Pseudotype Neutralization Assays: From Laboratory Bench to Data Analysis



Abstract
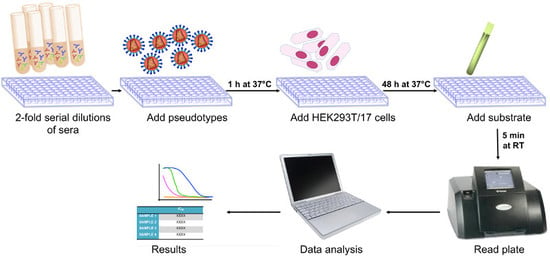
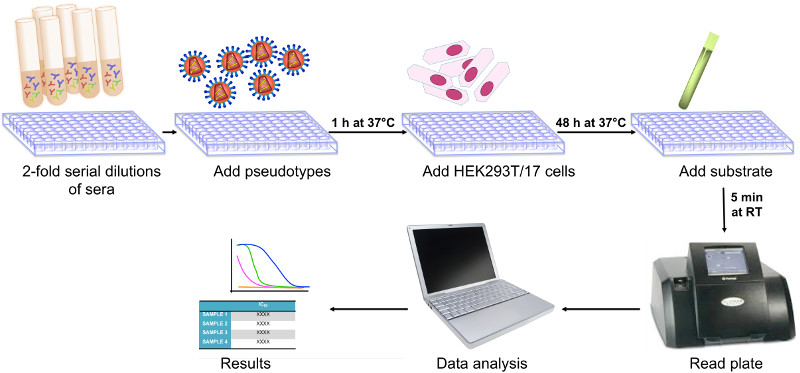
:
1. Introduction
2. Experimental Design
2.1. General Considerations
2.2. Materials
- 12-channel micro-pipette
- 8-channel micro-pipette
- Pipettes (10–200 µL)
- 200 µL MultiGuard NX Barrier tips (Sorenson BioScience, Salt Lake City, UT, USA; Cat. no.: 30550T)
- 20 µL MultiGuard Barrier tips (Sorenson BioScience; Cat. no. 35220)
- 10 µL MultiGuard E Barrier tips (Sorenson BioScience; Cat. no. 15020T)
- Dulbecco’s Modified Eagle Medium (DMEM) with high glucose and GlutaMAX (Thermo Fisher Scientific Inc., Waltham, MA, USA; Cat. no.: 31966-021; or Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: D6429; or PAN Biotech, Aidenbach, Bavaria, Germany; Cat. no.: P04 04510)
- Fetal Bovine Serum (FBS; Thermo Fisher Scientific Inc.; Cat. no.: 10500-064; or PAN Biotech; Cat. no.: P30 8500)
- Penicillin/streptomycin (Sigma-Aldrich; Cat. No: P4333).
- 0.05% (w/v) Trypsin 0.53 mM EDTA solution (Sigma-Aldrich; Cat. no.: T3924; or PAN Biotech; Cat. no.: P10 040100)
- Nunc F96 MicroWell white polystyrene plates (Thermo Fisher Scientific Inc.; Cat. no.: 136101)
- Reagent reservoirs (Corning Inc., Corning, NY, USA; Cat. no.: 4870; or Dutscher Scientific, Brumath, France; Cat. no.: 006793)
- Positive control sera (appropriate for the pseudotyped lentiviral vectors used, see Section 2.6)
- Negative control sera (appropriate for the pseudotyped lentiviral vectors used, see Section 2.6)
- Bright Glo Luciferase Assay System (Promega, Madison, WI, USA; Cat. no.: E2650)
2.3. Cell Line
2.4. Equipment and Software
- GloMax Multi detection system luminometer (Promega; Cat. no.: E7031 and E7041) or equivalent
- Centrifuge for 96-well plates
- Humidified CO2 Incubator
- Biological Safety Cabinet
- Automated cell counter (Bio-Rad Laboratories Inc., Hercules, CA, USA; Cat. no.: 1450102) or cell counting chamber slides
- VACUSIP (INTEGRA Biosciences, Hudson, NH, USA; Cat. no.: 159000) or vacuum pump or similar instrument (optional)
- GraphPad Prism version 6 Software (GraphPad Software Inc., La Jolla, CA, USA)
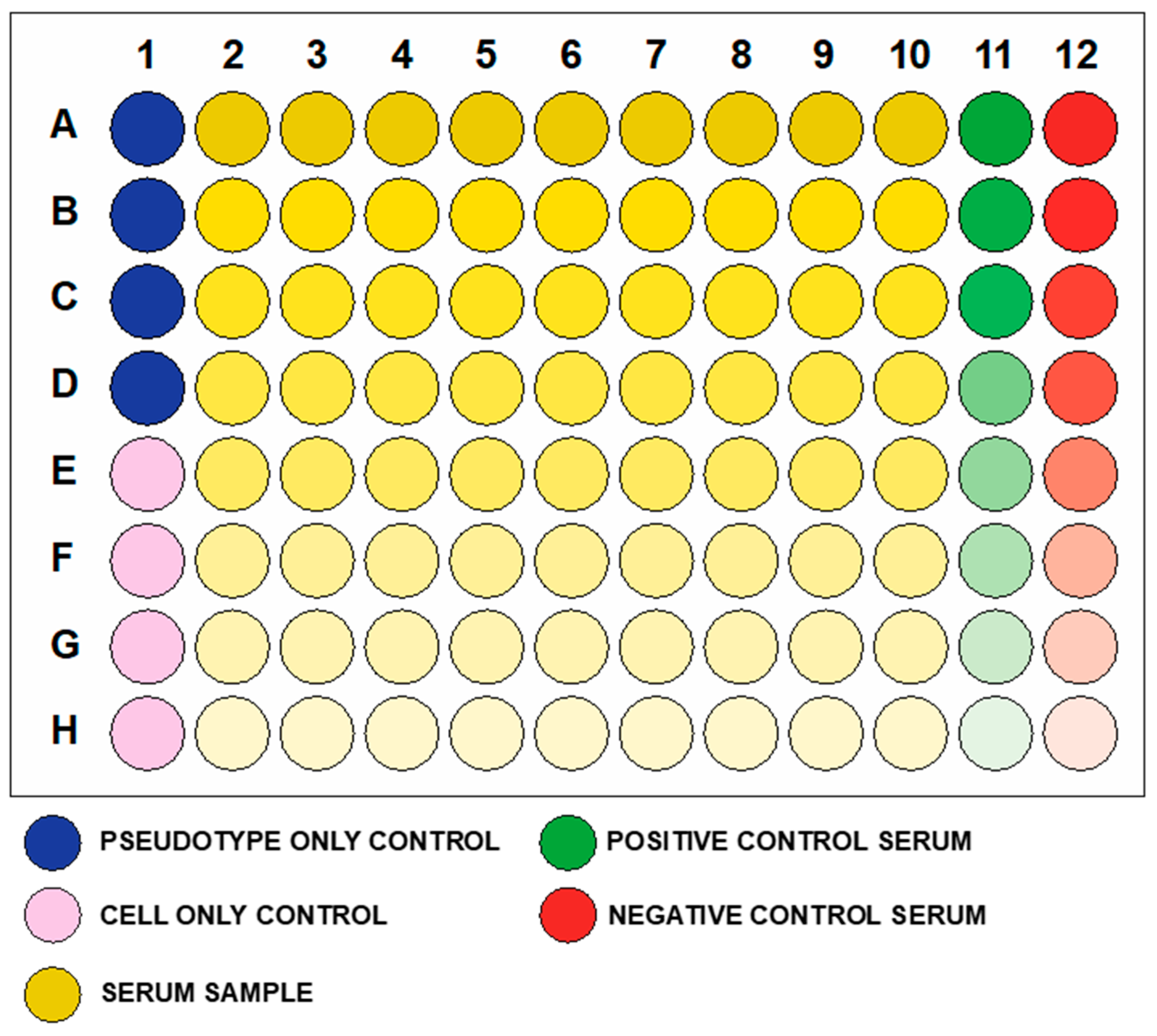
2.5. Plate Format
2.6. Sera
3. Procedure
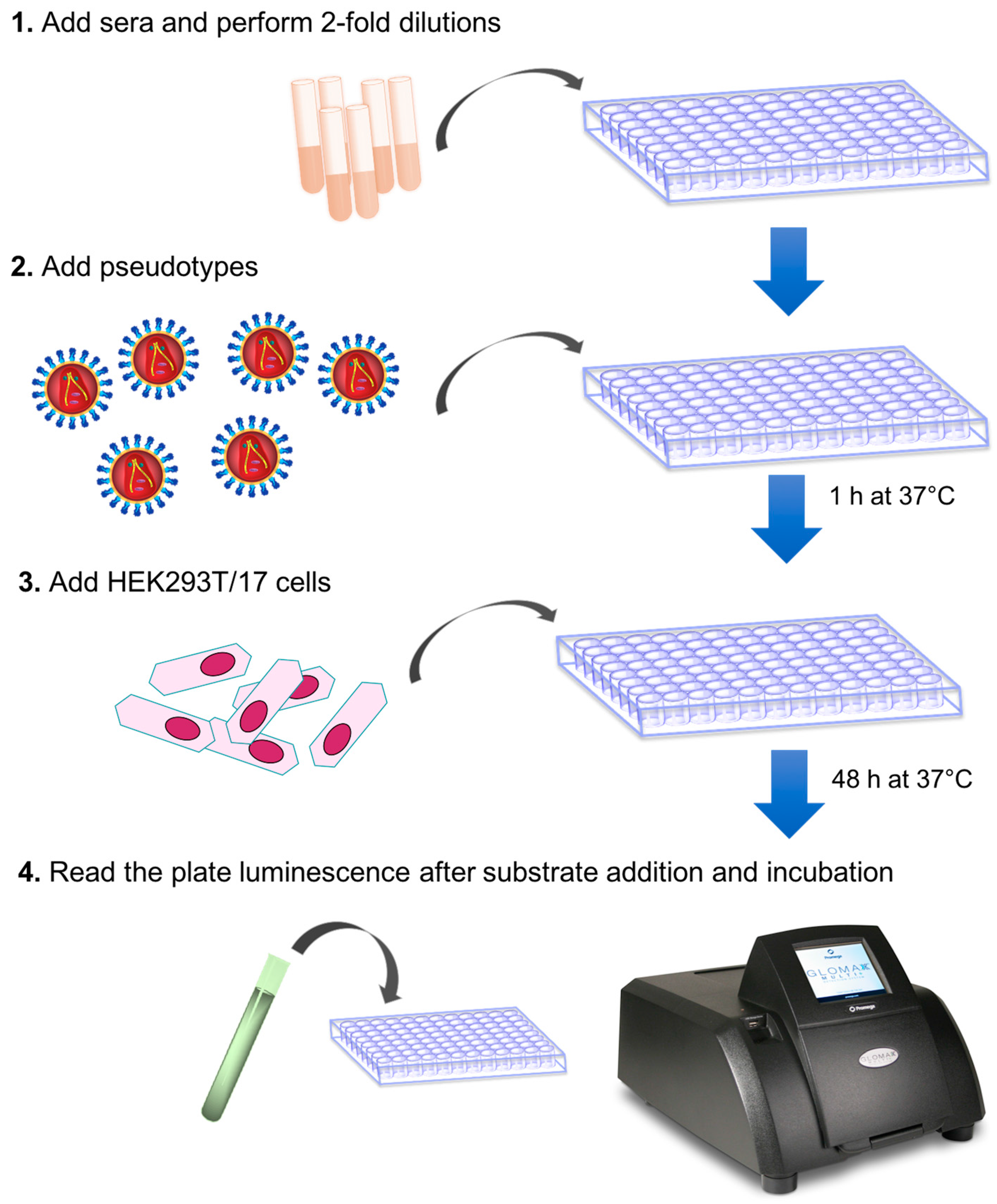
3.1. Assay Set-Up. Time for Completion: 2:30 h
- Before performing the assay, calculate the amount of influenza pseudotyped lentiviral vector that will be used.
- Firstly, calculate how many RLU are required for one assay, performed in one plate. In each well, we recommend a virus input that results in 1 × 106 RLU:
- Therefore, the volume of pseudotype (P) that should be used is calculated, as shown below:
- Considering that pseudotypes are added to each well at a volume of 50 µL for one plate, 5 mL (50 µL × 100) of pseudotype suspension should be prepared by adding to the volume calculated in b., bringing the complete medium to a total volume of 5 mL.
- Add 50 µL of complete medium to A1, B1, C1, D1.
- Add 100 µL to E1, F1, G1, H1 (Cell only control).
- Add 50 µL of medium to B2-H12 (A2–A12 should remain empty in this step).
