Abstract
A convenient and general method for the synthesis in 26–73% yields of a new series of 7-alkyl(aryl/heteroaryl)-2-amino-5-trifluoromethyl-1,8-naphthyridines from direct cyclocondensation reactions of 4-alkoxy-1,1,1-trifluoroalk-3-en-2-ones [CF3C(O)CH=C(R1)OR, where R1 = H, Me, Ph, 4-MePh, 4-OMePh, 4-FPh, 4-BrPh, 4-NO2Ph, 2-furyl, 2-thienyl and R = Me, Et] with 2,6-diaminopyridine (2,6-DAP), under mild conditions, is described. Another synthetic route also allowed the synthesis of 2-amino-5-trifluoromethyl-cycloalka[b][1,8]naphthyridines in 33–36% yields, from direct or indirect cyclo-condensation reactions of five-, six- and seven-membered 2-trifluoroacetyl-1-methoxy-cycloalkenes with 2,6-DAP.
1. Introduction
Among the nitrogenous heterocycles, naphthyridines and their derivatives represent an important class of organic molecules that attract the interest of both synthetic and medicinal chemists due to their exceptionally broad spectrum of biological activities as well as their use as important binding units in the molecular design of synthetic receptors [1]. Naphthyridine derivatives have attracted considerable attention primarily due to the presence of a 1,8-naphthyridine skeleton in many compounds which have been isolated from natural substances and exhibit various biological activities [2]. As a heterocyclic moiety, 1,8-naphthyridine also deserves special interest as in its molecule, the arrangement of the nitrogen atoms is optimal for chelation of various metal cations, including lanthanide ions [3].
In parallel to the growing interest in the synthesis of 1,8-naphthyridines to provide biologically active molecules, a large number of publications have reported that several of their derivatives possess antibacterial [4], antimycobacterial [5], antitumor [6], anti-inflammatory [7,8], analgesic [8], antiplatelet [9], gastric antisecretary [10], local anaesthetic [11], anticonvulsant [12] and antihypertensive activity [13,14], besides being associated with β-adrenergic blocking properties [15]. Some 1,8-naphthyridine compounds have been patented as fungicides, bactericides, insecticides, herbicides, anxiolytic, antihypertensives, antiarrhythmics and also as immunostimulants [2,16,17,18,19].
In addition, has been recognized that attachment of a trifluoromethyl group into heterocycles can be used to modulate the physical, chemical and biological properties. It is well documented that the influence of the trifluoromethyl substituent on physiological activity is due mainly to the increased lipophilicity of the molecules, causing greater cell permeability and resistance to enzyme degradation [20]. Consequently, synthetic methodology to incorporate fluorine and fluorous synthons must be improved in order to prepare sophisticated fluoroorganic molecules on a practical scale. One of the most satisfactory methods for introducing a CF3 group into heterocycles is via the trifluoromethylated building block approach. The trifluoroacetylation of enol ethers or acetals provided, in one step and in good yields, β-alkoxyvinyl trifluoromethyl ketones 1 which proved to be useful building blocks for the syntheses of many series of heterocyclic compounds [21].
Since the 50s various diamino-ketoester condensations involving reactions of cyclic and acyclic β-ketoesters or diketones with aminopyridines or diaminopyridines have been studied in an attempt to develop generalized predictions regarding the direction of ring closure to form diazepinones, naphthyridones, naphthyridines or pyrimidines [6,12,13,14,15,19,22]. Whereas a literature review shows that the synthesis of trifluoromethylated naphthyridines and derivatives has been little explored and that 1,8-naphthyridines trifluoromethylated described are associated with satisfactory biological activities [14], the incorporation of trifluoromethyl group in a variety of 1,8-naphthyridines would be expected to provide highly desirable intermediates for the synthesis of new drug candidates. So, due to the great biological importance and employment of amino-naphthyridines as starting material for the synthesis of new tri and tetracyclic heterocycles, the development of new synthetic approaches remains an active research area [19,23].
The use of diethyl ethoxymethylenemalonate (EMME) [24], Conrad-Limpach [25], Knorr [26] and Skraup [27] methods have been particularly successful in the synthesis of certain quinolines. The adaptation of these reactions to the synthesis of the corresponding naphthyridines by employing aminopyridines instead of anilines should furnish convenient methods for the preparation of these types of compounds since aminopyridines are readily available [19]. However, these methods have not been as satisfactory for the preparation of 1,8-naphthyridines as they are the preparation of quinolines. In contrast to aniline derivatives, 2-aminopyridine derivatives may cyclize in two ways, one of which leads to the formation of 1,8-naphthyridines and the other leads to the formation of pyrimidines, and the latter course of reaction has is observed with more frequency [28]. In both types of cyclization, the pyridine ring functions as the electron donor and the carbonyl group in the side chain serves as the electron acceptor.
The formation of pyrimidines should not be surprising, since the resonance structures existing in the structure of the 2-aminopyridine derivatives strongly favor cyclization leading to the pyrimidine ring. Nevertheless, although the formation of a pyrimidine often occurs, an investigation of the synthesis of certain 1,8-naphthyridines from aminopyridines with diethylmalonate, ethoxymethylidenemalonate or ethyl acetoacetate has been made [14,19,29]. It is known that the synthesis of 1,8-naphthyridines has been performed successfully when 6-methyl-2-aminopyridine or 2,6-diaminopyridine are used as precursors, since 6-methyl or 6-amino groups activate the 3-position leading to those molecules [14,29]. Thus, the great difference in behavior of 2-aminopyridine and 2,6-diaminopyridine, for example, has usually been attributed to activation of the 3-position by the electron releasing amine group.
Recently, we reported reactivity of the endocyclic nitrogen atom of the π-deficient pyridine ring towards the carbonyl group of the trichloroacetyl enamine derivates from the reactions employing 4-alkoxy-4-alkyl(aryl)-1,1,1-trichloroalk-3-en-2-ones and 2-aminopyridine in a molar ratio of 1:1, presenting a convenient method to obtain 4-oxo-4H-pyrido[1,2-a]pyrimidines in good yields (45–81%) [28]. We also reported the synthesis of 5H-thiazolo[3,2-a]pyrimidi-5-ones from the reactions of β-alkoxyvinyl trichloromethyl ketones and 2-aminothiazole [30]. Unfortunately, reactions using β-alkoxyvinyl trifluoromethyl ketones and 2-aminopyridine in an attempt to obtain the respective cyclic structure only resulted in the isolation of trifluoroacetylenamine derivatives [32]. On the other hand, reactions using β-alkoxyvinyl trifluoro(chloro)methyl ketones and 2,3-diaminopyridine have been successfully employed in the synthesis of 3H-pyrido[2,3-b][1,4]diazepinols [31,32] or diazepinones [31,33].
Although the reactions of 4-alkoxy-4-alkyl(aryl/heteroaryl)-1,1,1-trifluoroalk-3-en-2-ones with primary and secondary amines have been well documented [28,30,31,32,33,34,35,36,37,38,39,40,41], there are no reports in the literature dealing with β-alkoxyvinyl trifluoromethyl ketones as electrophilic precursors and 2,6-diaminopyridine (2,6-DAP) as nucleophilic precursor. Considering the importance of trifluoromethylated heterocycles, as an extension of our research the purpose of this paper is to report the results of a chemical behavior study of the reactions of 4-alkoxy-4-alkyl(aryl/heteroaryl)-1,1,1-trifluoroalk-3-en-2-ones and 2-trifluoroacetyl-1-methoxycycloalkenes 1 with 2,6-DAP, a symmetrical heteroaromatic diamine, aiming at the synthesis of new nitrogen-containing trifluoromethylated heterocycles with conventional procedures (Scheme 1). None of methods reported to date for the synthesis of naphthyridines employs the strategy adopted in this study. Our method allows the easier introduction of CF3 group at position 5 and of wide scope of both electron-donor and electron-withdrawing substituents at position 7 and fused cycloalkanes to the C6-C7 bond of the naphthyridines ring. Furthermore, the free amino group at position 2, in both cases, allows further important derivatizations.
2. Results and Discussion
Initially, a series of ten examples of 4-alkoxy-4-alkyl(aryl/heteroaryl)-1,1,1-trifluoroalk-3-en-2-ones 1a-j, which are readily available CCC synthetic blocks, were prepared from trifluoroacetylation reactions of enol ethers commercially available (for 1a-b) or generated in situ from the respective acetophenone dimethyl acetal (for 1c-j) with trifluoroacetic anhydride, respectively, in the presence of pyridine, as described in the literature [42,43,44,45].
Fortunately, we found that trifluoromethylated ketones 1a-j when added dropwise to 2,6-DAP at a molar ratio of 1:1 in methanol as solvent at 0 °C for 2 hours and after heating under reflux for 24 hours, produced 7-alkyl(aryl/heteroaryl)-2-amino-5-trifluoromethyl-1,8-naphthyridines 3a-j. These compounds were easily isolated from the one-step reaction mixtures in 51–73% yields, although 3a, 3b and 3i were obtained in lower yields 26, 39 and 38%, respectively (Scheme 1). This is not surprising since a detailed review of the literature shows that enaminone derivatives of β-ethoxyvinyl trifluoromethyl ketone (enone 1a) present a different chemical behavior from other enones [41], leading to heterocycles with lower yields [34] and the absence of cyclization has been reported in many papers [28,35]. The naphthyridine 3b was previously also synthesized in a low yield (10–23%) by Eichler et al. from the reaction of 2,6-DAP and 1,1,1-trifluoropentane-2,4-dione [22].
