biol. Int. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Larance, M.; Pourkarimi, E.; Wang, B.; Brenes Murillo, A.; Kent, R.; Lamond, A.I.; Gartner, A. Global Proteomics Analysis of the Response to Starvation in C. elegans. Mol. Cell Proteom. 2015, 14, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Boyd, W.A.; Crocker, T.L.; Rodriguez, A.M.; Leung, M.C.K.; Wade Lehmann, D.; Freedman, J.H.; van Houten, B.; Meyer, J.N. Nucleotide excision repair genes are expressed at low levels and are not detectably inducible in Caenorhabditis elegans somatic tissues, but their function is required for normal adult life after UVC exposure. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 683, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Pontoizeau, C.; Mouchiroud, L.; Molin, L.; Mergoud-dit-Lamarche, A.; Dallière, N.; Toulhoat, P.; Elena-Herrmann, B.; Solari, F. Metabolomics Analysis Uncovers That Dietary Restriction Buffers Metabolic Changes Associated with Aging in Caenorhabditis elegans. J. Proteom. Res. 2014, 13, 2910–2919. [Google Scholar] [CrossRef] [PubMed]
- McElwee, J.J.; Schuster, E.; Blanc, E.; Thomas, J.H.; Gems, D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 2004, 279, 44533–44543. [Google Scholar] [CrossRef] [PubMed]
- Golden, T.R.; Melov, S. Microarray analysis of gene expression with age in individual nematodes. Aging Cell 2004, 3, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar] [PubMed]
- Henderson, S.T.; Johnson, T.E. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001, 11, 1975–1980. [Google Scholar] [CrossRef]
- Mair, W.; Morantte, I.; Rodrigues, A.P.C.; Manning, G.; Montminy, M.; Shaw, R.J.; Dillin, A. Lifespan extension induced by AMPK and calcineurin is mediated by CRTC-1 and CREB. Nature 2012, 470, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Apfeld, J.; O’Connor, G.; McDonagh, T.; DiStefano, P.S.; Curtis, R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004, 18, 3004–3009. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, J.; Yan, X.-H. Cross-talk between AMPK and mTOR in regulating energy balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Chen, J. Regulation of peroxisome proliferator-activated receptor-γ activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. Interplay between FOXO, TOR, and Akt. Biochim. Biophys. Acta 2011, 1813, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, H.; Yu, S.; Xue, J.; Driscoll, M. Novel EGF pathway regulators modulate C. elegans healthspan and lifespan via EGF receptor, PLC-γ, and IP3R activation. Aging Cell 2010, 9, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Rogers, J.; Murphy, C.T.; Rongo, C. EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J. 2011, 30, 2990–3003. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.J.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Mariño, G.; Ugalde, A.P.; Salvador-Montoliu, N.; Varela, I.; Quirós, P.M.; Cadiñanos, J.; van der Pluijm, I.; Freije, J.M.P.; López-Otín, C. Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum. Mol. Genet. 2008, 17, 2196–2211. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Miller, E.A.; Morimoto, R.I. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. USA 2009, 106, 14914–14919. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Dillin, A. The insulin paradox: Aging, proteotoxicity and neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Chen, X.; Kang, J.H.; Stolz, D.B.; Liu, J.; Yin, X.-M. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol. Cancer Ther. 2009, 8, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Megalou, E.V.; Tavernarakis, N. Autophagy in Caenorhabditis elegans. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yin, X.M. Coordination of autophagy and the proteasome in resolving endoplasmic reticulum stress. Vet. Pathol. 2011, 48, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and Aging: The Importance of Maintaining “Clean” Cells. Autophagy 2014, 1, 131–140. [Google Scholar] [CrossRef]
- Kikis, E.A.; Gidalevitz, T.; Morimoto, R.I. Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 2010, 694, 138–159. [Google Scholar] [PubMed]
- Basaiawmoit, R.V.; Rattan, S.I.S. Cellular stress and protein misfolding during aging. Methods Mol. Biol. 2010, 648, 107–117. [Google Scholar] [PubMed]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread Protein Aggregation as an Inherent Part of Aging in C. elegans. PLoS Biol. 2010, 8, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.C. The role of α-synuclein in neurodegenerative diseases. Pharmacol. Ther. 2005, 105, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Cicardi, M.E.; Giorgetti, E.; Onesto, E.; Messi, E.; Poletti, A. ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 2014, 79, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Dimayuga, E.; Chen, Q.; Thorpe, J.; Gee, J.; Ding, Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int. J. Biochem. Cell Biol. 2004, 36, 2376–2391. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.-L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovács, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Meléndez, A.; Hansen, M. Autophagy links lipid metabolism to longevity in C. elegans. Autophagy 2012, 8, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Gelino, S.; Meléndez, A.; Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol. 2011, 21, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Tavernarakis, N.; Kroemer, G. Can autophagy promote longevity? Nat. Cell Biol. 2010, 12, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: Pathophysiological implications. Am. J. Physiol. Cell Physiol. 2010, 298, C973–C978. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Garcia-Garcia, A.; Panayiotidis, M.I.; Franco, R. DNA damage and autophagy. Mutat. Res. 2011, 711, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Buckley, K.M.; Melikian, H.E.; Provoda, C.J.; Waring, M.T. Regulation of neuronal function by protein trafficking: A role for the endosomal pathway. J. Physiol. 