- Add 5 µL (or 2.5 µL or 2 µL or less) of each serum sample to wells A2 to A10.Note: if the same serum will be analyzed multiple times to obtain technical replicates, we advise putting the replicates in adjacent columns to simplify the subsequent analysis.
- Add to A11, the same amount (5 µL or 2.5 µL or 2 µL or less) of positive control serum and to A12, the same amount of negative control serum.
- Make volume up to 100 µL with the complete medium in A2–A12.
- OPTIONAL STEP Spin the plate in a centrifuge for 1 min at 500× g.
- Take 50 µL from wells A2–A12 (samples and serum control) and serially dilute down the plate (from A to H) using a 12-channel pipettor.
- Spin the plate in a centrifuge for 1 min at 500× g.
- In a new reservoir, prepare the pseudotyped lentiviral vector on the basis of the calculation performed in step 1: add the pseudotype volume and then the complete medium, to give a final volume of 5 mL. Mix by gently moving the reservoir and by pipetting up and down with the multichannel pipette.
- Add 50 µL of the pseudotyped lentiviral vector to each well, with the exception of E1, F1, G1, H1 (cell only control).A1, B1, C1 and D1 will be the pseudotype only (no sera) control.
![Mps 01 00008 i001]() CRITICAL STEP To avoid carry over, change tips for each column or row of the plates when adding the pseudotyped lentiviral vector.
CRITICAL STEP To avoid carry over, change tips for each column or row of the plates when adding the pseudotyped lentiviral vector. - Spin the plate in a centrifuge for 1 min at 500× g.NOTE: Manual mixing is not required after this step.
- Incubate the serum-pseudotype mix for 1 h at 37 °C and 5% CO2.
- Fifteen minutes before the end of the 1-hour incubation time, prepare a HEK293T/17 cell suspension (assay cell suspension) directly in a sterile reservoir.
![Mps 01 00008 i002]() CRITICAL STEP Do not use tubes to prepare the cell suspension as HEK293T/17 can tend to clump, and this could result in a non-homogenous cell suspension. Keep cells in the tissue culture dish during the preparation of the cell suspension.
CRITICAL STEP Do not use tubes to prepare the cell suspension as HEK293T/17 can tend to clump, and this could result in a non-homogenous cell suspension. Keep cells in the tissue culture dish during the preparation of the cell suspension.- Remove media from a 95–100% confluent HEK293T/17 cell dish.
- To wash the cells and neutralize remaining serum, add 2 mL of Trypsin-EDTA, and then remove it.
- Add 2 mL of Trypsin-EDTA.
- Incubate the dish for 5 min at 37 °C and 5% CO2.Neutralize the Trypsin-EDTA by adding 6 mL of complete medium.
- Resuspend carefully, until a single cell suspension is obtained (this can require 2–3 min).
- Count the HEK293T/17 cells, using an appropriate cell counting chamber slide under the microscope, or a cell counter, following the manufacturer’s instructions. A concentration, expressed as cells/mL, should be obtained.
- Calculate the cells needed for one assay plate:
- Calculate the volume of cell suspension for one assay plate:
- Calculate the volume (x) of HEK293T/17 cells for assay cell suspension:
- Resuspend the HEK293T/17 cells in the dish and then add the volume of cells calculated (x) to a clean and sterile reservoir.