As an extension of this study we also developed the synthesis of compound 3b from the cyclocondensation reaction of enamino ketone intermediate 2b in methanol at reflux temperature for 24 hours (Scheme 1). The isolation of 2b was possible when the reaction of enone 1b with 2,6-DAP was carried out in methanol as solvent at 0 °C for 2 hours. Unfortunately, the enamino ketones 2a, 2c-j could be not isolated as pure compounds, under the same or similar reaction conditions.
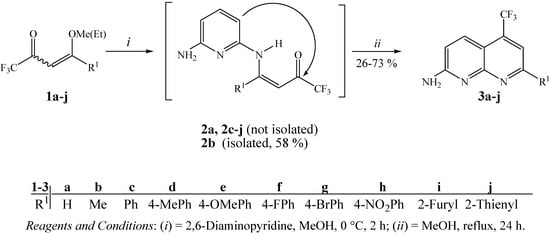
Scheme 1.
Synthesis of 7-alkyl(aryl/heteroaryl)-2-amino-5-trifluoromethyl-1,8-naphthyridines.
Scheme 1.
Synthesis of 7-alkyl(aryl/heteroaryl)-2-amino-5-trifluoromethyl-1,8-naphthyridines.
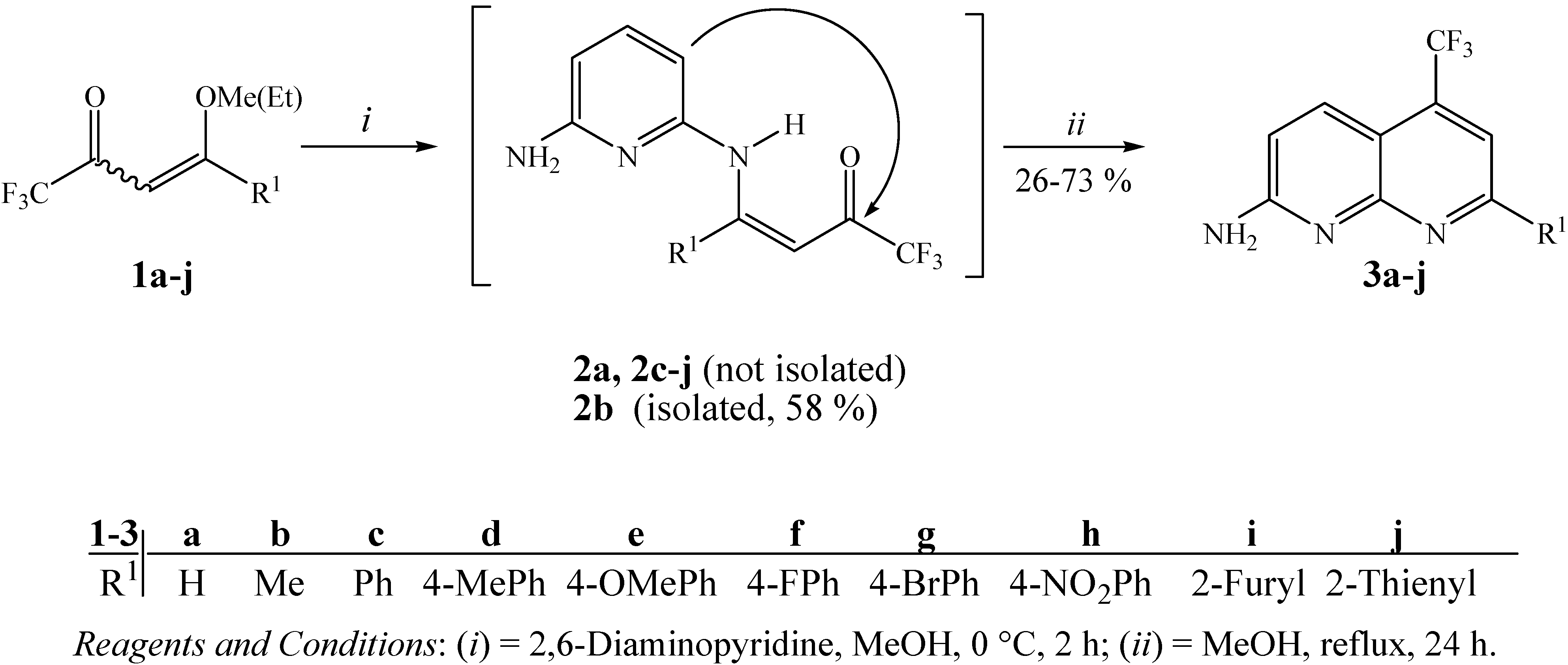
The structures of 3a-j were established on the basis of 1H- and 13C-NMR spectroscopy and literature data for similar compounds (chemical shifts and spin ± spin coupling constants) [5,9,12,14,22,41,46]. According to the literature, it is well known that the proton in the 4-position of the naphthyridine nucleus shows long-range coupling with fluorine atoms of the 5-trifluoromethyl substituent, but in some cases the outer signals of the quartets can appear as shoulders on the inner signals instead of as clearly resolved quartets [22]. This splitting of the H-4 signal is seen in all of the compounds having this structural feature and was clearly seen in 1H-NMR spectral data of compounds 3a-j.
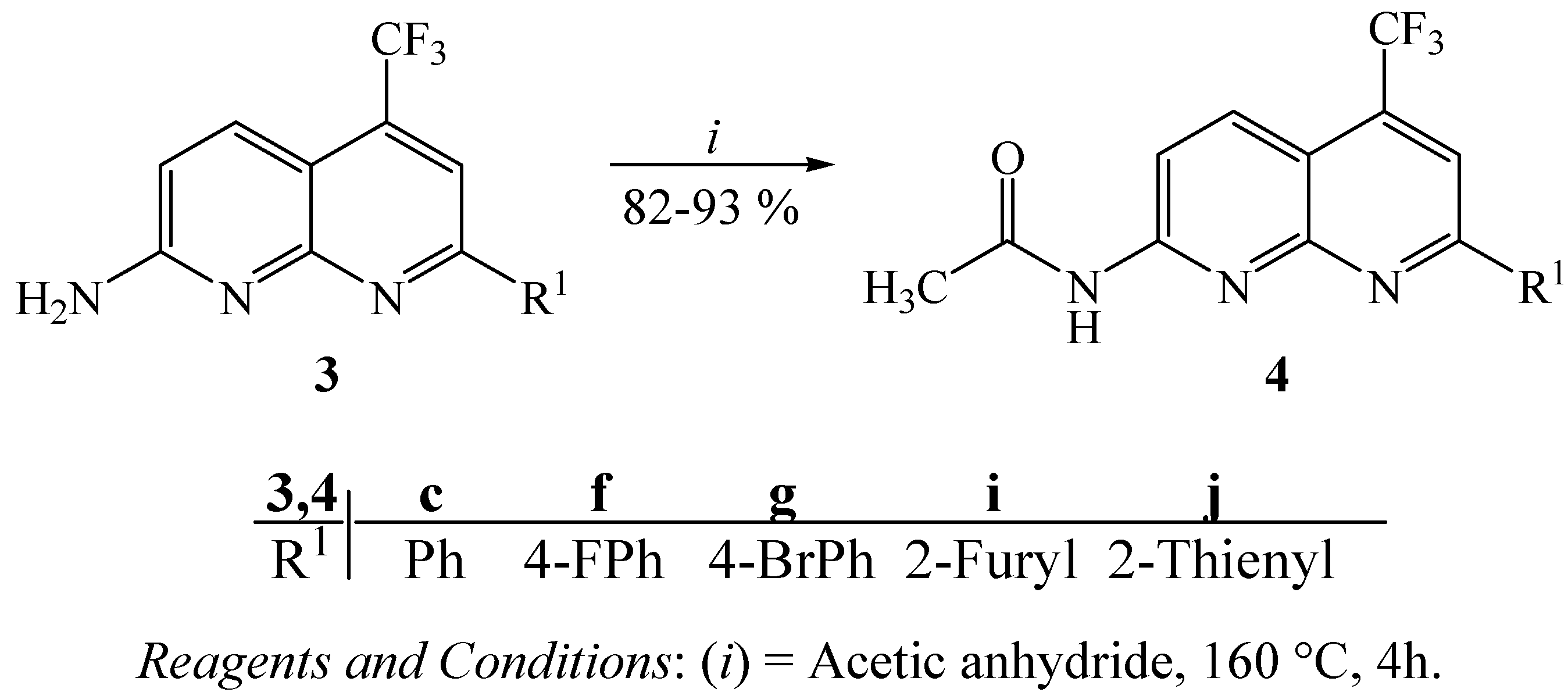
Furthermore, some examples of compounds 3 were converted in good yields (82–93%) to the corresponding 2-acetamide derivatives 4, by reaction with acetic anhydride under high temperature [14] (Scheme 2).
