Abstract
Pseudomonas is a phylogenetically diverse bacterial genus which is broadly distributed in different ecological niches, and whose taxonomy is continuously under revision. For that purpose, gyrB is one of the housekeeping genes routinely used for multilocus sequence analysis (MLSA). As we noticed that there was not a single primer pair available in the literature suitable for direct sequencing of this gene, we decided to design a unique oligonucleotide pair and to set up a polymerase chain reaction (PCR) protocol to obtain a single amplicon for the entire Pseudomonas genus. Based on the available gyrB sequence from 148 Pseudomonas species, we identified highly conserved regions to design oligonucleotides without fully degenerate positions. We then set up cycling conditions for achieving high specificity and yield of the PCR protocol. Then, we showed that the amplicons produced with this procedure were appropriate for direct sequencing with both primers, obtaining more than 95% of amplicons coverage. Finally, we demonstrated that a PCR-RFLP (restriction fragment length polymorphism) approach served to differentiate among Pseudomonas species, and even between members of the same species.
1. Introduction
DNA gyrase is an essential bacterial enzyme that catalyzes the ATP-dependent negative super-coiling of circular double-stranded DNA (dsDNA) [1]. Genes encoding the two subunits of this type II topoisomerase, gyrA and gyrB, have been widely used as markers in phylogenetic studies of different prokaryotic taxa [2,3,4]. Also, the sequences of gyrA and gyrB alleles from quinolone-resistant isolates have been characterized to understand the evolutionary dynamics of antibiotic resistance, and to promote development of novel drugs [5,6]. In particular, gyrB has been considered a useful housekeeping gene for multilocus sequence analysis (MLSA) in several bacterial genera [7,8,9,10,11]. These studies typically used gyrB polymerase chain reaction (PCR) primers that were designed on the basis of the amino acid sequence of GyrB polypeptides from different bacterial species, and therefore, they contain several degenerate bases [2,12,13]. For this reason, some authors had to utilize different primer pair combinations to obtain gyrB amplicons for phylogenetic analyses of a whole bacterial genus, such as Pseudomonas [14,15]. Other authors designed specific primers for their particular purposes based on the gyrB nucleotide sequence of certain bacterial species [16,17,18,19].
Pseudomonas is a phylogenetically diverse bacterial genus of high interest in clinical, environmental, and molecular sciences. This is mainly due to the fact that Pseudomonas species are broadly distributed in different ecological niches, they can be isolated with relative ease in a variety of selective media, there are hundreds of fully sequenced genomes available, and they are easy to manipulate with genetic engineering tools [20,21]. Since pseudomonads can behave as human, plant or insect pathogens, as well as plant growth-promoting rhizobacteria or key players in bioremediation processes [22], molecular methods that allow a rapid identification of isolates belonging to this genus with a single PCR reaction are useful and desired tools [20,23]. We noticed that there was not a single primer pair in literature that could be employed for the amplification of the entire Pseudomonas genus, but only specific primers for some species or groups [16,17,24,25]. In this report, we present a novel primer pair and a PCR protocol for the amplification of a larger fragment of the gyrB gene from representative species of the entire Pseudomonas genus that is suitable for downstream applications such as restriction fragment length polymorphism (RFLP) analysis or direct sequencing for taxonomic purposes.
2. Materials and Methods
2.1. Bacterial Strains
Type strains and isolates used to set up the PCR protocol are listed in Table 1. All strains were cultured on nutrient agar (NA; 40 g/L tryptone soy agar; 5 g/L yeast extract; Biokar, Beauvais, France) or nutrient yeast broth (NYB; 20 g/L nutrient broth; 5 g/L yeast extract; Biokar), for 24 h at 28 °C and 200 rpm, when required.

Table 1.
Pseudomonas strains used to set up the gyrB polymerase chain reaction (PCR) protocol in vitro.
2.2. Primer Design
Oligonucleotides targeting conserved regions within the gyrB gene were designed manually, based on the alignment of the whole gyrB gene sequence (2421 bp in P. aeruginosa PA01) of Pseudomonas species representing all taxonomic clusters based on the classification made by Gomila et al. [7]. Available sequences were retrieved from the GenBank database (as of September 2017; Table S1), for the 168 Pseudomonas species described at LPSN (list of Prokaryotic names with standing in Nomenclature) [41]. The alignment of the final set of 148 gyrB full sequences was performed with MEGA7 software [42], using the MUSCLE program of the alignment explorer option [43]. Different candidate primer pairs defining amplicons larger than 1200 bp were tested in silico using AmplifX v.1.7.0 [44] and FastPCR 3.3.21 [45]; the best primer pairs were evaluated in vitro by PCR, using a collection of target strains (Table 1). The properties of the novel chosen primers are shown in Table 2.