- Add the volume of complete medium needed to reach a total volume of 5 mL.
- Mix by moving the reservoir back and forward.
- After careful mixing of the assay cell suspension by pipetting up and down, add 50 µL of HEK293T/17 cell suspension to each well of the assay plate that was removed from the incubator.
![Mps 01 00008 i003]() CRITICAL STEP To avoid carry over, change tips for each column or row of the plates when adding the cells.
CRITICAL STEP To avoid carry over, change tips for each column or row of the plates when adding the cells. - Spin the plate in a centrifuge for 1 min at 500× g.
- Incubate plate for 48 h at 37 °C and 5% CO2.



3.2. Firefly Luciferase Read-Out. Time for Completion: 15 min
- 19.
- Assay firefly luciferase RLU, using a Promega GloMax Multi detection system luminometer or equivalent.
- Remove plate from the incubator.
- Add 50 µL of Bright-Glo, reconstituted following the manufacturer’s instructions, to each well of the 96-well plate.
- Incubate for 5 min at room temperature.
- Read, using the Bright-Glo protocol, pre-installed on the GloMax Multi detection system luminometer (or alternative machine, using readily available settings for firefly luciferase).
- Export the data onto a USB stick for further analysis on a PC.
OR- f.
- Remove plate from the incubator.
- g.
- Remove the medium from each well of the plate (vacuum pump or similar instrument).
![Mps 01 00008 i004]() CRITICAL STEP To avoid carry over, change the tips for each column or row of the plates when removing the media.
CRITICAL STEP To avoid carry over, change the tips for each column or row of the plates when removing the media. - h.
- Add 50 µL of a reading solution, previously prepared by a 1:2 dilution of Bright Glo in complete medium (i.e., 2.5 mL of Bright Glo and 2.5 mL of complete medium).
- i.
- Incubate for 5 min at room temperature.
- j.
- Read using the Bright-Glo protocol pre-installed on the GloMax Multi detection system luminometer (or alternative machine, using readily available settings for firefly luciferase)
- k.
- Export the data onto a USB stick for further analysis on a PC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.3. Data Analysis. Time for Completion: 01:30 h
- 20.
- Open the raw data file using Microsoft Excel and calculate the average values of the pseudotype only control wells and of the cell only wells.
- 21.
- Open GraphPad Prism version 6.
- 22.
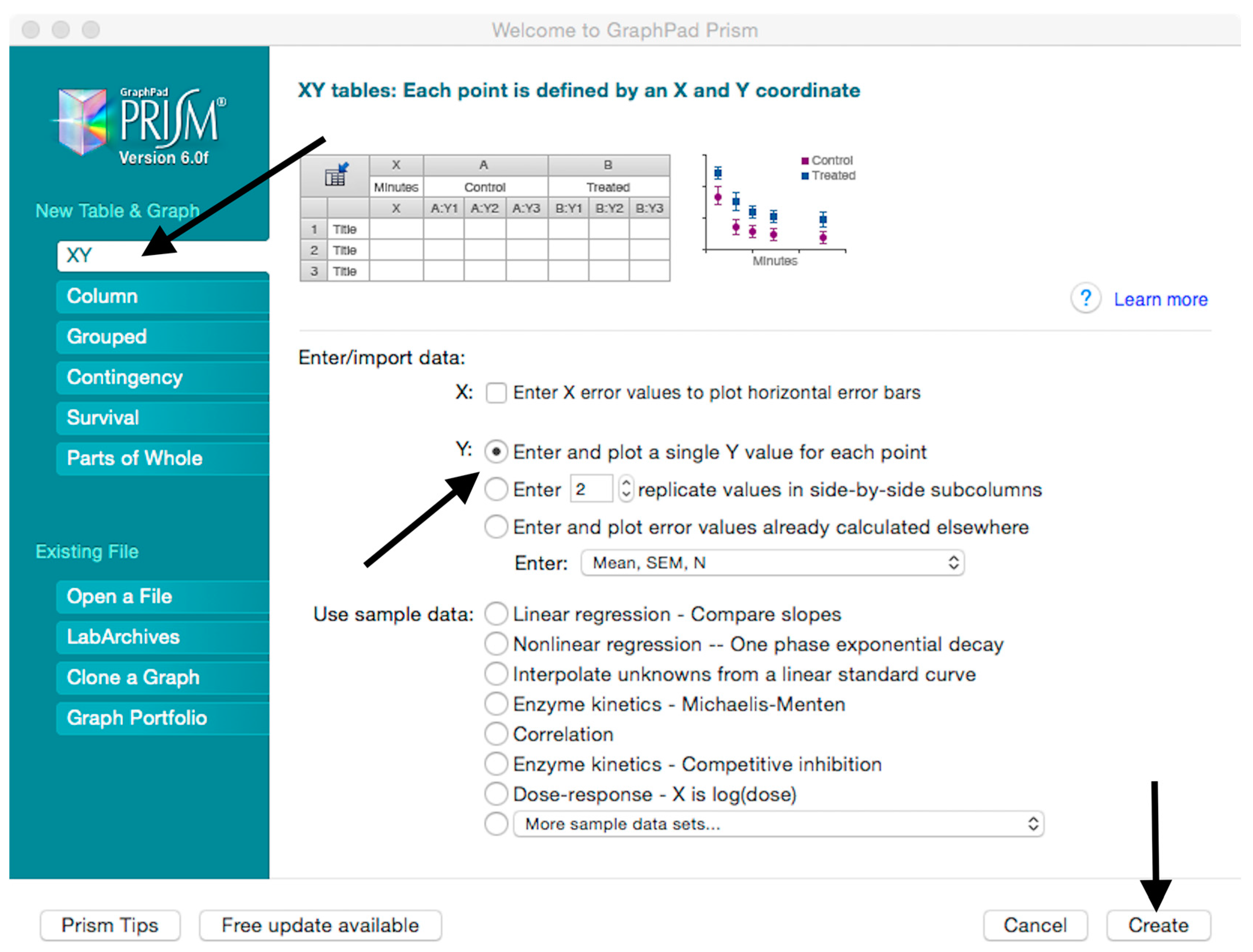
- Create a New project file selecting “XY” table and “Enter and plot a single value Y for each point” and then press the “Create button” (Figure 3).If multiple replicates for the same serum were performed on the same plate, select “Enter in … replicate values side-by-side subcolumns” and in the box, insert how many replicates were performed. Be careful to put replicates next to each other when the data are copied.If replicates were performed on different plates, we advise processing each replicate with “Enter in side-by-side subcolumns”, and then average the ID50/IC50 obtained.
- 23.