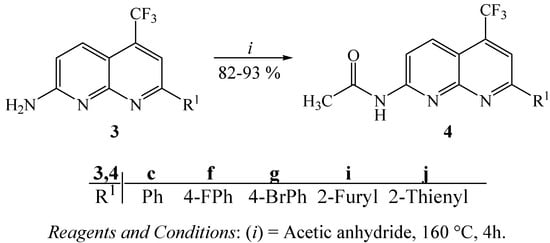
Scheme 2.
Synthesis of 2-acetamide derivatives.
Scheme 2.
Synthesis of 2-acetamide derivatives.
As a second extension, applying a similar cyclization method to that previously described by us for the preparation of some trifluoromethyl-substituted benzo[h]quinolines [35], dihydrobenzo[c]acridines [36], cycloalka[b]quinolines [37], 1,2,3,4-tetrahydroacridines [38], 7-aminoquinolines and 1,7-phenanthrolines [41], to investigate the chemical behavior of enamino ketone intermediate 2b in polyphosphoric acid medium (PPA), we found that a 1:1 mixture of isomers 3b:5b is obtained when 2b is heated at 90 °C for 20 h (Scheme 3). These compounds were easily identified since CH3 and CF3 groups have different chemical shifts in the 13C-NMR spectra of each of the isomers. Thus, 7-methyl-2-amino-5-trifluoromethyl-1,8-naphthyridine (3b) showed chemical shifts at 133.8 ppm (C5, q, 2JCF 31 Hz) and 24.6 ppm for the CH3 group, while the isomer 5-methyl-2-amino-7-trifluoromethyl-1,8-naphthyridine (5b) showed chemical shifts at 148.1 ppm (C7, q, 2JCF 34 Hz) and 17.5 ppm for the CH3 group.
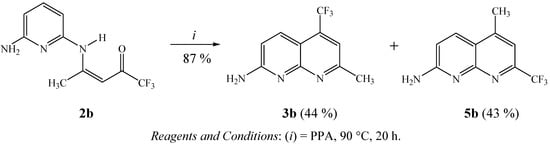
Scheme 3.
Synthesis of isomers 3b:5b.
Scheme 3.
Synthesis of isomers 3b:5b.
As an second extension of this study, we also developed the synthesis of trifluoromethyl substituted cycloalka[b][1,8]naphthyridines 3 from the reactions of 2-trifluoroacetyl-1-methoxycycloalkenes 1 with 2,6-DAP (Scheme 4). Firstly, three examples of methoxycycloalkenes 1k-m were obtained by a direct acylation reaction of the cycloalkane dimethyl acetals with trifluoracetic anhydride in the presence of pyridine, as described in the literature [47,48]. Subsequently, the intramolecular cyclization reactions of trifluoroacetylated cycloalkenes 1k-m were carried out, applying the same conditions described for the preparation of 3a-j. This reaction condition allows to isolate, in one-pot, only the respective cycloalka[b][1,8]naphthyridines 3l, 3m in 30–33% yields because the cyclization of 1k did not take place. This reaction condition allowed the isolation of enaminone2k in 43% yield, derived from 2-trifluoroacetyl-1-methoxycyclopentene. The synthesis of 3k, in 78% yield, was only possible from intramolecular cyclization reaction of enaminone 2k in polyphosphoric acid medium (PPA), as shown in Scheme 4. The structures of compounds 2k, 3k-m were easily established on the basis of 1H- and 13C-NMR spectroscopy and literature data for similar compounds, such as trifluoromethyl-containing cycloalka[b]quinolines [37].
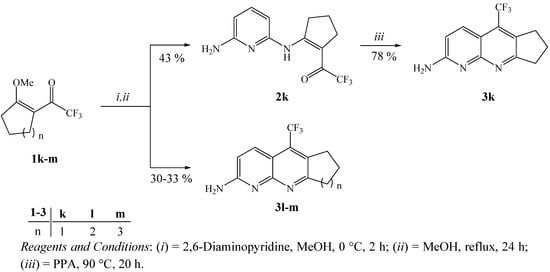
Scheme 4.
Synthesis of trifluoromethyl substituted cycloalka[b][1,8]naphthyridines.
Scheme 4.
Synthesis of trifluoromethyl substituted cycloalka[b][1,8]naphthyridines.
Any attempt to explain the low yield for the synthesis of compounds 3l and 3m and the absence of cyclization for 1k requires an examination of the structural effects on internal cycloalkenes 1k-m. Information from the literature for precursors of these compounds [47], obtained by calculation using AM1 semi-empirical method, indicate the possibility of non-planar conformations for these molecules, which would explain the difficulty in achieving [3+3] intramolecular cyclization in the case of 1k [37,48].
3. Experimental
3.1. General
Unless otherwise indicated all common reagents and solvents were used as obtained from commercial suppliers without further purification. The melting points were determined using a Kofler Reichert-Thermovar and Electrothermal Mel-Temp 3.0 apparatus. 1H- and 13C-NMR spectra were acquired on a Bruker DPX 200 spectrometer (1H at 200.13 MHz) and Bruker DPX 400 (1H at 400.13 MHz, 13C at 100.32 MHz) spectrometer, 5 mm sample tubes, 298 K, digital resolution ±0.01 ppm, in DMSO-d6, in CDCl3 for 3a, 3m, 4i, 4j and CDCl3 + TFA for 4g, using TMS as internal reference. Mass spectra were registered in a HP 6890 GC connected to a HP 5973 MSD and interfaced by a Pentium PC. The GC was equipped with a split-splitless injector, autosampler, cross-linked HP-5 capillary column (30 m, 0.32 mm of internal diameter), and helium was used as the carrier gas. The CHN elemental analyses were performed on a Perkin-Elmer 2400 CHN elemental analyzer (São Paulo University, USP/Brazil).
3.2. General procedure for the synthesis of (Z)-N(5,5,5-Trifluoro-4-oxo-2-penten-2-yl)-2,6-diamino-pyridine (2b)
To a magnetically stirred solution of 2,6-diaminopyridine (0.54 g, 5 mmol) in methanol (25 mL), a solution of 1b (0.84 g, 5 mmol) in methanol (25 mL) was added dropwise at 0 °C over a period of 2 h. After the end of the reaction, the solvent was evaporated under reduced pressure. Then, the crude oily product was dissolved in hot ethanol and subsequently cooled (4–8 °C, 24 h) to give the title compound 2b (0.28 g, 58% yield) as a brown solid, m.p. 130–132 °C.1H-NMR (200 MHz, DMSO-d6): δ = 12.69 (s, 1H, NH), 7.40 (t, J= 8 Hz, 1H, H-10), 6.33 (d, J= 8 Hz, 1H, H-9), 6.34 (d, J= 8 Hz, 1H, H-11), 5.49 (s, 1H, H-3), 5.20 (s, 2H, NH2), 2.53 (s, 3H, CH3). 13C-NMR (100 MHz, DMSO-d6): δ = 175.3 (q, 2J= 31 Hz, C=O), 166.5 (C-2), 157.7 (C-8), 149.0 (C-6), 138.7 (C-10), 116.6 (q, 1J = 288 Hz, CF3), 104.5, 103.8 (C-9, C-11), 91.8 (C-3), 21.7 (CH3). GC-MS (EI, 70 eV) m/z: 245 (M+, 100), 228 (15), 176 (22), 148 (15), 69 (5%). Anal. Calcd. For C10H10F3N3O (245.08): C, 48.98; H, 4.11; N, 17.14%. Found: C, 48.82; H, 4.03; N, 17.28%.
3.3. General procedure for the synthesis of 2-(6-Aminopyridin-2-ylamino)1-trifluoroacetyl-cyclopent-1-ene (2k)
To a magnetically stirred solution of 2,6-diaminopyridine (0.22 g, 2 mmol) in methanol (20 mL), a solution of 1k (0.38 g, 2 mmol) in methanol (20 mL) was added dropwise at 0 °C over a period of 2 h. The mixture was refluxed for an additional 24 h. After the end of the reaction, the solvent was evaporated under reduced pressure. Then, the crude oily product was dissolved in hot ethanol and subsequently cooled (4–8 °C, 24 h) to give the title compound 2k (0.22 g, 43% yield) as a brown solid, m.p. 139–141 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 11.85 (s, 1H, NH), 6.03 (s, 2H, NH2); Py: 7.39 (t, J= 8 Hz, 1H, H-4), 6.32 (d, J= 8 Hz, 1H, H-5), 6.27 (d, J= 8 Hz, 1H, H-3); c-Pent: 3.28–3.24 (m, 2H, CH2), 2.67–2.64 (m, 2H, CH2), 1.96–1.88 (m, 2H, CH2). 13C-NMR (100 MHz, DMSO-d6): δ = 172.5 (q, 2J= 33 Hz, C=O), 171.3 (C-2), 101.0 (C-1), 117.2 (q, 1J= 289 Hz, CF3); Py: 158.8 (C-6), 149.6 (C-2), 139.0 (C-4), 104.1, 103.5 (C-3, C-5); c-Pent: 34.1 (CH2), 26.3 (q, 4J= 3 Hz, CH2), 22.1 (CH2). GC-MS (EI, 70 eV) m/z: 271 (50), 256 (5), 202 (25), 174 (M+, 100%). Anal. Calcd. For C12H12F3N3O (271.09): C, 53.14; H, 4.46; N, 15.49%. Found: C, 53.02; H, 4.38; N, 15.61%.