Table 2.
Novel oligonucleotides targeting the Pseudomonas spp. gyrB gene.
2.3. PCR Reaction
Polymerase chain reaction conditions for the new primer pair reported in this study were optimized to obtain a single amplicon. Typically, 25 μL-reaction mixtures contained 1× buffer (TransGen Biotech, Beijing, China, containing 2.0 mM MgCl2), 0.2 mM deoxyribonucleotide triphosphates (dNTPs) (PB-L, Bernal, Argentina), 0.2 μM of each primer (Invitrogen, Carlsbad, CA, USA), 5% (v/v) dimethyl sulfoxide (DMSO, Sigma Aldrich, St Louis, Missouri, USA), and 1 U of EasyTaq DNA polymerase (TransGen Biotech, China). The PCR cycle consisted of an initial denaturation step of 5 min at 94 °C, followed by 35 cycles of 0.5 min at 94 °C, 0.5 min at 57 °C, and 1 min at 72 °C; and a final extension step at 72 °C for 5 min. As DNA template, we used 1 μL of a thermal cell lysate of each strain. Thermal lysates were obtained by treating a 2–3 mm colony resuspended in 100 μL of ultrapure H2O (PCR grade) at 100 °C for 15 min. After amplification, 10 μL of each reaction was run in 1% agarose gels in 0.5× Tris–borate–EDTA (TBE) at 10 V/cm for 50 min. The gel was stained with ethidium bromide and visualized under UV light.
Polymerase chain reactions with the previously described primers [2,13] were performed with 1 μL of template DNA from thermal lysates of reference pseudomonads from our strain collection (Table 1). We tested two primer combinations, based on the recommended reactions described by Mulet and colleagues [14]. We followed the instructions reported for the reaction mixes and cycling conditions corresponding to each primer pair [2,13] in a final volume of 25 μL. Electrophoresis and staining conditions were the same as described previously.
When corresponding, PCR products from representative pseudomonads were sequenced in Macrogen Inc. (Seoul, Korea) from both ends with each one of the primers designed in this study. Reconstruction of the gyrB fragment and control of the sequences’ quality were performed manually with the BlastN tool [48]. Sequences from isolates of our strain collection were submitted to Genbank database, and the corresponding accession numbers are detailed in File S1.
2.4. PCR-RFLP
Restriction analysis of the gyrB sequences was set up in silico with Serial Cloner v2.6.1 [49] using the amplicon sequences obtained by a virtual PCR reaction run with the designed primers (Table 2) on a group of representative strains as templates (Table 1). A set of classical enzymes was tested to select those ones producing differential RFLP patterns for each reference Pseudomonas species. To confirm the in silico results, in vitro PCR-RFLP assays were carried out in a final volume of 20 μL, containing 10 μL of PCR mix and 2 U of the endonuclease TaqI (Thermo Fisher, Waltham, MA, USA). Reactions were incubated at 62 °C for 3 h. The restriction products were separated by electrophoresis in 2% agarose gels in 0.5× TBE at 6 V/cm for 2 h. Gels were stained with ethidium bromide and DNA banding patterns were visualized under UV light.
3. Results
3.1. Primer Pair Design and In Silico Tests
Using the most employed gyrB oligonucleotides reported in literature, we failed to obtain single bands of the expected size and with uniform yield (Figure 1). We observed a low specificity in the reaction with the APrU/BAUP2 primer pair, and a low yield, or no reaction, with the APrU/UP1-E primer pair (Figure 1). This outcome hampers direct sequencing of the amplicons and forces introduction of a gel extraction step for purification of the expected amplicon before sequencing. These results prompted us to design a novel primer pair targeting gyrB based on the high number of Pseudomonas genomes that are currently available in public databases (Table S1), and we set up a PCR protocol aiming to obtain a single amplicon suitable for downstream applications, such as RFLP or direct sequencing (Table 2).
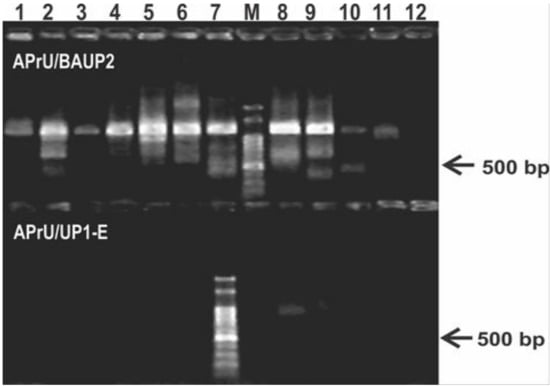
Figure 1.