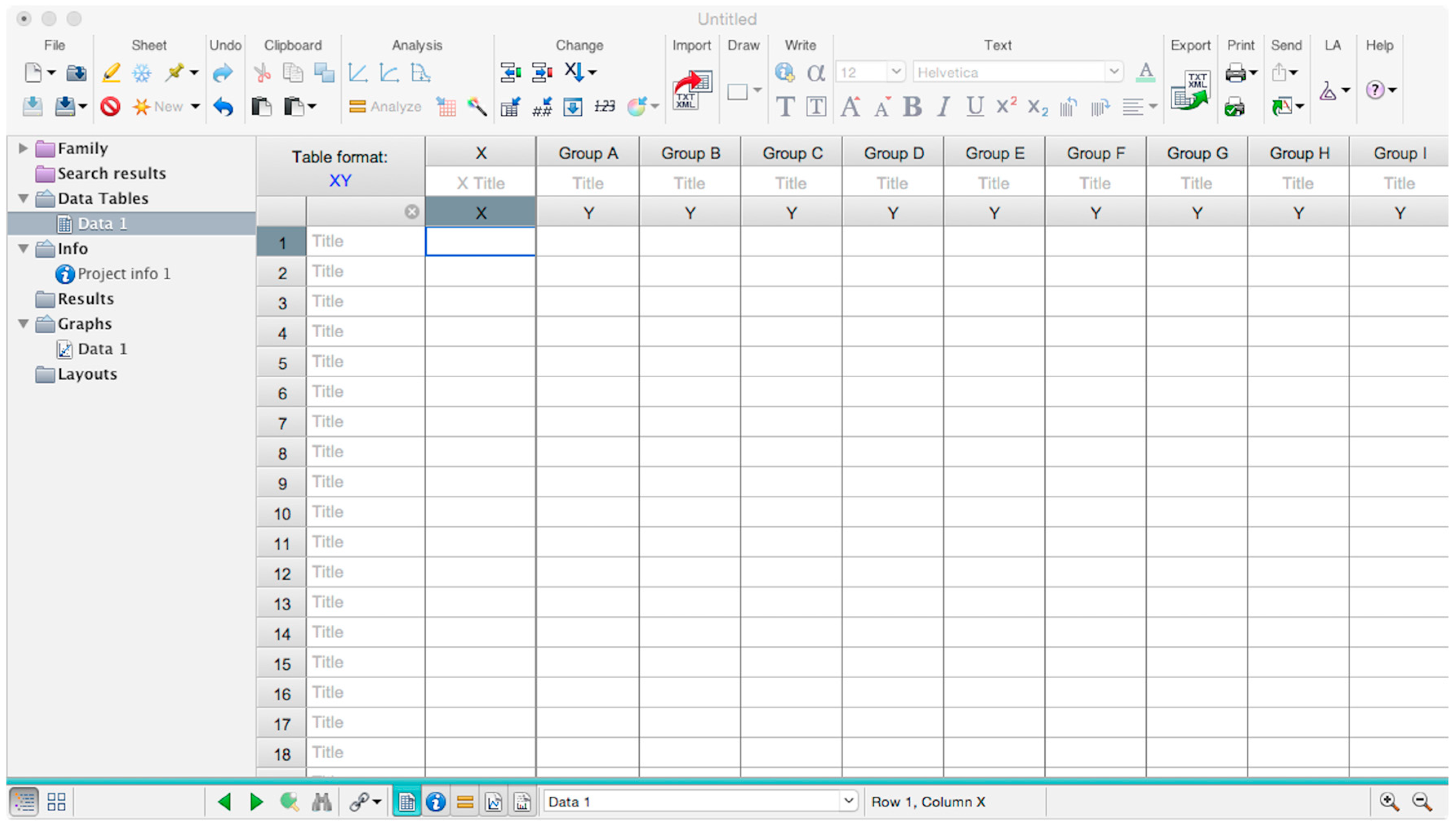
- A window like the one reported in Figure 4 will appear.
- 24.
- Insert assay dilution factors in the first column (40–5120, for example, if the serum sample volume was 5 µL).
- 25.
- Insert logarithmic dilution factors (which can be calculated using Microsoft Excel using the LOG10 function) in the X column.
- 26.
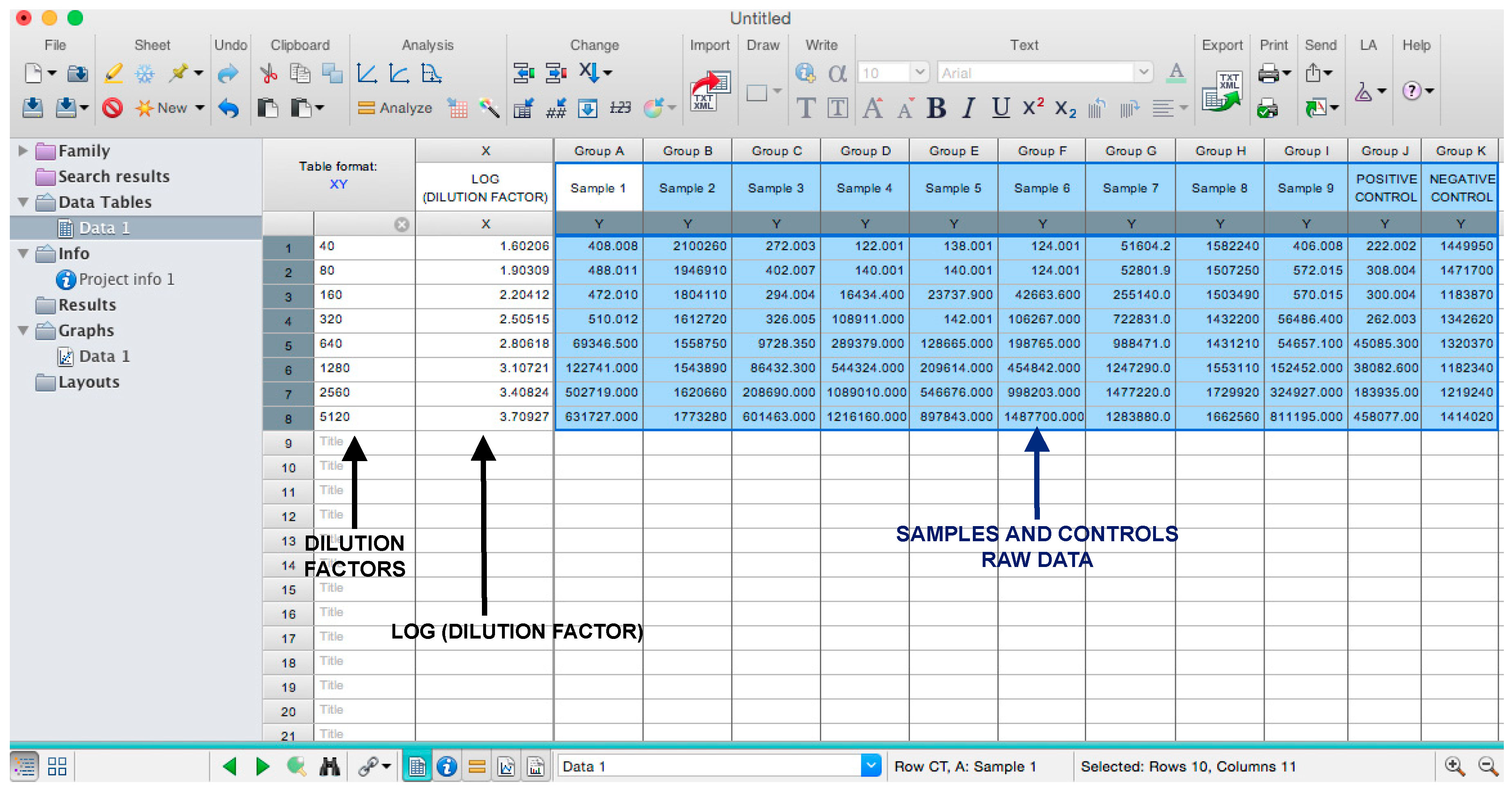
- Copy the samples, positive and negative serum control raw data (with the exception of pseudotype and cell only values) into the other columns; each column should represent a sample/control (Figure 5).
- 27.
- To transform the data into percentage neutralization, click on “Analyze”.
- 28.