3.4. General procedure for the synthesis of 7-alkyl(aryl/heteroaryl)-2-amino-5-trifluoromethyl-1,8-naphthyridines 3a-j
To a magnetically stirred solution of 2,6-diaminopyridine (1.08 g, 10 mmol) in methanol (40 mL), a solution of 1a-j (10 mmol) in methanol (40 mL) was added dropwise at 0 °C over a period of 2 h. The mixture was refluxed for an additional 24 h. After the end of the reaction, the solvent was evaporated under reduced pressure. The crude product was dissolved in ethanol and cooled (4–8 °C, 24 h). The solids 3b-j were isolated from the cooled solution by filtration under reduced pressure. The compound 3a was purified by flash chromatography eluting with ethyl acetate/n-hexane (1:2); yields: 26–73%.
2-Amino-5-trifluoromethyl-1,8-naphthyridine (3a): Brown solid; yield 26%; m.p. 126–128 °C. 1H-NMR(200 MHz, CDCl3): δ = 8.91 (d, J= 5 Hz, 1H, H-7), 8.08 (dq, J1= 2, J2= 9 Hz, 1H, H-4), 7.41 (d, J= 5 Hz, 1H, H-6), 6.94 (d, J= 9 Hz, 1H, H-3), 6.43 (s, 2H, NH2). 13C-NMR (100 MHz, CDCl3): δ = 159.9 (C-7), 156.8 (C-8a), 152.0 (C-2), 135.1 (q, 2J= 31 Hz, C-5), 134.2 (C-4), 123.1 (q, 1J= 275 Hz, CF3), 114.8 (C-3), 114.5 (q, 3J= 5 Hz, C-6), 112.5 (C-4a). GC-MS (EI, 70 eV) m/z: 214 (M+, 100), 194 (52), 69 (10%). Anal. Calcd. For C9H6F3N3 (213.05): C, 50.71; H, 2.84; N, 19.71%. Found: C, 50.77; H, 2.92; N, 19.81%.
2-Amino-5-trifluoromethyl-7-methyl-1,8-naphthyridine (3b): Yellow solid; yield 39%; m.p. 200–202 °C (lit. [22] 195–197 °C). 1H-NMR(200 MHz, DMSO-d6): δ = 8.01 (dq, J1 = 2, J2= 9 Hz, 1H, H-4), 7.43 (s, 1H, H-6), 7.16 (s, 2H, NH2), 6.97 (d, J = 9 Hz, 1H, H-3), 2.64 (s, 3H, CH3). 13C-NMR (100 MHz, DMSO-d6): δ = 160.8 (C-7), 160.7 (C-8a), 157.0 (C-2), 132.9 (q, 2J = 31 Hz, C-5), 132.4 (C-4), 123.5 (q, 1J = 275 Hz, CF3), 114.1 (C-3), 113.6 (q, 3J = 5 Hz, C-6), 108.8 (C-4a), 24.8 (CH3). GC-MS (EI, 70 eV) m/z 227 (M+, 100), 210 (3), 200 (37), 158 (3), 131 (5%).
2-Amino-5-trifluoromethyl-7-phenyl-1,8-naphthyridine (3c): Beige solid; yield 69%; m.p. 255–257 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 8.28 (dd, J1 = 2, J2 = 8 Hz, 2H, Ph), 8.06 (dq, J1 = 2, J2= 9 Hz, 1H, H-4), 8.03 (s, 1H, H-6), 7.58-7.53 (m, 3H, Ph), 7.18 (s, 2H, NH2), 7.05 (d, J = 9 Hz, 1H, H-3). 13C-NMR (100 MHz, DMSO-d6): δ = 161.0 (C-7), 157.5 (C-8a), 157.2 (C-2), 137.7 (C-Ph), 134.0 (q, 2J = 31 Hz, C-5), 132.3 (q, 4J = 2 Hz, C-4), 129.8, 128.5, 127.0 (5 C-Ph), 123.2 (q, 1J = 275 Hz, CF3), 114.8 (C-3), 110.2 (q, 3J = 5 Hz, C-6), 110.0 (q, 3J = 2 Hz, C-4a). GC-MS (EI, 70 eV) m/z: 290 (M+, 100), 270 (23%). Anal. Calcd. For C15H10F3N3 (289.08): C, 62.28; H, 3.48; N, 14.53%. Found: C, 62.12; H, 3.60; N, 14.30%.
2-Amino-5-trifluoromethyl-7-(4-methylphenyl)-1,8-naphthyridine (3d): Yellow solid; yield 51%; m.p. 219–221 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 8.19 (d, J = 8 Hz, 2H, Ph), 8.09 (dd, J1 = 2, J = 9 Hz, 1H, H-4), 7.98 (s, 1H, H-6), 7.35 (d, J = 8 Hz, 2H, Ph), 7.20 (s, 2H, NH2), 7.08 (d, J = 9 Hz, 1H, H-3), 2.40 (s, 3H, CH3).13C-NMR (100 MHz, DMSO-d6): δ = 160.9 (C-7), 157.4 (C-8a), 157.2 (C-2), 139.5, 134.9 (2 C-Ph), 133.8 (q, 2J = 31 Hz, C-5), 132.1 (C-4), 129.1, 126.8 (4 C-Ph), 123.2 (q, 1J = 275 Hz, CF3), 114.5 (C-3), 109.8 (q, 3J = 5 Hz, C-6), 99.5 (C-4a), 20.5 (CH3). GC-MS (EI, 70 eV) m/z: 303 (M+, 100), 276 (12), 234 (14%). Anal. Calcd. For C16H12F3N3 (303.1): C, 63.36; H, 3.99; N, 13.86%. Found: C, 63.44; H, 4.06; N, 14.01%.
2-Amino-5-trifluoromethyl-7-(4-methoxyphenyl)-1,8-naphthyridine (3e): Beige solid; yield 53%; m.p. 210–212 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 8.32 (d, J = 8 Hz, 2H, Ph), 8.10 (dq, J1 = 2, J2= 9 Hz, 1H, H-4), 8.03 (s, 1H, H-6), 7.44 (s, 2H, NH2), 7.13 (d, J = 8 Hz, 2H, Ph), 7.10 (d, J = 9 Hz, 1H, H-3), 3.89 (s, 3H, CH3). 13C-NMR (100 MHz, DMSO-d6): δ = 161.2 (C-7), 160.9 (C-8a), 157.4 (C-Ph), 157.3 (C-2), 134.0 (q, 2J = 31 Hz, C-5), 132.5 (C-4), 130.2, 128.7 (3 C-Ph), 123.4 (q, 1J = 275 Hz, CF3), 114.5 (C-3), 114.1 (2 C-Ph), 109.8 (q, 3J = 5 Hz, C-6), 109.6 (C-4a), 55.2 (OCH3). GC-MS (EI, 70 eV) m/z: 320 (M+, 100), 300 (22%). Anal. Calcd. For C16H12F3N3O (319.09): C, 60.19; H, 3.79; N, 13.16%. Found: C, 59.90; H, 3.89; N, 13.07%.
2-Amino-5-trifluoromethyl-7-(4-fluorophenyl)-1,8-naphthyridine (3f): Yellow solid; yield 73%; m.p. 260–262 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 8.36–8.32 (m, 2H, FPh), 8.06 (dq, J1 = 2, J2= 9 Hz, 1H, H-4), 8.01 (s, 1H, H-6), 7.38–7.33 (m, 2H, FPh), 7.14 (s, 2H, NH2), 7.02 (d, J = 9 Hz, 1H, H-3). 13C- NMR (100 MHz, DMSO-d6): δ = 163.4 (d, 1J = 247 Hz, C-FPh), 161.2 (C-7), 157.3 (C-8a), 156.5 (C-2), 134.2 (d, 4J = 3 Hz, C-FPh), 134.1 (q, 2J = 31 Hz, C-5), 132.4 (C-4), 129.5 (d, 3J = 9 Hz, 2 C-FPh), 123.3 (q, 1J = 275 Hz, CF3), 115.6 (d, 2J = 21 Hz, 2 C-FPh), 115.1 (C-3), 110.2 (q, 3J = 5 Hz, C-6), 110.0 (q, 3J = 2 Hz, C-4a). GC-MS (EI, 70 eV) m/z: 307 (M+, 100), 238 (25), 164 (2), 143 (4%). Anal. Calcd. For C15H9F4N3 (307.07): C, 58.64; H, 2.95; N, 13.68%. Found: C, 58.48; H, 3.11; N, 13.24%.