2000, 525, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Reenstra, W.R.; Yaar, M.; Gilchrest, B.A. Aging affects epidermal growth factor receptor phosphorylation and traffic kinetics. Exp. Cell Res. 1996, 227, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Deák, F. Neuronal vesicular trafficking and release in age-related cognitive impairment. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, Y.; Zhang, X.; Bu, G.; Xu, H.; Zhang, Y.-W. Trafficking regulation of proteins in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Hunn, B.H. M.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.-G.; Wade-Martins, R. Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci. 2015, 38, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Thathiah, A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015, 589, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Torres, K.; Liu, X.; Liu, C.-G.; Pollock, R.E. An Overview of Chromatin-Regulating Proteins in Cells. Curr. Protein Pept. Sci. 2016, 17, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Marteijn, J.A.; Schumacher, B.; Hoeijmakers, J.H.J.; Jansen, G.; Vermeulen, W. Involvement of Global Genome Repair, Transcription Coupled Repair, and Chromatin Remodeling in UV DNA Damage Response Changes during Development. PLoS Genet. 2010, 6, e1000941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feser, J.; Tyler, J. Chromatin structure as a mediator of aging. FEBS Lett. 2011, 585, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Tyler, J.K. Histone exchange and histone modifications during transcription and aging. Biochim. Biophys. Acta 2013, 1819, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yip, R.K.; Zhou, Z. Chromatin remodeling, DNA damage repair and aging. Curr. Genomics 2012, 13, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Daniilidou, M.; Koutroumani, M.; Tsolaki, M. Epigenetic mechanisms in Alzheimer’s disease. Curr. Med. Chem. 2011, 18, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-F.; Zhang, Y.-W.; Xu, H.; Bu, G. Transcriptional regulation and its misregulation in Alzheimer’s disease. Mol. Brain 2013, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Ammal Kaidery, N.; Tarannum, S.; Thomas, B. Epigenetic landscape of Parkinson’s disease: Emerging role in disease mechanisms and therapeutic modalities. Neurotherapeutics 2013, 10, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Riedel, C.G.; Dowen, R.H.; Lourenco, G.F.; Kirienko, N.V.; Heimbucher, T.; West, J.A.; Bowman, S.K.; Kingston, R.E.; Dillin, A.; Asara, J.M.; et al. DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat. Cell Biol. 2013, 15, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ebata, A.; Dong, Y.; Rizki, G.; Iwata, T.; Lee, S.S. Caenorhabditis elegans HCF-1 functions in longevity maintenance as a DAF-16 regulator. PLoS Biol 2008, 6, e233. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Ma, H.; Burch, D.; Maciel, G.A.; Hunter, T.; Dillin, A. SMK-1, an Essential Regulator of DAF-16-Mediated Longevity. Cell 2006, 124, 1039–1053. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model Organism | Stress Condition | Study | Affected Processes |
|---|---|---|---|
| Saccharomyces cerevisiae | DNA-damaging agents (MMS,4NQO,T-BUOOH and UV) | Begley et al. 2002 [27] Begley et al. 2004 [28] Said et al. 2004 [29] | Chromatin remodeling Nucleo-cytoplasmic transport of RNA and proteins Macromolecular trafficking Cytoskeleton remodeling Protein and Lipid metabolism |
| Caenorhabditis elegans | UV irradiation upon NER deficiency | Edifizi et al. 2017 [10] | Chromatin remodeling |
| Protein homeostasis | |||
| Protein refolding and degradation | |||
| Macromolecular trafficking | |||
| Fatty and amino acid metabolism | |||
| Insulin-, EGF-, and AMPK-like signaling pathways | |||
| Heat, osmotic, and oxidative-stress | Horikawa et al. 2009 [16] Liang et al. 2014 [19] | Fatty-acid metabolism Protein homeostasis | |
| Aging | Copes et al. 2015 [31] Walther et al. 2015 [17] Narayan et al. 2016 [18] | Fatty and amino acid metabolism Protein homeostasis Protein refolding and degradation Peroxisomal enzymes Insulin-like signaling pathway | |
| Mus musculus/ Rattus norvegicus | Heat and chronic stress | Ippolito et al. 2014 [32] | Fatty and amino acid metabolism |
| Oliveira et al 2015 [15] | |||
| Nutrient stress | Magliarelli et al. 2016 [33] | Post-translational modifications | |
| Macromolecular trafficking | |||
| Copper oxide nanoparticles | Triboulet et al. 2015 [34] | Oxidative stress response | |
| Macrophage immune responses | |||
| Aging | Chakravarti et al. 2009 [35] Stauch et al. 2015 [36] | Protein refolding and degradation Macromolecular trafficking Cellular metabolism | |
| Homo sapiens | Nutrient stress coupled to physical exercise | Chorell et al. 2009 [37] | Fatty and amino acid metabolism |
| Aging / aging-related diseses | Valdes et al. 2013 [38] Montoliou et al. 2014 [25] | Fatty and amino acid metabolism Oxidative stress response Protein refolding and degradation Macromolecular trafficking |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edifizi, D.; Schumacher, B. Omics Approaches for Identifying Physiological Adaptations to Genome Instability in Aging. Int. J. Mol. Sci. 2017, 18, 2329. https://doi.org/10.3390/ijms18112329
Edifizi D, Schumacher B. Omics Approaches for Identifying Physiological Adaptations to Genome Instability in Aging. International Journal of Molecular Sciences. 2017; 18(11):2329. https://doi.org/10.3390/ijms18112329
Chicago/Turabian StyleEdifizi, Diletta, and Björn Schumacher. 2017. "Omics Approaches for Identifying Physiological Adaptations to Genome Instability in Aging" International Journal of Molecular Sciences 18, no. 11: 2329. https://doi.org/10.3390/ijms18112329
APA StyleEdifizi, D., & Schumacher, B. (2017). Omics Approaches for Identifying Physiological Adaptations to Genome Instability in Aging. International Journal of Molecular Sciences, 18(11), 2329. https://doi.org/10.3390/ijms18112329