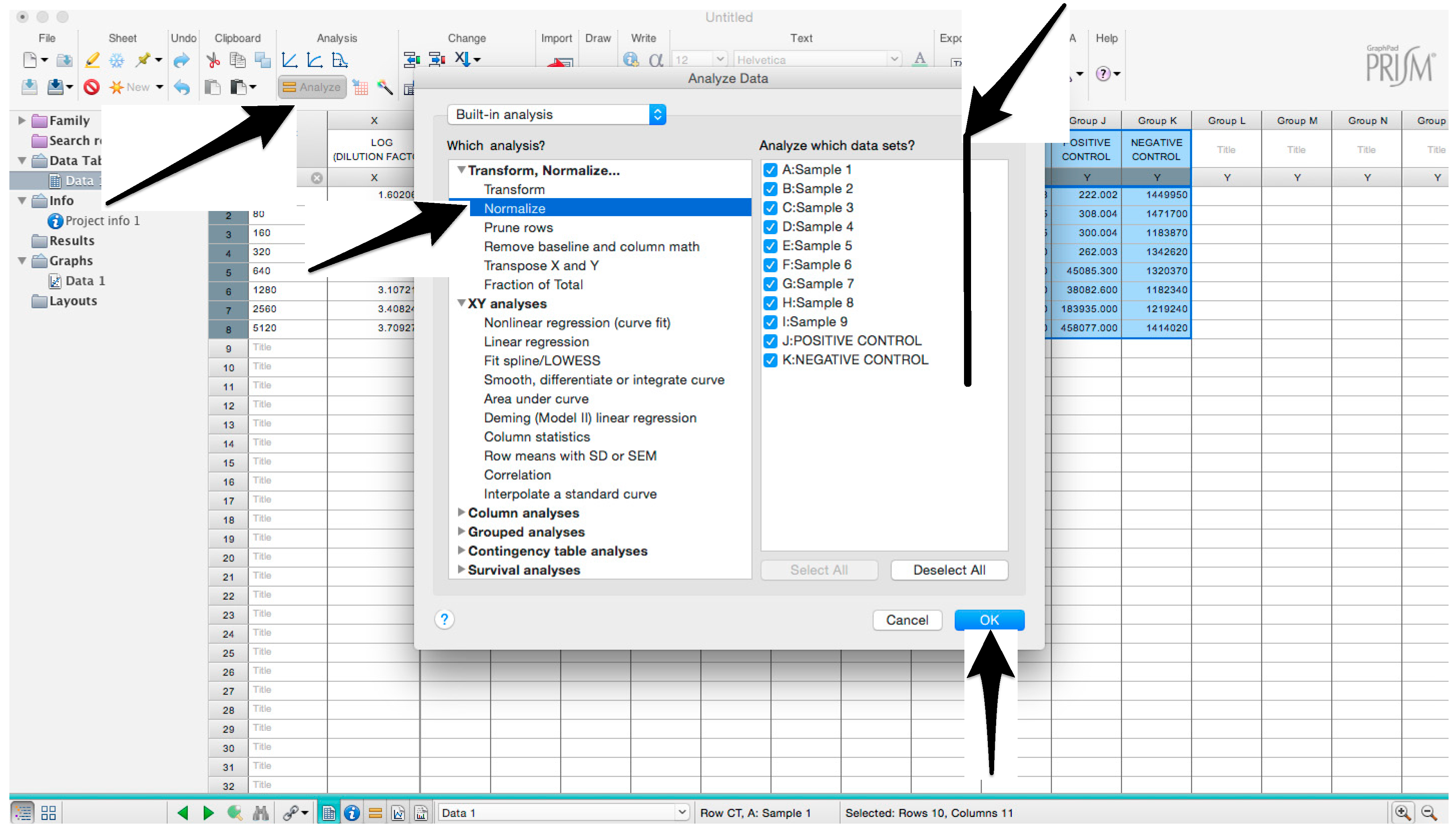
- In the new window that opens, select “Normalize” and be sure that all the data sets are ticked; then click “OK” (Figure 6).
- 29.
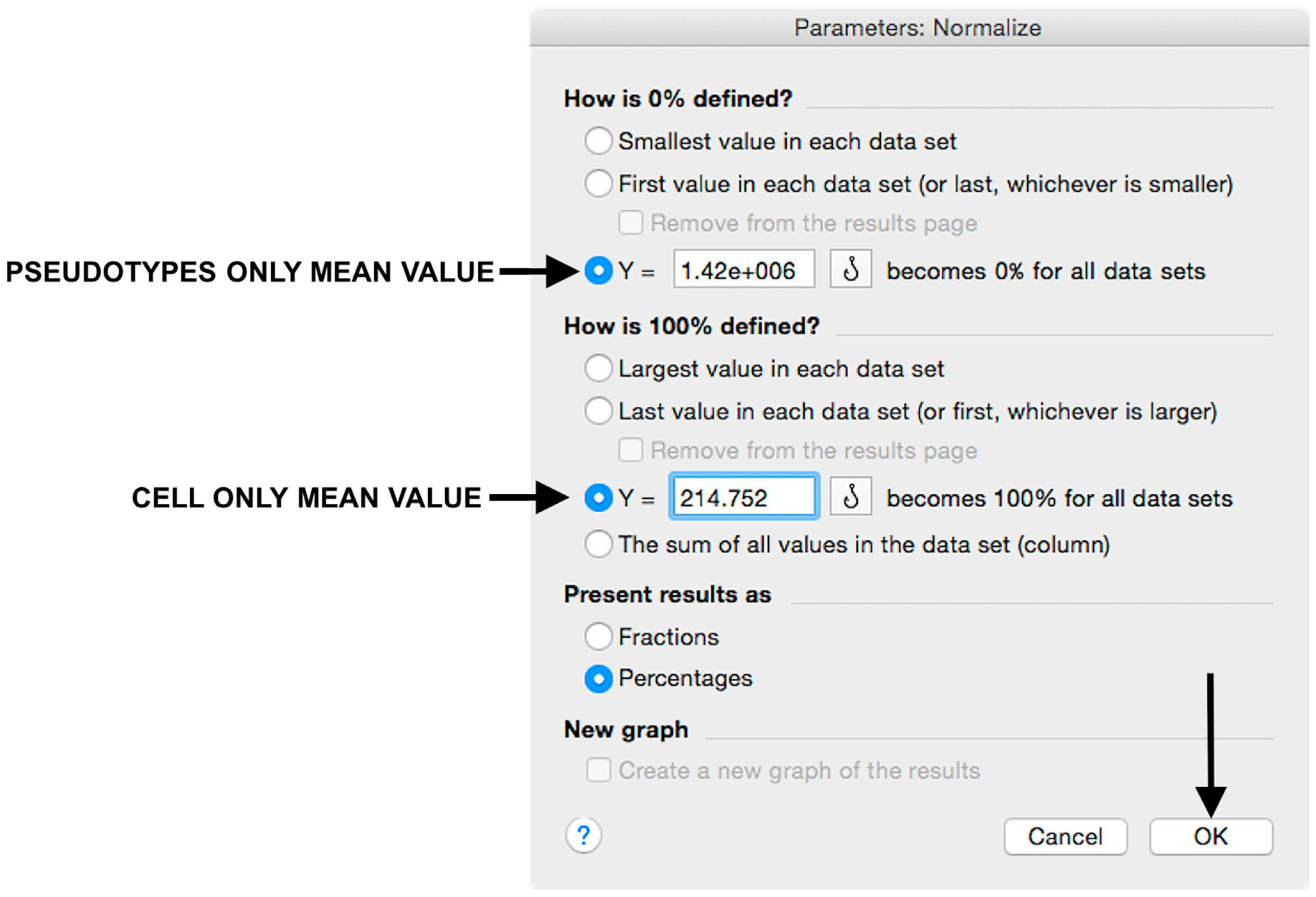
- Insert the pseudotype only mean value (which was calculated in step 20) in “How is 0% defined?” and cell-only mean value in “How is 100% defined?”. Be sure that the “Y=” and the “Percentages” options are selected. Then click “OK” (Figure 7).
- 30.
- “Normalize of Data 1” table will be created. From now on, this table will be used for analysis (Figure 8).
- 31.
- Now click again on “Analyze”.
- 32.