2-Amino-7-(4-bromophenyl)-5-trifluoromethyl-1,8-naphthyridine (3g): Yellow solid; yield 73%; m.p. 285–287 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 8.23 (dd, J1 = 2, J2 = 8 Hz, 2H, Ph), 8.08 (dq, J1 = 2, J2= 9 Hz, 1H, H-4), 8.02 (s, 1H, H-6), 7.72 (dd, J1 = 2, J2 = 8 Hz, 2H, Ph), 7.18 (s, 2H, NH2), 7.07 (d, J = 9 Hz, 1H, H-3). 13C-NMR (100 MHz, DMSO-d6):δ = 161.3 (C-7), 157.3 (C-8a), 156.4 (C-2), 136.9 (C-Ph), 134.2 (q, 2J = 31 Hz, C-5), 132.5 (C-4), 131.7, 129.2, 123.9 (5 C-Ph), 123.4 (q, 1J = 275 Hz, CF3), 115.3 (C-3), 110.4 (C-4a), 110.2 (q, 3J = 5 Hz, C-6). GC-MS (EI, 70 eV) m/z: 367 (M+, 100), 298 (12), 288 (50), 144 (17%). Anal. Calcd. For C15H9BrF3N3 (366.99): C, 48.94; H, 2.46; N, 11.41%. Found: C, 49.04; H, 2.54; N, 11.37%.
2-Amino-5-trifluoromethyl-7-(4-nitrophenyl)-1,8-naphthyridine (3h): Yellow solid; yield 63%; m.p. > 340 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 8.53 (d, J = 8 Hz, 2H, Ph), 8.35 (d, J = 8 Hz, 2H, Ph), 8.06 (dd, J1 = 2, J = 9 Hz, 1H, H-4), 8.02 (s, 1H, H-6), 7.20 (s, 2H, NH2), 7.06 (d, J = 9 Hz, 1H, H-3). 13C-NMR (100 MHz, DMSO-d6): δ = 161.1 (C-7), 157.1 (C-8a), 155.0 (C-2), 148.0, 143.4 (2 C-Ph), 134.3 (q, 2J = 31 Hz, C-5), 132.1 (C-4), 128.2, 123.5 (4 C-Ph), 123.0 (q, 1J = 275 Hz, CF3), 115.7 (C-3), 110.9 (C-4a), 109.8 (q, 3J = 2 Hz, C-6). GC-MS (EI, 70 eV) m/z: 334 (M+, 100), 288 (97), 144 (17%). Anal. Calcd. For C15H9F3N4O2 (334.07): C, 53.90; H, 2.71; N, 16.76%. Found: C, 53.85; H, 2.78; N, 16.52%.
2-Amino-5-trifluoromethyl-7-(2-furyl)-1,8-naphthyridine (3i): Yellow solid, yield 38%; m.p. 230–232 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 7.98 (d, J = 4 Hz, 1H, furyl), 7.89 (dq, J1 = 2, J2 = 9 Hz, 1H, H-4), 7.80 (s, 1H, H-6), 7.40 (d, J = 5 Hz, 1H, furyl), 7.17 (s, 2H, NH2), 7.04 (d, J = 9 Hz, 1H, H-3), 6.75 (t, J = 4 Hz, 1H, furyl). 13C-NMR (100 MHz, DMSO-d6): δ = 160.9 (C-7), 157.0 (C-8a), 152.3 (C-2), 149.5, 145.1, (2 C-furyl), 133.9 (q, 2J = 31 Hz, C-5), 132.3 (q, 4J = 2 Hz, C-4), 123.0 (q, 1J = 275 Hz, CF3), 114.5 (C-3), 112.4, 111.1 (2 C-furyl), 109.8 (q, 3J = 2 Hz, C-4a), 108.6 (q, 3J = 5 Hz, C-6). GC-MS (EI, 70 eV) m/z: 279 (M+, 100), 251 (25), 223 (12%). Anal. Calcd. For C13H8F3N3O (279.06): C, 55.92; H, 2.89; N, 15.05%. Found: C, 55.48; H, 2.88; N, 14.72%.
2-Amino-5-trifluoromethyl-7-(2-thienyl)-1,8-naphthyridine (3j): Yellow solid; yield 60%; m.p. 260–262 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 8.12 (d, J = 4 Hz, 1H, thienyl), 8.07 (dq, J1 = 2, J2 = 9 Hz, 1H, H-4), 8.04 (s, 1H, H-6), 7.78 (d, J = 5 Hz, 1H, thienyl), 7.26 (t, J = 4 Hz, 1H, thienyl), 7.23 (s, 2H, NH2), 7.07 (d, J = 9 Hz, 1H, H-3). 13C-NMR (100 MHz, DMSO-d6): δ = 161.1 (C-7), 157.0 (C-8a), 153.2 (C-2), 143.9 (C-thienyl), 133.9 (q, 2J = 31 Hz, C-5), 132.3 (q, 4J = 2 Hz, C-4), 130.0, 128.3, 127.5 (3 C-thienyl), 123.1 (q, 1J = 275 Hz, CF3), 114.3 (C-3), 109.9 (q, 3J = 2 Hz, C-4a), 109.3 (q, 3J = 5 Hz, C-6). GC-MS (EI, 70 eV) m/z: 295 (M+, 100), 268 (25), 226 (4%). Anal. Calcd. For C13H8F3N3S (295.04): C, 52.88; H, 2.73; N, 14.23%. Found: C, 52.94; H, 2.85; N, 14.10%.
3.5. General procedure for the synthesis of 2-amino-5-trifluoromethyl-7,8-dihydro-6H-cyclopenta[b][1,8]-naphthyridine (3k)
To a stirred mixture of H3PO4 (0.8 mL) and P2O5 (1.2 g) (PPA) at 90 °C, 2k (0.27 g, 1 mmol) was added. The reaction mixture was stirred for an additional 20 h. After cooling, the reaction mixture was treated with crushed ice and with concentrated NH4OH until the pH was 8. The compound 3k wasisolated of the solution by filtration at reduced pressure as a brown solid, in 78% yield, m.p. 172–174 ºC. 1H-NMR (200 MHz, DMSO-d6): δ = 8.00 (dq, J1 = 2, J2 = 9 Hz, 1H, H-4), 6.91 (d, J = 9 Hz, 1H, H-3), 6.80 (s, 2H, NH2), 3.18–3.14 (m, 2H, CH2), 3.02 (t, J = 9 Hz, 2H, CH2), 2.15–2.07 (m, 2H, CH2). 13C-NMR (100 MHz, DMSO-d6): δ = 170.0 (C-9a), 159.8 (C-2), 156.5 (C-10a), 132.5 (q, 4J = 3 Hz, C-4), 128.0 (q, 3J = 3 Hz, C-5a), 127.6 (q, 2J = 30 Hz, C-5), 124.2 (q, 1J = 276 Hz, CF3), 113.0 (C-3), 109.2 (q, 3J = 2 Hz, C-4a), 33.7, 29.7 (q, 4J = 2 Hz), 21.9 (3 CH2). GC-MS (EI, 70 eV) m/z: 253 (M+, 100), 237 (5), 184 (20%). Anal. Calcd. For C12H10F3N3 (253.08): C, 56.92; H, 3.98; N, 16.59%. Found: C, 56.80; H, 3.84; N, 16.73%.
3.6. General procedure for the synthesis of 2-amino-5-trifluoromethyl-cycloalka[b][1,8]-naphthyridines 3l, 3m
To a magnetically stirred solution of 2,6-diaminopyridine (0.22 g, 2 mmol) in methanol (20 mL), a solution of 1k-l (2 mmol) in methanol (20 mL) was added drop wise at 0 °C over a period of 2 h. The mixture was refluxed for an additional 24 h. After the end of the reaction, the solvent was evaporated under reduced pressure. The compounds 3k-l were purified by flash chromatography eluting with ethyl acetate/n-hexane (1:2); yields: 30–33%.
2-Amino-5-trifluoromethyl-6,7,8,9-tetrahydrobenzo[b][1,8]naphthyridine (3l): Brown solid, yield 33%; m.p. 128–130 °C.1H-NMR (200 MHz, DMSO-d6): δ = 8.07 (dq, J1 = 2, J2 = 9 Hz, 1H, H-4), 6.93 (d, J = 9 Hz, 1H, H-3), 6.85 (s, 2H, NH2), 2.99–2.96 (m, 4H, CH2), 1.90–1.78 (m, 4H, CH2). 13C-NMR (100 MHz, DMSO-d6): δ = 160.9 (C-2), 159.8 (C-10a), 154.6 (C-9a), 132.7 (q, 4J = 3 Hz, C-4), 130.3 (q, 2J = 28 Hz, C-5), 124.7 (q, 1J = 278 Hz, CF3), 124.5 (C-5a), 114.0 (C-3), 110.5 (q, 3J = 2 Hz, C-4a), 33.8, 25.6, 22.0, 21.2 (4 CH2). GC-MS (EI, 70 eV) m/z: 268 (M+, 100), 248 (20), 198 (9%). Anal. Calcd. For C13H12F3N3 (267.1): C, 58.42; H, 4.53; N, 15.72%. Found: C, 58.59; H, 4.65; N, 15.61%.