Typical results of the PCR protocol targeting the gyrB gene using primers reported in literature. The upper panel of the agarose gel corresponds to PCR reactions using the primer pair APrU/BAUP2 [13]; the lower panel corresponds to PCR reactions using the primer pair APrU/UP1-E [2]. Strains were as follows: 1, P. stutzeri ATCC 17588; 2, P. putida KT2440; 3, P. protegens CHA0; 4, P. fluorescens SBW25; 5, P. syringae pv. tomato DC3000; 6, P. aeruginosa PA01; 7; P. alkylphenolica KL28; 8, P. sihuiensis 2013; 9, P. donghuensis SVBP6; 10, P. chlororaphis SMMP3; 11, P. sp. 1008; 12, negative control (without DNA). Molecular weight markers (M) correspond to the 100-bp DNA ladder (Embiotec, Buenos Aires, Argentina).
The sequence alignment of the entire gyrB gene (ranging 2412–2424 bp long depending on the species) from the 148 available Pseudomonas genomes revealed several conserved regions with over 90% of nucleotide identity distributed along the coding sequence (Figure 2A). A desired aspect for comparative taxonomic purposes is to obtain as much sequence information as possible of the housekeeping gene; to this end, we focused in conserved sequence regions separated by at least 1200 bp. Inspection of the 5′ region of the alignment did not reveal a fully conserved stretch with sufficient length for a forward PCR primer (Figure 2A). From the best candidate regions, the chosen one is shown in Figure 2C, which corresponds to positions 46–65 of the P. aeruginosa PA01 gyrB allele. Based on this 20 bp stretch, a forward primer containing 3 partially degenerate positions was designed, avoiding the incorporation of fully degenerate bases (Table 2). Towards the 3′ region of the gyrB alignment, we found a segment of 21 bp (positions 1498–1518 of the P. aeruginosa PA01 gyrB allele) that is highly conserved among all retrieved sequences and that is ideal for the reverse primer (Figure 2B,D). In silico analyses of the PCR reaction with AmplifX and FastPCR software confirmed the compatibility of the chosen primer pair and provided the optimal annealing temperatures (Table 2). The expected size for the gyrB amplicon would be 1461–1467 bp, depending on the target Pseudomonas species (Table 2). Previously, reported oligonucleotides for amplification of the gyrB gene in several pseudomonads [2,14], anneal in different regions from those targeted by our newly designed ones, and were expected to generate an amplicon of shorter lengths and less informative content (Figure 2A). Although the forward primer zone has a conservation level similar to the forward primer we designed, the reverse oligonucleotide is in a poorly conserved region of the gyrB gene compared with our new reverse primer (Figure 2A).
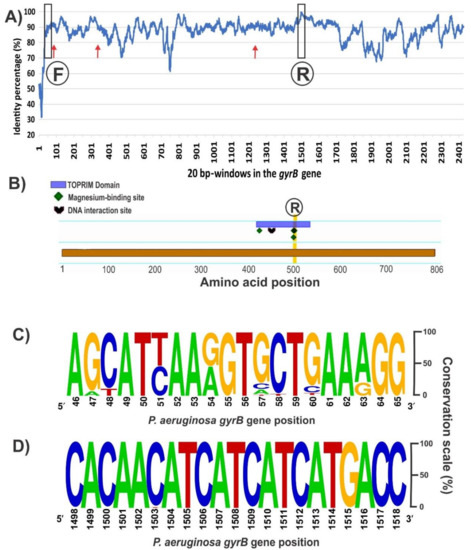
Figure 2.
Conservation of the gyrB gene among the 148 sequences aligned from all the Pseudomonas genomes accessible in Genbank on September 2017 (A), domain and conserved sites in the GyrB monomer (B), and frequency plots of conserved regions selected for the primers design (C,D). (A) Conservation of the gyrB sequence among the 148 genomes analyzed. The percentage of every position was calculated with Jalview software v. 2.1.3 [52] based on the alignment performed in MEGA7 with MUSCLE. Then, an average percentage value was obtained from 20 bp windows, as that is the standard primer size. The positions where the forward (F) and reverse (R) primers were selected are marked. Red arrows show the annealing positions of the previously described forward primers BAUP2 (position 63) and UP-1E (position 335), and the reverse APrU (position 1234); (B) GyrB features in the amino acid sequence from P. aeruginosa PA01 obtained with the Feature Viewer of UniProtKB database (Q9I7C2). The TOPRIM (topoisomerase-primase) domain contains three key amino acids involved in the binding of two magnesium cations and a DNA interaction site. Positions 500 and 502, where the second magnesium binds, matches the highly conserved nucleotide region selected for the design of the novel reverse primer; (C) Forward primer was designed based on this conserved region of 20 bp at the beginning of the gyrB gene (between positions 46 and 65 of the sequence from P. aeruginosa PA01 strain). As we show in Table 2, positions 51, 54 and 63, where the frequency was distributed between two nucleotides, were replaced by the corresponding partially degenerate IUPAC (International Union of Pure and Applied Chemistry) base codes, trying to avoid fully degenerate bases; (D) The reverse primer of 21 bp was chosen as part of a sequence region with the highest identity along the gyrB gene. The analyses of (c) and (d) were performed with the Weblogo software v2.8.2 [53].