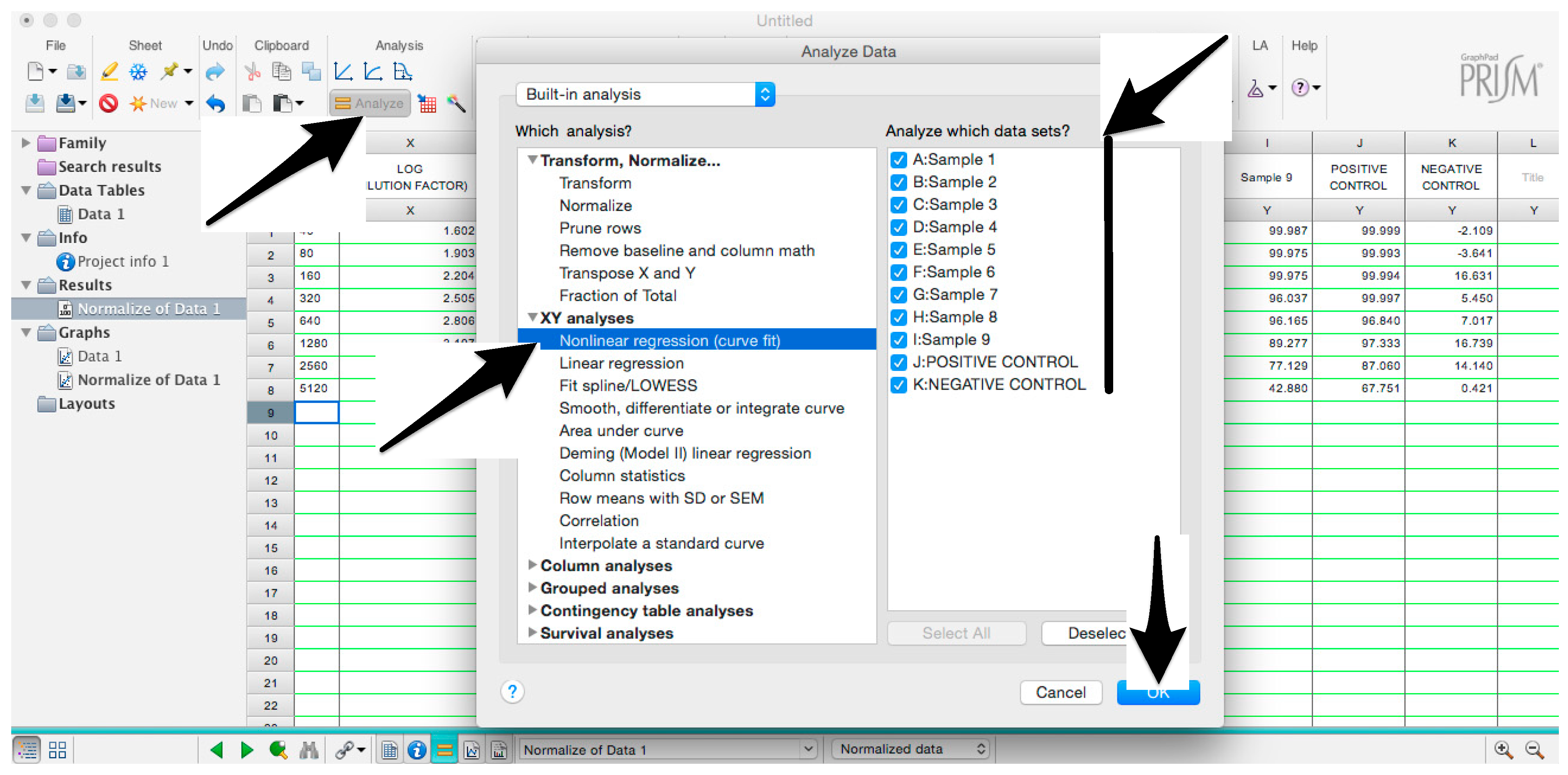
- Select “Nonlinear regression (curve fit)” and be sure that all the data sets are ticked; then click “OK” (Figure 9).
- 33.
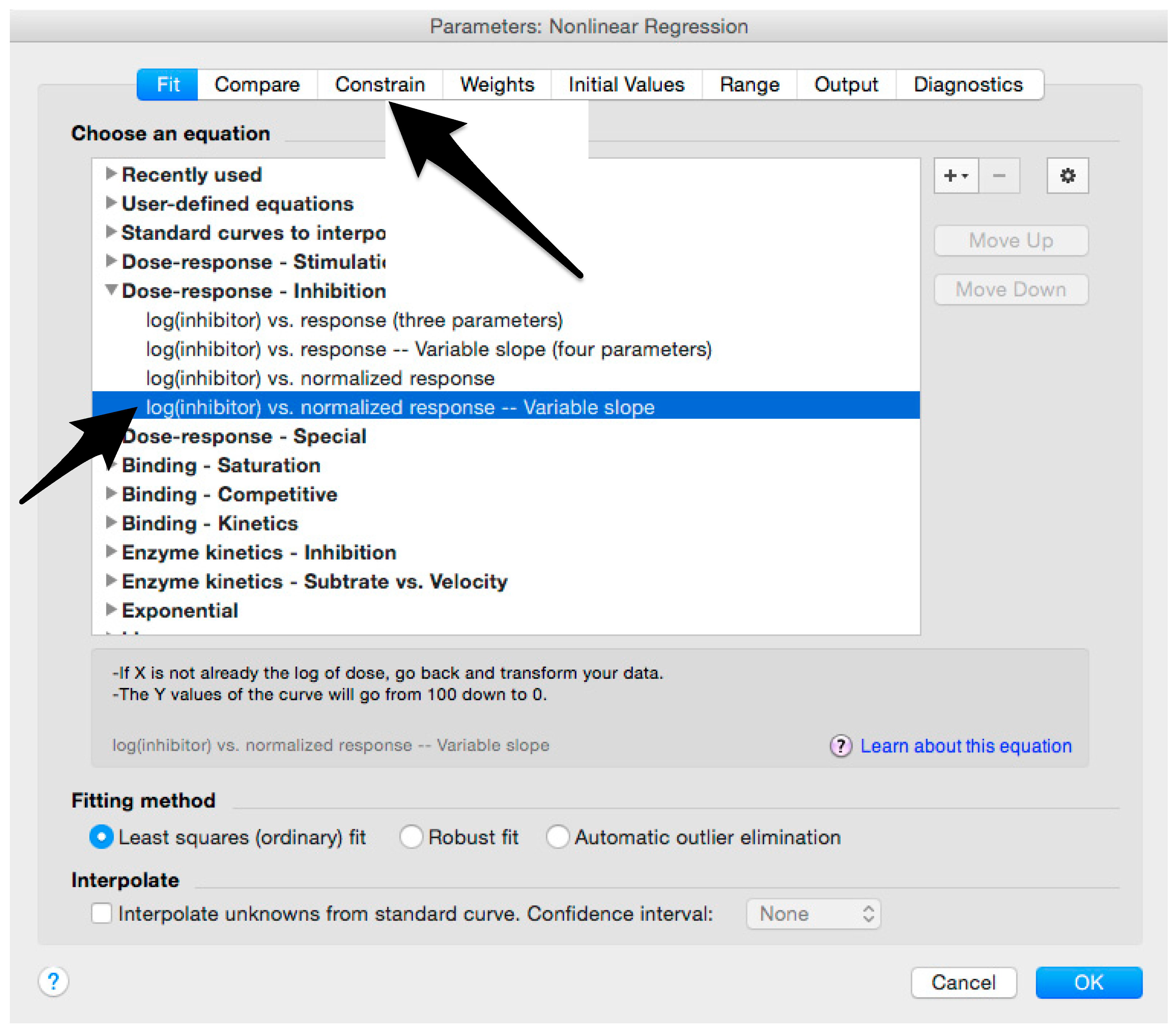
- Select “log(inhibitor) vs. normalized response—Variable slope” from “Dose-response-Inhibition”; then click on “Constrain” (Figure 10).NOTE: If ID20 (or IC20) or ID80 (or IC80) need to be reported, create and use a user-defined equation based on “Dose-response—Special: log(agonist) vs. response—FindECanything” (see Figure 10); after double-clicking, press “clone this equation”, change the name and edit the formula by putting “Bottom” as 0 and “Top” as 100. In “Rules for Initial Values”, be sure that the variables are as follows: “logECF” should be “*(Value of X at YMID)”, “HillSlope” should be “SIGN(YATXMAX- YATXMIN)” and F should be set as “(Initial Value, to be fit)”In “Default Constraints”, select “Must be less than” 0 for the “Hill-Slope” (see also step 34). Constrain the F parameter to 20 if IC20/ID20 needs to be calculated, or to 80 for the IC80/ID80 calculation, or any other number between 0 and 100.In “Transform to Report” change “ECF” to “ICF”.