2-Amino-5-trifluoromethyl-7,8,9,10-tetrahydro-6H-cyclohepta[b][1,8]naphthyridine (3m): Brown solid; yield 30%; m.p. 93–95 °C. 1H-NMR (200 MHz, CDCl3): δ = 8.20 (d, J = 9 Hz, 1H, H-4), 7.75 (d, J = 9 Hz, 1H, H-3), 7.19 (s, 2H, NH2), 3.21–3.00 (m, 4H, CH2), 1.79–1.68 (m, 6H, CH2). 13C-NMR (100 MHz, CDCl3): δ = 158.8 (C-2), 156.5 (C-11a), 154.5 (C-10a), 140.7 (C-5a), 134.6 (C-4), 131.2 (q, 2J = 30 Hz, C-5), 123.9 (q, 1J = 278 Hz, CF3), 113.5 (C-3), 97.3 (C-4a), 39.0, 38.9, 29.3, 27.5, 26.2 (5 CH2). GC-MS (EI, 70 eV) m/z: 281 (M+, 100), 265 (15), 252 (30), 212 (4%). Anal. Calcd. For C14H14F3N3 (281.1): C, 59.78; H, 5.02; N, 14.94%. Found: C, 59.69; H, 4.88; N, 15.01%.
3.7. General procedure for the synthesis of 2-acetylamino-7-(aryl/heteroaryl)-5-trifluoromethyl-1,8-naphthyridines 4
A suspension of 2 mmol of amino derivatives 3c, 3f,3g, 3i or 3j in 5 mL of acetic anhydride was refluxed at 160 °C for 2 h. After cooling, the solids were collected and washed with water to give their acetamide derivatives 4, in 82–93% yields.
2-Acetylamino-5-trifluoromethyl-7-phenyl-1,8-naphthyridine (4c): Beige solid; yield 74%; m.p. 224–226 °C. 1H-NMR (200 MHz, DMSO-d6): δ = 11.34 (s, 1H, NH), 8.54-850 (m, 2H, H-3, H-4), 8.40 (s, 1H, H-6), 8.37-835 (m, 2H, Ph), 760-7.58 (m, 3H, Ph), 2.22 (CH3). 13C-NMR (100 MHz, DMSO-d6): δ = 171.6 (C=0), 170.0 (C-8a), 158.7 (C-7), 155.5 (C-2), 136.9 (C-Ph), 135.4 (q, 2J = 33 Hz, C-5), 134.4 (C-4), 130.4, 128.7, 127.3 (5 C-Ph), 122.9 (q, 1J = 275 Hz, CF3), 115.9 (C-3), 114.4 (q, 3J = 5 Hz, C-6), 113.1 (C-4a), 23.9 (CH3). GC-MS (EI, 70 eV) m/z: 331 (40), 288 (M+, 100), 262 (15), 219 (9%). Anal. Calcd. For C17H12F3N3O (331.09) C, 61.63; H, 3.65; N, 12.68%. Found: C, 61.49; H, 3.50; N, 12.78%.
2-Acetylamino-5-trifluoromethyl-7-(4-fluorophenyl)-1,8-naphthyridine (4f): Beige solid; yield 83%; m.p. 263–265 °C. 1H-NMR (200 MHz, CDCl3): δ = 11.21 (s, 1H, NH), 8.50 (d, J = 9 Hz, 1H, H-3), 8.45 (d, J = 9 Hz, 1H, H-4), 8.40-8.37 (m, 2H, FPh), 8.33 (s, 1H, H-6), 7.39-7.35 (m, 2H, FPh), 2.23 (s, 3H, CH3). 13C-NMR (100 MHz, DMSO-d6): δ = 170.0 (C=0), 163.6 (d, 1J = 248 Hz, C-FPh), 157.6 (C-8a), 155.0 (C-7), 154.9 (C-2), 135.5 (q, 2J = 33 Hz, C-5), 134.3 (C-4), 133.4 (d, 4J = 3 Hz, C-FPh), 129.6 (d, 3J = 9 Hz, 2 C-FPh), 122.7 (q, 1J = 275 Hz, CF3), 115.8 (C-6), 115.6 (d, 2J = 21 Hz, 2 C-FPh), 114.2 (C-3), 113.0 (C-4a), 23.8 (CH3). GC-MS (EI, 70 eV) m/z: 349 (45), 307 (M+, 100), 280 (25), 238 (15%). Anal. Calcd. For C17H11F4N3O (349.08): C, 58.46; H, 3.17; N, 12.03%. Found: C, 58.28; H, 3.02; N, 12.15%.
2-Acetylamino-7-(4-bromophenyl)-5-trifluoromethyl-1,8-naphthyridine (4g): Beige solid; yield 93%; m.p. 222–224°C. 1H-NMR (200 MHz, CDCl3 + TFA): δ = 11.34 (s, 1H, NH), 8.90 (d, J = 9 Hz, 1H, H-3), 8.45 (s, 1H, H-6), 8.18 (d, J = 9 Hz, 2H, Ph), 7.80 (d, J = 9 Hz, 2H, Ph), 7.74 (d, J = 9 Hz, 1H, H-4), 2.54 (s, 3H, CH3). 13C-NMR (100 MHz, CDCl3 + TFA): δ = 176.0 (C=0), 169.8 (C-8a), 152.1 (C-7), 145.4 (C-2), 143.0 (C-Ph), 137.7 (q, 2J = 33 Hz, C-5), 133.5 (C-4), 133.0, 129.7, 128.8, (5 C-Ph), 121.6 (q, 1J = 275 Hz, CF3), 118.2 (q, 3J = 5 Hz, C-6), 114.1 (C-3), 113.3 (C-4a), 24.2 (CH3). GC-MS (EI, 70 eV) m/z: 409 (42), 367 (M+, 100), 339 (5), 296 (6), 288 (42%). Anal. Calcd. For C17H11BrF3N3O (409.1): C, 49.78; H, 2.70; N, 10.24%. Found: C, 49.61; H, 2.58; N, 10.33%.
2-Acetylamino-5-trifluoromethyl-7-(2-furyl)-1,8-naphthyridine (4i): Yellow solid; yield 82%; m.p. 221–223 °C. 1H-NMR (200 MHz, CDCl3): δ = 9.74 (s, 1H, NH), 8.60 (d, J = 9 Hz, 1H, H-3), 8.41 (d, J = 9 Hz, 1H, H-4), 8.10 (s, 1H, H-6), 7.65 (s, 1H, furyl), 7.45 (d, J = 3 Hz, 1H, furyl), 6.62 (t, J = 2 Hz, 1H, furyl), 2.29 (s, 3H, CH3). 13C-NMR (100 MHz, CDCl3): δ = 176.2 (C=0), 169.8 (C-8a), 152.5 (C-7), 151.8 (C-2), 145.3 (2 C-furyl), 136.0 (q, 2J = 32 Hz, C-5), 135.9 (C-4), 122.8 (q, 1J = 275 Hz, CF3), 115.8 (C-3), 114.3 (C-4a), 113.8 (q, 3J = 5 Hz, C-6), 112.9, 112.7 (2 C-furyl), 24.5 (CH3). GC-MS (EI, 70 eV) m/z: 321 (50), 306 (5), 279 (M+, 100), 251 (10%). Anal. Calcd. For C15H10F3N3O2 (321.07) C, 56.08; H, 3.14; N, 13.08%. Found: C, 56.00; H, 3.03; N, 13.18%.
2-Acetylamino-5-trifluoromethyl-7-(2-thienyl)-1,8-naphthyridine (4j): Beige solid; yield 90%; m.p. 225–227 °C. 1H-NMR (200 MHz, CDCl3): δ = 9.76 (s, 1H, NH), 8.59 (d, J = 9 Hz, 1H, H-3), 8.40 (d, J = 9 Hz, 1H, H-4), 7.98 (s, 1H, H-6), 7.84 (s, 1H, thienyl), 7.54 (d, J = 4 Hz, 1H, thienyl), 7.16 (d, J = 3 Hz, 1H, thienyl), 2.29 (s, 3H, CH3). 13C-NMR (100 MHz, CDCl3): δ = 176.1 (C=0), 169.8 (C-8a), 155.3 (C-7), 154.9 (C-2), 143.4 (C-thienyl), 135.9 (q, 2J = 33 Hz, C-5), 135.7 (C-4), 130.9, 128.4, 127.9 (3 C-thienyl), 122.9 (q, 1J = 275 Hz, CF3), 115.8 (C-3), 114.3 (C-4a), 114.1 (q, 3J = 5 Hz, C-6), 24.7 (CH3). GC-MS (EI, 70 eV) m/z: 337 (43), 322 (5), 295 (M+, 100), 268 (25%). Anal. Calcd. For C15H10F3N3OS (337.05): C, 53.41; H, 2.99%; N, 12.46. Found: C, 53.33; H, 2.81; N, 12.54%.
3.8. General procedure for the synthesis of 2-amino-5-trifluoromethyl-7-methyl-1,8-naphthyridine (3b) and 2-amino-7-trifluoromethyl-5-methyl-1,8-naphthyridine (5b)
These compounds were obtained as a yellow solid, a 1:1 mixture of 3b:5b in 87% yield, m.p. 179–181 °C. To a stirred mixture of H3PO4 (0.8 mL) and P2O5 (1.2 g) (PPA) at 90 °C, 2b (0.24 g, 1 mmol) was added. The reaction mixture was stirred for an additional 20 h. After cooling, the reaction mixture was treated with crushed ice and with concentrated NH4OH until the pH was 8. The solution was then extracted with ethyl acetate (3 × 20 mL), the combined extracts were dried (MgSO4) and evaporated to dryness under a vacuum to obtain the solid mixture of isomers 3b:5b.