3.2. In Vitro Assay with Target Microorganisms
We set up the PCR protocol by optimizing the reaction mix and cycling conditions, so that all tested samples resulted in a single PCR product of the expected size (Figure 3). The addition of DMSO to the PCR mix improved the amplification yield and specificity, as previously reported for other protocols [20,50]. A gradient PCR test with annealing temperatures between 49 °C and 57 °C was performed to find the optimal condition where we could obtain a single band and a good yield (Figure S1). After confirming that the amplification was successful at up to 57 °C, an additional PCR gradient between 57 °C and 60 °C was performed to check if we could improve the PCR specificity without losing yield. We found that 57 °C was the optimal annealing temperature (Figure 3A), in agreement with the in silico prediction. Once set up, we confirmed the ability of the optimized PCR protocol using this unique primer pair (Table 2) to amplify a 1.4 kb internal portion of the gyrB gene from a range of Pseudomonas strains representing most of the different species subgroups (Table 1 and Figure S2). As shown in Figure 3B, all thermal lysates of the tested pseudomonads resulted in a single amplicon of the expected size, without any secondary product. Also, we observed that this reaction protocol allows performing colony PCR instead of preparing thermal lysates or DNA extraction as template (Figure S1). Amplicon sequencing using either of both novel primers allowed us to reconstruct a sequence fragment of around 1400 bp long, thus covering over 95% of the amplicon size obtained in the PCR reaction (File S1). Nonetheless, the gyrB amplicon sequences of the environmental isolate Pseudomonas sp. N23 [37] and of the clinical isolate Pseudomonas sp. 2013, the latter previously assigned to the species P. mendocina [20], enabled us to reassign those isolates to the recently described species P. sihuiensis [51], as the sequence identity raised 99% with 100% of coverage in the BlastN analysis with the sequence from P. sihuiensis KCTC 32246 (Genbank accession number LT629797.1) (Figure S2 and File S1). Thus, in a single PCR round, the newly designed primers provided amplicons that are suitable for direct sequencing and taxonomic analysis.
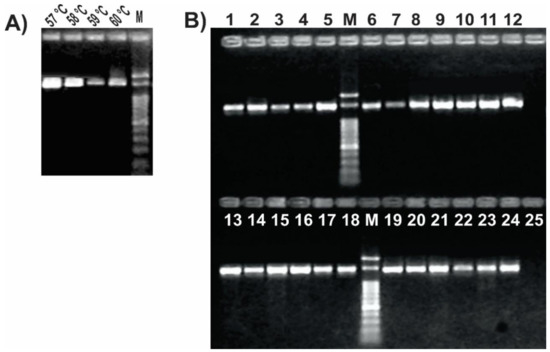
Figure 3.
Set up of the PCR protocol targeting Pseudomonas gyrB gene. 12.5 μL of each reaction was run in 1% agarose gels in 0.5× Tris–borate–EDTA (TBE) at 10 V/cm for 50 min. (A) Gradient PCR of using a P. fluorescens SBW25 lysate as template. Reactions were carried out with primers listed in Table 2 with the annealing temperatures shown above each lane; (B) PCR reactions for the pseudomonads listed in Table 1. Strains were as follows: 1, P. stutzeri ATCC 17588; 2, P. stutzeri 2014; 3, P. stutzeri 2018; 4, P. putida KT2440; 5, P. putida GR12-2; 6, P. protegens CHA0; 7, P. protegens Pf-5; 8, P. fluorescens 2–79; 9, P. fluorescens SBW25; 10, P. simiae WCS417; 11, P. syringae pv. tomato DC3000; 12, P. syringae pv. maculicola ES4326; 13, P. aeruginosa PA01; 14; P. aeruginosa Hex1T; 15, P. aeruginosa DN; 16, P. alkylphenolica KL28; 17, P. sihuiensis 2013; 18, Pseudomonas sp. 2019; 19, P. donghuensis SVBP6; 20, P. chlororaphis SMMP3; 21, P. sihuiensis N23; 22, Pseudomonas sp. CF5; 23, Pseudomonas sp. LDe; 24, P. sp. 1008; 25, negative control (without DNA). Molecular weight markers (M) correspond to the 100-bp DNA ladder (Embiotec).