- 34.
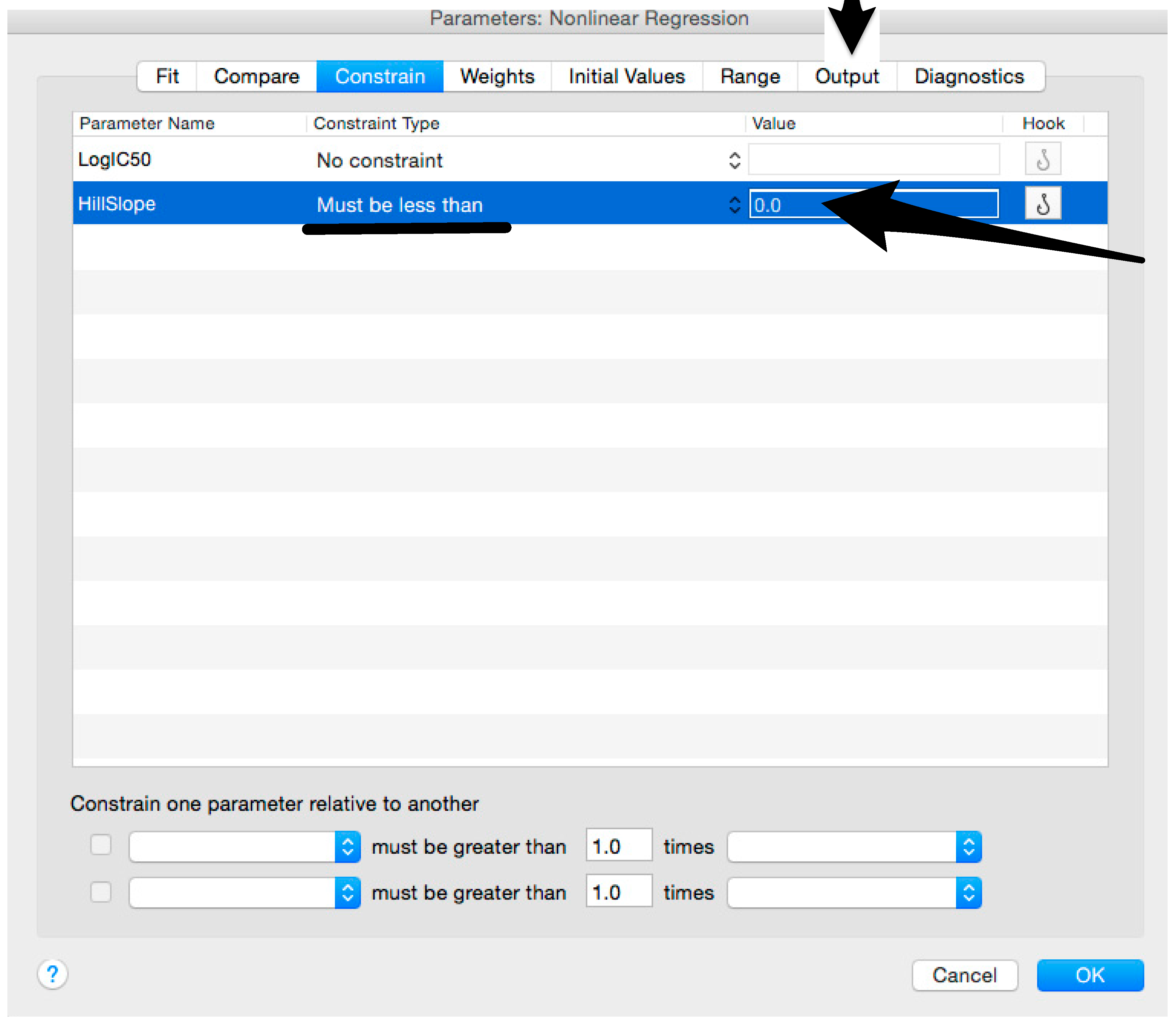
- Constrain the “Hill Slope” to be less than 0, as shown below; then click on “Output” (Figure 11).NOTE 1: Constraining the hill slope usually improves the data fit, forcing the software to consider the data points as part of inhibitory curves. However, if done improperly, it can block the IC50 calculation.NOTE 2: It is possible to use a pre-modified “log(inhibitor) vs. normalized response—Variable slope” function, in which the hill slope is already constrained. Please see Prism Manual Online to learn how to modify an already existing equation (or see step 33 NOTE as example).
- 35.
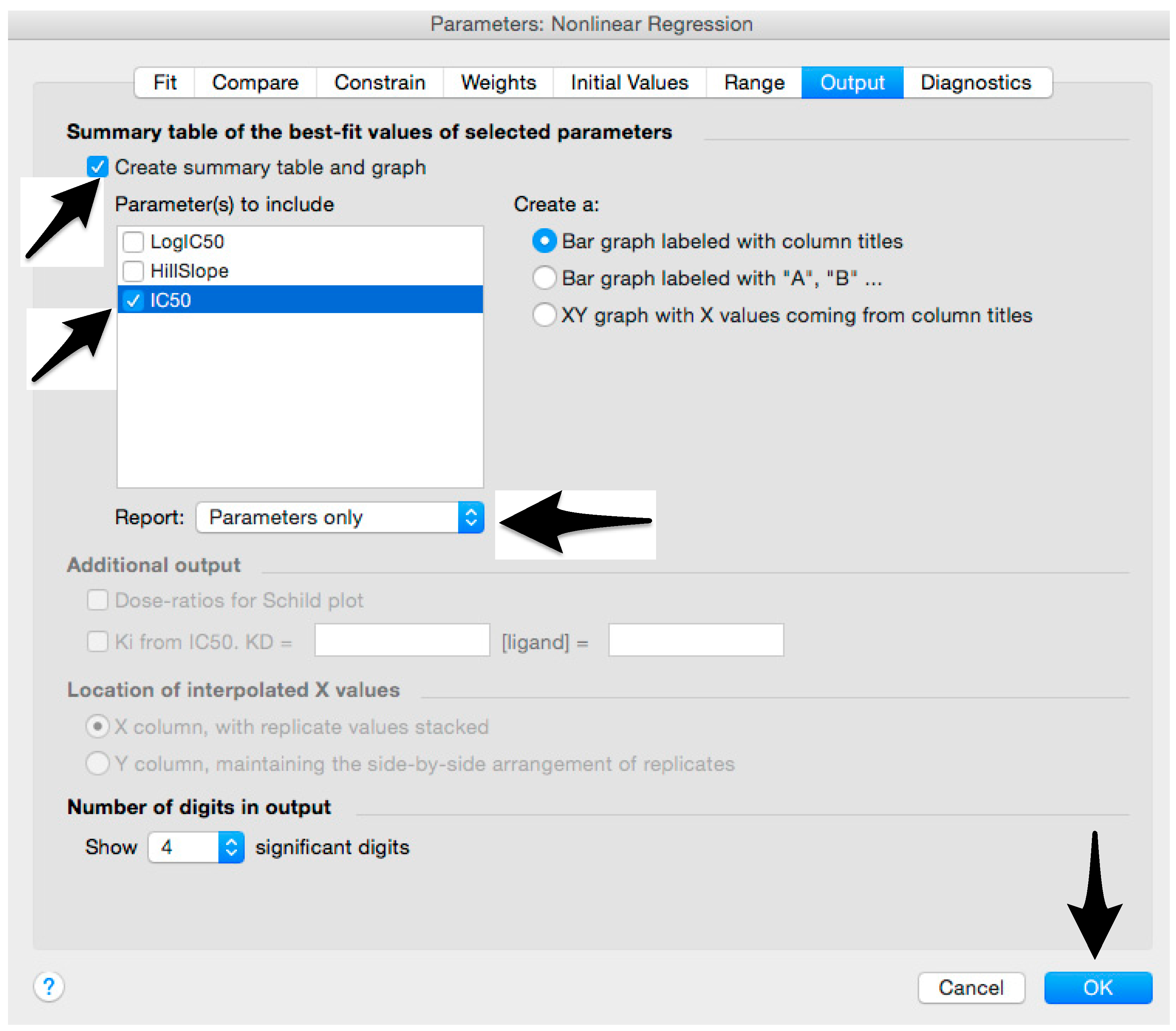
- Tick “Create summary table and graph”, and then remove all the ticks apart from “IC50” (Figure 12).