2-Amino-5-trifluoromethyl-7-methyl-1,8-naphthyridine (3b):Yield 44%. 1H-NMR (200 MHz, CDCl3): δ = 8.08 (d, J = 9 Hz, 1H, H-4), 7.36 (s, 1H, H-6), 6.98 (d, J = 9 Hz, 1H, H-3), 6.19 (s, 2H, NH2), 2.74 (s, 3H, CH3). 13C-NMR (100 MHz, DMSO-d6): δ = 160.5 (C-7), 159.8 (C-8a), 156.5 (C-2), 133.8 (q, 2J = 31 Hz, C-5), 133.0 (C-4), 122.6 (q, 1J = 275 Hz, CF3), 114.2 (C-3), 113.8 (q, 3J = 5 Hz, C-6), 112.9 (C-4a), 24.6 (CH3). GC-MS (EI, 70 eV) m/z 227 (M+, 100), 210 (3), 200 (37), 158 (3), 131 (5%).
2-amino-7-trifluoromethyl-5-methyl-1,8-naphthyridine (5b): Yield 43%.1H-NMR (200 MHz, CDCl3): δ = 8.08 (d, J = 9 Hz, 1H, H-4), 7.33 (s, 1H, H-6), 6.90 (d, J = 9 Hz, 1H, H-3), 5.98 (s, 2H, NH2), 2.67 (s, 3H, CH3). 13C-NMR (100 MHz, DMSO-d6): δ = 160.3 (C-7), 159.8 (C-8a), 155.2 (C-2), 148.1 (q, 2J = 34 Hz, C-7), 146.9 (C-5), 132.7 (C-4), 120.8 (q, 1J = 275 Hz, CF3), 114.2 (C-3), 113.8 (q, 3J = 5 Hz, C-6), 112.9 (C-4a), 17.5 (CH3). GC-MS (EI, 70 eV) m/z: 227 (M+, 100), 210 (3), 200 (37), 158 (3), 131 (5%).
4. Conclusions
In summary, we have developed a new, simple and convenient route for the preparation of a new series of 7-alkyl(aryl/heteroaryl)-2-amino-5-trifluoromethyl-1,8-naphthyridines 3a-jin moderate to good yields, derived from direct cyclocondensation reactions employing acyclic β-alkoxyvinyl trifluoromethyl ketones 1a-j and 2,6-DAP, under mild conditions by a conventional one-pot procedure. We also developed the synthesis of new 2-amino-5-trifluoromethyl-cycloalka[b][1,8]naphthyridines 3l-m as fused heteropolycycles from direct or indirect cyclocondensation reactions of 2-trifluoroacetyl-1-methoxycycloalkenes 1l-m and 2,6-DAP. Furthermore, we have been able to use acyclic and cyclic ketones 1, for the first time, in the synthesis of trifluoromethylated 2-amino-1,8-naphthyridines, which possess a free amino group for further important derivatizations.
Acknowledgements
The authors thank the Conselho Nacional de Desenvolvimento Científico, CNPq, for the financial support (Proc. Nr. 303.296/2008-9). Fellowships from CAPES and CNPq are also acknowledged.
References and Notes
- Goswami, S.; Mukherjee, R.; Mukherjee, R.; Jana, S.; Maity, A.C.; Adak, A.K. Simple and efficient synthesis of 2,7-difunctionalized-1,8-naphthyridines. Molecules 2005, 10, 929–936. [Google Scholar] [CrossRef]
- Mekheimer, R.A.; Hameed, A.M.A.; Sadek, K.U. 1,8-Naphthyridines II: Synthesis of novel polyfunctionally substituted 1,8-naphthyridinones and their degradation to 6-aminopyridones. ARKIVOC 2007, xiii, 269–281. [Google Scholar]
- He, C.; Lippard, S.J. Design and synthesis of multidentate dinucleating ligands based on 1,8-naphthyridine. Tetrahedron 2000, 56, 8245–8252. [Google Scholar] [CrossRef]
- Bouzard, D.; DiCesare, P.; Essiz, M.; Jacquet, J.P.; Ledoussal, B.; Remuzon, P.; Kessler, R.E.; Fung-Tomc, J. Fluoronaphthyridines as antibacterial agents. 4. Synthesis and structure-activity relationships of 5-substituted-6-fluoro-7-(cycloalky1amino)-l,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acids. J. Med. Chem. 1992, 35, 518–525. [Google Scholar]
- Ferrarini, P.L.; Manera, C.; Mori, C.; Badawneh, M.; Saccomanni, G. Synthesis and evaluation of antimycobacterial activity of 4-phenyl-1,8-naphthyridine derivatives. Farmaco 1998, 53, 741–746. [Google Scholar] [CrossRef]
- Tsuzuki, Y.; Tomita, K.; Sato, Y.; Kashimoto, S.; Chiba, K. Synthesis and structure–activity relationships of 3-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridines as novel antitumor agents. Bioorg. Med. Chem. Lett. 2004, 14, 3189–3193. [Google Scholar]
- Dianzani, C.; Collino, M.; Gallicchio, M.; Di Braccio, M.; Roma, G.; Fantozzi, R. Effects of anti-inflammatory [1,2,4]triazolo[4,3-a][1,8]naphthyridine derivatives on human stimulated PMN and endothelial cells: An in vitro study. J. Inflamm. 2006, 3, 4. [Google Scholar] [CrossRef]
- Roma, G.; Di Braccio, M.; Grossi, G.; Piras, D.; Ballabeni, V.; Tognolini, M.; Bertoni, S.; Barocelli, E. 1,8-Naphthyridines VIII. Novel 5-aminoimidazo[1,2-a][1,8]naphthyridine-6-carboxamide and 5-amino[1,2,4]triazolo[4,3-a][1,8] naphthyridine-6-carboxamide derivatives showing potent analgesic or anti-inflammatory activity, respectively, and completely devoid of acute gastrolesivity. Eur. J. Med. Chem. 2010, 45, 352–366. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Badawneh, M.; Franconi, F.; Manera, C.; Miceli, M.; Mori, C.; Saccomanni, G. Synthesis and antiplatelet activity of some 2,7-di(N-cycloamino)-3-phenyl-1,8-naphthyridine derivatives. Farmaco 2001, 56, 311–318. [Google Scholar] [CrossRef]
- Santilli, A.A.; Scotese, A.C.; Bauer, R.F.; Bell, S.C. 2-Oxo-1,8-naphthyridine-3-carboxylic acid derivatives with potent gastric antisecretory properties. J. Med. Chem. 1987, 30, 2270–2277. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Tellini, N. Synthesis and local anesthetic activity of (E)- and (Z)-diethylaminoethyliminothers of 1,8-naphtyridine. Farmaco 1990, 45, 385–389. [Google Scholar]
- Leonard, J.T.; Gangadhar, R.; Gnanasam, S.K.; Ramachandran, S.; Saravanan, M.; Sridhar, S.K. Synthesis and pharmacological activities of 1,8-naphthyridine derivatives. Biol. Pharm. Bull. 2002, 25, 798–802. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Calderone, V.; Calzolari, L.; Nieri, P.; Martinotti, E.; Saccomanni, G. Synthesis of 1,8-naphthyridine derivatives: Potential antihypertensive agents – Part VIII. Eur. J. Med. Chem. 1999, 34, 505–513. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Badawneh, M.; Calderone, V.; Calzolari, L.; Loffredo, T.; Martinotti, E.; Saccomanni, G. Synthesis of l,8-naphthyridine derivatives: Potential antihypertensive agents – Part VII. Eur. J. Med. Chem. 1998, 33, 383–397. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Badawneh, M.; Calderone, V.; Greco, R.; Manera, C.; Martinelli, A.; Nieri, P.; Saccomanni, G. Synthesis and β-blocking activity of (R,S)-(E)-oximeethers of 2,3-dihydro-1,8-naphthyridine and 2,3-dihydrothiopyrano[2,3-b]pyridine: Potential antihypertensive agents – Part IX. Eur. J. Med. Chem. 2000, 35, 815. [Google Scholar] [CrossRef]
- Graf, H.; Franz, L.; Sauter, H.; Ammermann, E.; Pommer, E-H. Substituted 1,8-naphthyridine derivatives and fungicides containing them. U.S. Patent 4,801,592, 31 January 1989. [Google Scholar]
- Saupe, T.; Schaefer, P.; Meyer, N.; Wuerzer, B.; Westphalen, K.O. Substituted 1,8-naphthyridines, their preparation and their use as antidotes. U.S. Patent 5,258,356, 2 November 1993. [Google Scholar]
- Cotrel, C.; Guyon, C.; Roussel, G.; Taurand, G. Anxiolytic amides derived from certain 1,8-naphthyridine-2-amines. U.S. Patent 4,753,933, 28 June 1988. [Google Scholar]
- Litvinov, V.P.; Roman, S.V.; Dyachenko, V.D. Naphthyridines. Structure, physicochemical properties and general methods of synthesis. Russ. Chem. Rev. 2000, 69, 201–220. [Google Scholar] [CrossRef]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluorine Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Druzhinin, S.V.; Balenkova, E.S.; Nenajdenko, V.G. Recent advances in the chemistry of α,β-unsatured trifluoromethylketones. Tetrahedron 2007, 63, 7753–7808. [Google Scholar]