3.3. PCR-RFLP
The RFLP patterns obtained for a subset of tested strains matched the expected profiles deduced from the in silico analysis of gyrB sequenced amplicons (Figure 4A). In addition, the RFLP banding patterns obtained after the digestion with TaqI were able to distinguish different pseudomonas species (Figure 4B). Moreover, the RFLP profile of different representatives of the same P. fluorescens species showed differential patterns with this enzyme (Figure 4).
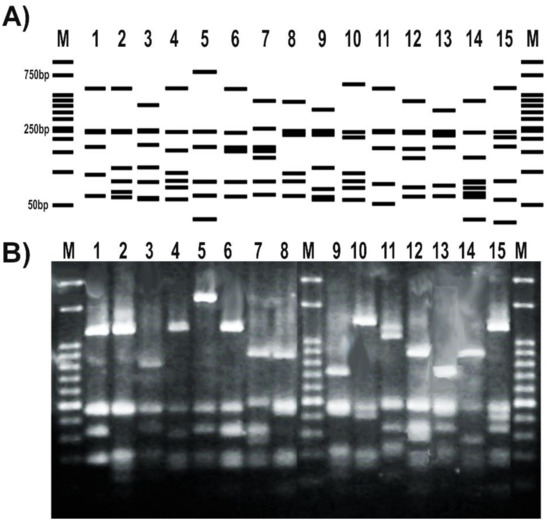
Figure 4.
Set up of a PCR-RFLP (restriction fragment length polymorphism) protocol targeting Pseudomonas gyrB gene. (A) RFLP patterns obtained from the in silico analysis of the amplicons sequenced at Macrogen S.A. and digested with TaqI; (B) RFLP patterns of representatives from our pseudomonads collection. Strains were as follows: 1, P. fluorescens 2–79; 2, P. sp. 1008; 3, P. fluorescens SBW25; 4, P. protegens CHA0; 5, P. simiae WCS417; 6, P. chlororaphis SMMP3; 7, P. syringae pv. syringae DC3000; 8, P. putida KT2440; 9, P. aeruginosa PA01; 10, P. alkylphenolica KL28; 11, P. stutzeri ATCC 17588; 12, P. stutzeri 2018; 13, P. sp. CF5; 14, P. sihuiensis N23; 15, P. donghuensis SVBP6. Markers (M) are 50-bp DNA ladder (Embiotec).
4. Discussion
The gyrB sequence is a useful tool for MLSA in bacteria [4,8,9,10,11] that improves the results obtained with classical approaches, such as DNA–DNA hybridization (technically challenging) or 16S ribosomal DNA (rDNA) gene sequencing (nowadays with a low resolving power for some taxa) [10]. Particularly, it is broadly employed for identification and phylogenetic analyses of members of a complex bacterial genus such as Pseudomonas [7,54,55]. The taxonomy of pseudomonads is continuously being revised, due to the discovery of new species, the reclassification of some of them, or reorganization of groups [7,56,57,58,59,60]. When we looked for a primer pair available in literature to obtain a single gyrB amplicon from diverse Pseudomonas isolates, we noticed that there was not a single one that could be employed for the amplification of representatives of the entire genus, but only specific primers for some species or groups [16,17,18,19]. Besides, when we tested published primer pairs, we failed to obtain a single band suitable for direct sequencing (Figure 1). When those oligonucleotides were designed, they were based on amino acid sequences from different bacterial groups [2,12,13,61], and the availability of gyrB nucleotide sequences was low. Thus, we considered that a new protocol was needed to improve obtaining a unique and specific band from the gyrB gene. A simple and rapid MLSA method could be performed by amplifying and sequencing directly the selected amplicons [10]. Also, a one-step procedure avoids additional investment of laboratory material and person-hours needed to obtain the sequences. Certainly, as many reported primer pairs for MLSA were designed when a much lower number of sequences were available in databases than today, we consider that there might be other housekeeping genes, such as rpoB, rpoD, gyrA or recA, whose sequences may require a similar analysis to eventually upgrade their PCR primers for MLSA, to confirm if the recently described Pseudomonas species are included as targets.