- 36.
- Select “Parameter only” and then click on “OK” (Figure 12).
- 37.
- Analyses will be performed, and new sheets will appear in results.
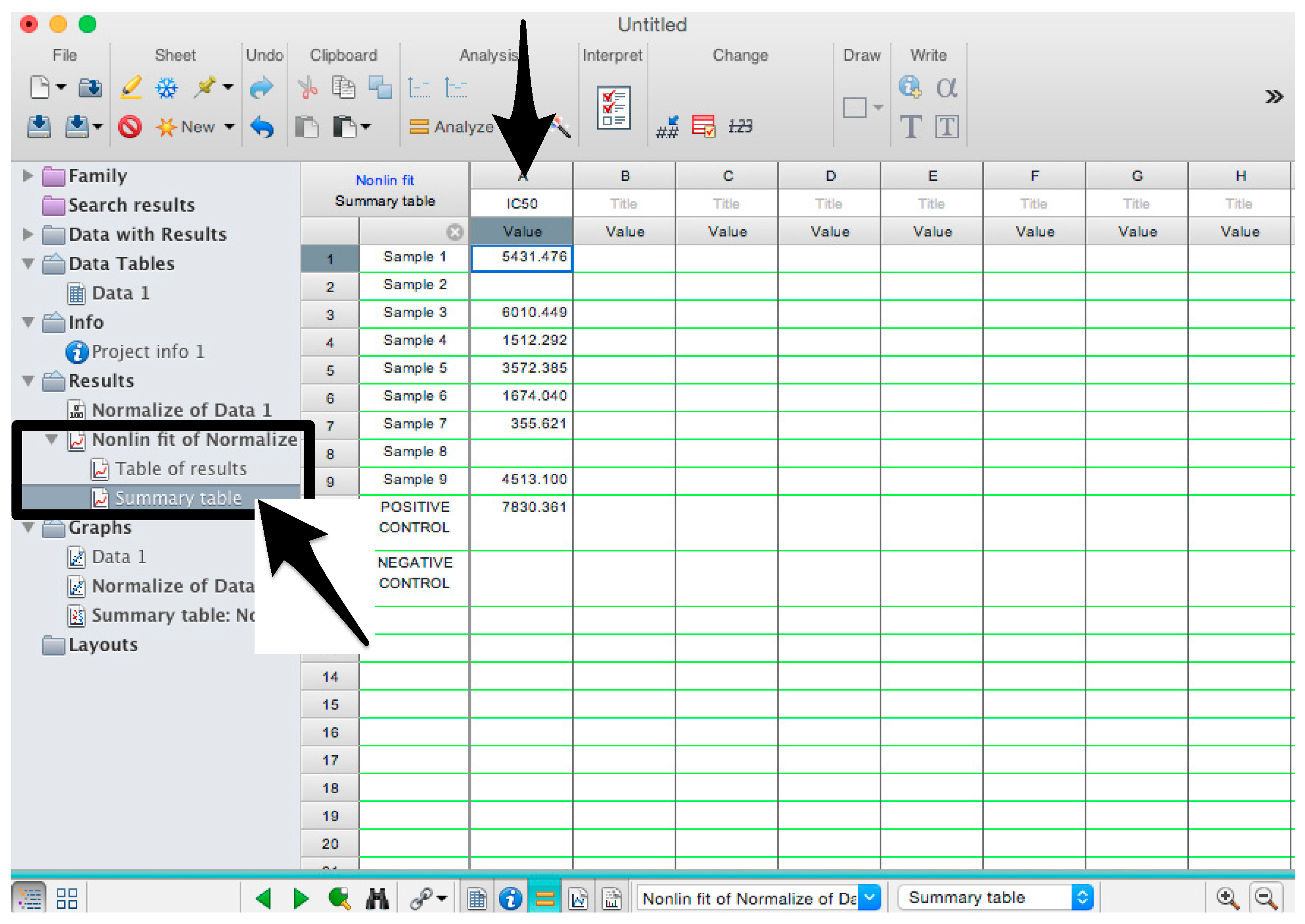
- 38.
- Check the “Summary table” for IC50 values (Figure 13).
- 39.
- Save the file.NOTE: If additional plates from the same experiment need to be analysed, the same file could be used; a new table should be added, and then proceed with analysis from point 3.
4. Expected Results
5. Conclusions and Future Work
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Storch, G.A.; Wang, D. Diagnostic Virology and Virus Discovery. In Fields Virology, 6th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1, pp. 414–452. ISBN 978-1451105636. [Google Scholar]
- King, B.; Temperton, N.J.; Grehan, K.; Scott, S.D.; Wright, E.; Tarr, A.W.; Daly, J.M. Technical considerations for the generation of novel pseudotyped viruses. Future Virol. 2016, 11, 47–59. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Q.; Huang, W.; Li, X.; Wang, Y. Current status on the development of pseudoviruses for enveloped viruses. Rev. Med. Virol. 2017, 13, e1963. [Google Scholar] [CrossRef] [PubMed]
- Steffen, I.; Simmons, G. Pseudotyping Viral Vectors with Emerging Virus Envelope Proteins. Curr. Gene Ther. 2016, 16, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Molesti, E.; Temperton, N.J. The application of pseudotypes to influenza pandemic preparedness. Future Virol. 2015, 10, 731–749. [Google Scholar] [CrossRef]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A Neutralizing Antibody Selected from Plasma Cells That Binds to Group 1 and Group 2 Influenza A Hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Palese, P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 2013, 3, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Carnell, G.W.; Ferrara, F.; Grehan, K.; Thompson, C.P.; Temperton, N.J. Pseudotype-based neutralization assays for influenza: A systematic analysis. Front. Immunol. 2015, 6, 161. [Google Scholar] [CrossRef] [PubMed]
- Temperton, N.J.; Hoschler, K.; Major, D.; Nicolson, C.; Manvell, R.; Hien, V.M.; Ha do, Q.; de Jong, M.; Zambon, M.; Takeuchi, Y.; et al. A sensitive retroviral pseudotype assay for influenza H5N1-neutralizing antibodies. Influza Respir Viruses 2007, 1, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Molesti, E.; Böttcher-Friebertshäuser, E.; Cattoli, G.; Corti, D.; Scott, S.D.; Temperton, N.J. The human Transmembrane Protease Serine 2 is necessary for the production of Group 2 influenza A virus pseudotypes. J. Mol. Genet. Med. 2012, 7, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 2014, 409, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Temperton, N. Chimeric influenza haemagglutinins: Generation and use in pseudotype neutralization assays. MethodsX 2017, 4, 11–24. [Google Scholar] [CrossRef] [PubMed]














© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrara, F.; Temperton, N. Pseudotype Neutralization Assays: From Laboratory Bench to Data Analysis. Methods Protoc. 2018, 1, 8. https://doi.org/10.3390/mps1010008
Ferrara F, Temperton N. Pseudotype Neutralization Assays: From Laboratory Bench to Data Analysis. Methods and Protocols. 2018; 1(1):8. https://doi.org/10.3390/mps1010008
Chicago/Turabian StyleFerrara, Francesca, and Nigel Temperton. 2018. "Pseudotype Neutralization Assays: From Laboratory Bench to Data Analysis" Methods and Protocols 1, no. 1: 8. https://doi.org/10.3390/mps1010008
APA StyleFerrara, F., & Temperton, N. (2018). Pseudotype Neutralization Assays: From Laboratory Bench to Data Analysis. Methods and Protocols, 1(1), 8. https://doi.org/10.3390/mps1010008