- Eichler, E.; Rooney, C.S.; Williams, H.W.R. 1,8-Naphthyridines. Part I. Synthesis of some trifluoromethyl-1,8-naphthyridines derivatives. J. Heterocycl. Chem. 1976, 13, 41–42. [Google Scholar] [CrossRef]
- Naik, T.R.R.; Naik, H.S.B.; Raghavendra, M.; Naik, S.G.K. Synthesis of thieno[2,3-b]benzo[1,8]naphthyridine-2-carboxylic acids under microwave irradiation and interaction with DNA studies. ARKIVOC 2006, xv, 84–94. [Google Scholar]
- Chen, S.; Chen, R.; He, M.; Pang, R.; Tan, Z.; Yang, M. Design, synthesis, and biological evaluation of novel quinoline derivatives as HIV–1 Tat–TAR interaction inhibitors. Bioorg. Med. Chem. 2009, 17, 1948–1956. [Google Scholar] [CrossRef]
- Pizzio, L.; Romanelli, G.; Vázquez, P.; Autino, J.; Blanco, M.; Cáceres, C. Keggin heteropolyacid-based catalysts for the preparation of substituted ethyl β-arylaminocrotonates, intermediates in the synthesis of 4-quinolones. Appl. Catal. A 2006, 308, 1531–1560. [Google Scholar]
- Hauser, C.R.; Reynolds, G.A. Reactions of β-keto esters with aromatic amines. Syntheses of 2- and 4-hydroxyquinoline derivatives. J. Am. Chem. Soc. 1948, 70, 2402–2404. [Google Scholar]
- Manske, R.H.F.; Kulka, M. The Skraup synthesis of quinolines. Org. React. 1953, 7, 59. [Google Scholar]
- Bonacorso, H.G.; Righi, F.J.; Rodrigues, C.A.; Cechinel, C.A.; Costa, M.B.; Wastowski, A.D.; Martins, M.A.P.; Zanatta, N. New efficient approach for the synthesis of 2-alkyl(aryl)substituted 4H-pyrido[1,2-a]pyrimidin-4-ones. J. Heterocycl. Chem. 2006, 43, 229–233. [Google Scholar] [CrossRef]
- Brown, E.V. 1,8-Naphthyridines. I. Derivatives of 2- and 4-methyl-1,8-naphthyridines. J. Org. Chem. 1965, 30, 1607–1610. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Lourega, R.V.; Wastowski, A.D.; Flores, A.F.C.; Zanatta, N.; Martins, M.A.P. β-Alkoxyvinyl trichloromethyl ketones as N-heterocyclic acylating agent. A new access to 5H-thiazolo[3,2-a]pyrimidin-5-ones. Tetrahedron Lett. 2002, 43, 9315–9318. [Google Scholar]
- Bonacorso, H.G.; Lourega, R.V.; Deon, E.D.; Zanatta, N.; Martins, M.A.P. The first synthesis of dihydro-3H-pyrido[2,3-b][1,4]diazepinols and a new alternative approach for diazepinone analogues. Tetrahedron Lett. 2007, 48, 4835–4838. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Lourega, R.V.; Righi, F.J.; Deon, E.D.; Zanatta, N.; Martins, M.A.P. Preparation of new 2-amino- and 2,3-diamino-pyridine trifluoroacetyl enamine derivatives and their application to the synthesis of trifluoromethyl-containing 3H-pyrido[2,3-b][1,4]diazepinols. J. Heterocycl. Chem. 2008, 45, 1679–1686. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Lourega, R.V.; Porte, L.M.F.; Deon, E.D.; Flores, A.F.C.; Zanatta, N.; Martins, M.A.P. Regiospecific synthesis of 3H-pyrido[2,3-b][1,4]diazepin-4(5H)-ones via haloform reaction with the isolation of N3-[3-oxo-4,4,4-trichloroalk-1-en-1-yl]-2,3-diaminopyridine intermediates. J. Heterocycl. Chem. 2009, 46, 603–609. [Google Scholar] [CrossRef]
- Gerus, I.I.; Gorbunova, M.G.; Kukhar, V.P. Ethoxyvinyl polyfluoroalkyl ketones – versatile synthones in fluoroorganic chemistry. J. Fluorine Chem. 1994, 69, 195–198. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Duarte, S.H.G.; Zanatta, N.; Martins, M.A.P. Regiospecific Synthesis of 3-alkyl-2-aryl-4-trifluoromethylbenzo[h]quinolines by intramolecular cyclization of N-(2-alkyl-1-aryl-3-oxo-4,4,4-trifluorobut-1-en-1-yl)-1-naphthylamines. Synthesis 2002, 1037–1042. [Google Scholar]
- Bonacorso, H.G.; Drekener, R.L.; Rodríguez, I.R.; Vezzosi, R.P.; Costa, M.B.; Martins, M.A.P.; Zanatta, N. Synthesis of new fluorine-containing dihydrobenzo[c]acridines from trifluoroacetyl dihydronaphthalene and substituted anilines. J. Fluorine Chem. 2005, 126, 1384–1389. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Moraes, T.S.; Zanatta, N.; Martins, M.A.P.; Flores, A.F.C. Synthesis of new trifluoromethyl-containing cycloalka[b]quinolines derived from alkoxycycloalkenes. ARKIVOC 2008, xvi, 75–83. [Google Scholar]
- Bonacorso, H.G.; Moraes, T.S.; Zanatta, N.; Martins, M.A.P. Synthesis of new fluorine-containing 1,2,3,4-tetrahydroacridines. Synth. Commun. 2009, 39, 3677–3686. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Bittencourt, S.R.T.; Lourega, R.V.; Flores, A.F.C.; Zanatta, N.; Martins, M.A.P. A Convenient synthetic method for fully conjugated 3-alkyl- and 3-aryl-5-trifluoromethyl-1-methyl-1,2-thiazine 1-oxide from β-alkoxyvinyl trifluoromethyl ketones. Synthesis 2000, 1431–1434. [Google Scholar]
- Bonacorso, H.G.; Wentz, A.P.; Bittencourt, S.T.R.; Marques, L.M.L.; Zanatta, N.; Martins, M.A.P. Synthesis of some N-[1-alkyl(aryl)-3-oxo-4,4,4-trichloro(trifluoro)-1-buten-1-yl]-o-aminophenols and o-phenylenediamines as potential anticancer agents. Synth. Commum. 2002, 32, 335–341. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Andrighetto, R.; Zanatta, N.; Martins, M.A.P. The unexpected cyclization routes of N,N’-bis(oxotrifluoroalkenyl)-1,3-phenylenediamines in polyphosphoric acid medium. Tetrahedron Lett. 2010, 51, 3752–3755. [Google Scholar] [CrossRef]
- Effenberger, F.; Maier, R.; Schonwalder, K.H.; Ziegler, T. Enolether, XIII. Die acylierung von enolethern mit reaktiven carbonsäure-chloriden. Chem. Ber. 1982, 115, 2766–2782. [Google Scholar] [CrossRef]
- Kamitori, Y.; Hojo, M.; Masuda, R.; Fujitani, T.; Kobuchi, T.; Nishigaki, T. A new convenient synthetic method for 3-allyl-1,1,1-trifluoroacetylacetone and its derivatives. Synthesis 1986, 340–342. [Google Scholar]
- Hojo, M.; Masuda, R.; Okada, E. A useful one-step synthesis of β-trihaloacetylvinyl ethers and trihaloacetylketene acetals. Synthesis 1986, 1013–1014. [Google Scholar]
- Colla, A.; Martins, M.A.P.; Clar, G.; Krimmer, S.; Fischer, P. Trihaloacetylated enol ethers - general synthetic procedure and heterocyclic ring closure reactions with hydroxylamine. Synthesis 1991, 483–486. [Google Scholar]
- Ezell, E.L.; Thummel, R.P.; Martin, G.E. Correlation of resonances of strongly coupled spin systems via responses due to strong coupling in homonuclear two-dimensional J-resolved spectra: Total assignment of the 1H-NMR spectrum of 2-(2'-pyridyl)-1,8-naphthyridine. J. Heterocycl.Chem. 1984, 21, 817–823. [Google Scholar] [CrossRef]
- Flores, A.F.C.; Siqueira, G.M.; Freitag, R.; Zanatta, N.; Martins, M.A.P. Síntese de 2-trialoacetil-cicloexanonas e -pentanonas: Um estudo comparativo dos rendimentos de reação de enoléteres, cetais e enaminas frente à trialometilacetilantes. Quim. Nova 1994, 17, 298–300. [Google Scholar]
- Bonacorso, H.G.; Costa, M.B.; Moura, S.; Pizzuti, L.; Martins, M.A.P.; Zanatta, N.; Flores, A.F.C. Synthesis, 17O NMR spectroscopy and structure of 2-trifluoroacetyl-1-methoxycycloalkenes. J. Fluorine Chem. 2005, 126, 1396–1402. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2b, 2k, 3a-m, 4c, 4f, 4g, 4i, 4j are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).