In silico analyses allowed us to find conserved regions of the gyrB gene from available Pseudomonas genomes (Figure 2). The region of the gene on which our reverse primer was designed determines a central role in the enzymatic activity of GyrB, thus explaining the strong sequence conservation (Figure 2B). Amino acid residues in positions 500 and 502, whose reverse complement codons are part of our reverse primer (Figure 2D), bind each one a magnesium cation that acts as cofactor, forming salt bridges with both the protein and the DNA [62]. Based on the characteristics of this sequence region, we selected and designed the forward primer to be compatible with. This methodology allowed us to obtain a robust primer pair that specifically amplify a long gyrB fragment, avoiding additional steps such as gel extraction and purification (Figure 3). Although members of the P. pertucinogena and P. straminea groups were not available in our strain collection (Table 1) to carry out the in vitro tests, both computational tools Amplifx and FastPCR confirmed that this newly designed primer pair amplifies the gyrB sequences from species included in these groups (Figure S2). Moreover, as we targeted strongly conserved regions to design the oligonucleotides, this primer pair could also be used for amplification of gyrB sequences from other Proteobacteria; amplicons with the expected size were obtained in our lab for Escherichia coli K12, Vibrio harveyii BB170, Pantoea spp., Chromobacterium violaceum VIR07 and Salmonella spp. (Figure S3). Polymerase chain reaction coupled with RFLP is a sensitive and economical approach that could be carried out in any molecular biology laboratory. Several PCR-RFLP assays based on other genes have been previously described as a useful and rapid tool to distinguish environmental and clinical isolates of this bacterial genus, and also to analyze pseudomonads communities under different treatments [20,63,64,65,66,67,68]. Here, we demonstrated that the housekeeping gene gyrB is also a suitable target for such kind of comparative taxonomic analyses (Figure 4). This may enhance the applicability of this method as a broad analysis tool, not only for the Pseudomonas group [69].
5. Conclusions
The gyrB gene sequence has become a useful tool for phylogenetic analyses of the Pseudomonas genus as part of MLSA approaches [7,14,60]. Polymerase chain reaction primers available in the literature before this study were designed based on the GyrB amino acid sequences of different bacterial taxa, thus they often result in unspecific amplification or lack of amplicons for some pseudomonas isolates (Figure 1). Here, we present a robust novel pair of primers targeting a larger and more informative region of the gyrB gene with broad amplification success across the pseudomonads clade (Table 2, Figure 2 and Figure S2). We demonstrated in silico that this primer pair is able to amplify the gyrB gene from all the Pseudomonas species described at the moment [41]. In vitro, we confirmed the efficiency and specificity of the novel primer pair and of the PCR protocol for a subset of type strains and isolates from the Pseudomonas genus (Table 1, Figure 3 and Figures S2). In addition, the gyrB amplicons were successfully sequenced from both ends using each of the same primers (Figures S2 and File S1) and served as templates for RFLP assays to distinguish among Pseudomonas species (Figure 4).
Supplementary Materials
The following are available online at http://www.mdpi.com/2409-9279/1/3/24/s1, File S1: gyrB sequences obtained after sequencing the PCR products of the reference pseudomonads (Table 1) representing the main taxonomic groups of this genus, Figure S1. Gradient PCR of gyrB gene, Figure S2. Evolutionary relationships of Pseudomonas species based on the amplicon sequences of the gyrB gene, Supplementary Figure S3. Polymerase chain reaction targeting the gyrB gene of non-pseudomonads strains of our laboratory collection, with the newly designed primers described in this study, Table S1. Sequences of gyrB gene from the Pseudomonas species included in the analysis, Table S2. Coverage and accuracy of the sequencing of gyrB gene from the Pseudomonas strains tested in vitro.
Author Contributions
Conceived and designed the experiments: B.C.A. and C.V. Performed the experiments and wrote the paper: B.C.A. Analyzed the data: B.C.A. Contributed reagents/materials/analysis tools: B.C.A. and C.V. All authors read and approved the final manuscript.
Funding
This work was supported by grants PPUNQ 1411/15 (Universidad Nacional de Quilmes, Argentina) and PICT 2016 N° 4632 (FONCyT, Agencia Nacional de Investigaciones Científicas y Técnicas).
Acknowledgments
We thank Marisa Díaz and Gustavo González Anta (Rizobacter Argentina S.A.), Claudia Casalongué (CONICET-Universidad Nacional de Mar del Plata, Argentina), Alexandre Jousset (Utrecht University, The Netherlands) and Kyoung Lee (Changwon National University, South Korea) for providing us with Pseudomonas sp. 1008, P. syringae DC3000, P. simiae WCS417 and P. alkylphenolica KL28 strains, respectively.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Reece, R.J.; Maxwell, A. DNA Gyrase: Structure and Function. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 335–375. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Kasai, H.; Arnold, D.L.; Jackson, R.W.; Vivian, A.; Harayama, S. Phylogeny of the genus Pseudomonas: Intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 2000, 146 Pt 1, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Huard, R.C.; Fabre, M.; De Haas, P.; Lazzarini, L.C.O.; Van Soolingen, D.; Cousins, D.; Ho, J.L. Novel genetic polymorphisms that further delineate the phylogeny of the Mycobacterium tuberculosis complex. J. Bacteriol. 2006, 188, 4271–4287. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Murcia, A.J.; Monera, A.; Saavedra, M.J.; Oncina, R.; Lopez-Alvarez, M.; Lara, E.; Figueras, M.J. Multilocus phylogenetic analysis of the genus Aeromonas. Syst. Appl. Microbiol. 2011, 34, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Avalos, E.; Catanzaro, D.; Catanzaro, A.; Ganiats, T.; Brodine, S.; Alcaraz, J.; Rodwell, T. Frequency and geographic distribution of gyrA and gyrB mutations associated with fluoroquinolone resistance in clinical Mycobacterium tuberculosis isolates: A systematic review. PLoS ONE 2015, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- Gomila, M.; Peña, A.; Mulet, M.; Lalucat, J.; García-Valdés, E. Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.; Cleenwerck, I.; Venter, S.; Vancanneyt, M.; Swings, J.; Coutinho, T. Phylogeny and identification of Pantoea species associated with plants, humans and the natural environment based on multilocus sequence analysis (MLSA). Syst. Appl. Microbiol. 2008, 31, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Macián, M.C.; Arahal, D.R.; Garay, E.; Pujalte, M.J. Multilocus sequence analysis of the central clade of the genus Vibrio by using the 16S rRNA, recA, pyrH, rpoD, gyrB, rctB and toxR genes. Int. J. Syst. Evol. Microbiol. 2010, 60, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Dawyndt, P.; Coopman, R.; Gillis, M.; De Vos, P.; Willems, A. Advantages of multilocus sequence analysis for taxonomic studies: A case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int. J. Syst. Evol. Microbiol. 2008, 58, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; Park, D.C.; Shearman, H.M.; Fargier, E. A multilocus sequence analysis of the genus Xanthomonas. Syst. Appl. Microbiol. 2008, 31, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Harayama, S. PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains. Appl. Environ. Microbiol. 1995, 61, 1104–1109. [Google Scholar] [PubMed]
- Santos, S.R.; Ochman, H. Identification and phylogenetic sorting of bacterial lineages with universally conserved genes and proteins. Environ. Microbiol. 2004, 6, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Mulet, M.; Lalucat, J.; García-Valdés, E. DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 2010, 12, 1513–1530. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Moreira, I.; Nunes, O.C.; Manaia, C.M. Diversity and antibiotic resistance in Pseudomonas spp. from drinking water. Sci. Total Environ. 2012, 426, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Rico, A.; López, R.; Asensio, C.; Aizpún, M.T.; Asensio-S-Manzanera, M.C.; Murillo, J. Nontoxigenic strains of Pseudomonas syringae pv. phaseolicola are a main cause of halo blight of beans in Spain and escape current detection methods. Phytopathology 2003, 93, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Wetzel, K.; Buckley, T.; Wozniak, D.; Lee, J. Rapid and sensitive detection of Pseudomonas aeruginosa in chlorinated water and aerosols targeting gyrB gene using real-time PCR. J. Appl. Microbiol. 2011, 111, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Brettar, I.; Luo, C.; Auchtung, J.; Konstantinidis, K.T.; Rodrigues, J.L.M.; Höfle, M.; Tiedje, J.M. Stability, genotypic and phenotypic diversity of Shewanella baltica in the redox transition zone of the Baltic Sea. Environ. Microbiol. 2014, 16, 1854–1866. [Google Scholar] [CrossRef] [PubMed]
- Goss, E.M.; Kreitman, M.; Bergelson, J. Genetic diversity, recombination and cryptic clades in Pseudomonas viridiflava infecting natural populations of Arabidopsis thaliana. Genetics 2005, 169, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Agaras, B.C.; Wall, L.G.; Valverde, C. Specific enumeration and analysis of the community structure of culturable pseudomonads in agricultural soils under no-till management in Argentina. Appl. Soil Ecol. 2012, 61, 305–319. [Google Scholar] [CrossRef]
- Schweizer, H.P.; de Lorenzo, V. Molecular Tools for Genetic Analysis of pseudomonads. In Pseudomonas; Springer US: Boston, MA, USA, 2004; pp. 317–350. [Google Scholar]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.C.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed]
- Mulet, M.; Bennasar, A.; Lalucat, J.; García-Valdés, E. An rpoD-based PCR procedure for the identification of Pseudomonas species and for their detection in environmental samples. Mol. Cell. Probes 2009, 23, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Andreani, N.A.; Martino, M.E.; Fasolato, L.; Carraro, L.; Montemurro, F.; Mioni, R.; Bordin, P.; Cardazzo, B. Reprint of ’Tracking the blue: A MLST approach to characterise the Pseudomonas fluorescens group. Food Microbiol. 2015, 45, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, J.A.; Morrissey, J.P.; Coppoolse, E.R.; Stiekema, W.J.; Gara, F.O.; Boyd, E.F. Evolutionary History of the phl Gene Cluster in the Plant-Associated Bacterium Pseudomonas fluorescens. Appl. Environ. Microbiol. 2009, 75, 2122–2131. [Google Scholar] [CrossRef] [PubMed]
- Pezza, R.J.; Smania, A.M.; Barra, J.L.; Argaraña, C.E. Nucleotides and heteroduplex DNA preserve the active conformation of Pseudomonas aeruginosa MutS by preventing protein oligomerization. Biochem. J. 2002, 361, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Holloway, B.W. Genetics of Pseudomonas. Bacteriol. Rev. 1969, 33, 419–443. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.J.; Kim, J.H.; Kim, C.K.; Hwang, I.; Lee, K. 3- and 4-alkylphenol degradation pathway in Pseudomonas sp. strain KL28: Genetic organization of the lap gene cluster and substrate specificities of phenol hydroxylase and catechol 2,3-dioxygenase. Microbiology 2003, 149, 3265–3277. [Google Scholar] [CrossRef] [PubMed]
- Agaras, B.C.; Scandiani, M.; Luque, A.; Fernández, L.; Farina, F.; Carmona, M.; Gally, M.; Romero, A.; Wall, L.G.; Valverde, C. Quantification of the potential biocontrol and direct plant growth promotion abilities based on multiple biological traits distinguish different groups of Pseudomonas spp. isolates. Biol. Control 2015, 90, 173–186. [Google Scholar] [CrossRef]
- Weller, D.M.; Cook, R.J. Suppression of Take-All of wheat by seed treatments with fluorescent Pseudomonads. Phytopathology 1983, 73, 463–469. [Google Scholar] [CrossRef]
- Bailey, M.J.; Lilley, A.K.; Thompson, I.P.; Rainey, P.B.; Ellis, R.J. Site directed chromosomal marking of a fluorescent pseudomonad isolated from the phytosphere of sugar beet; stability and potential for marker gene transfer. Mol. Ecol. 1995, 4, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Stutz, E.W.; Défago, G.; Kern, H. Naturally ocurring fluorescent pseudomonads involved in supression of black root rot of tobacco. Phytopathology 1986, 76, 181–185. [Google Scholar] [CrossRef]
- Howell, C.R.; Stipanovic, R.D. Control of Rhizoctonia solani on cotton seedlings with Pseudomonas fluorescens and with an antibiotic produced by the bacterium by the soil tube method described previously. Phytopathology 1979, 69, 480–482. [Google Scholar] [CrossRef]
- Stanier, R.Y.; Palleroni, N.J.; Doudoroff, M. The aerobic pseudomonads: A taxonomic study. J. Gen. Microbiol. 1966, 43, 159–271. [Google Scholar] [CrossRef] [PubMed]
- Lifshitz, R.; Kloepper, J.W.; Scher, F.M.; Tipping, E.M.; Laliberté, M. Nitrogen-fixing pseudomonads isolated from roots of plants grown in the canadian high arctic. Appl. Environ. Microbiol. 1986, 51, 251–255. [Google Scholar] [PubMed]
- Bagdasarian, M.M.; Lurz, R.; Rückert, B.; Franklin, F.C.; Bagdasarian, M.M.; Frey, J.; Timmis, K.N. Specific-purpose plasmid cloning vectors II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 1981, 16, 237–247. [Google Scholar] [CrossRef]
- Ordoñez, O.F.; Flores, M.R.; Dib, J.R.; Paz, A.; Farías, M.E. Extremophile culture collection from Andean lakes: extreme pristine environments that host a wide diversity of microorganisms with tolerance to UV radiation. Microb. Ecol. 2009, 58, 461–73. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, R.L.; van Verk, M.C.; Stringlis, I.A.; Zamioudis, C.; Tommassen, J.; Pieterse, C.M.J.; Bakker, P.A.H.M. Unearthing the genomes of plant-beneficial Pseudomonas model strains WCS358, WCS374 and WCS417. BMC Genomics 2015, 16, 539. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.R.; Schott, E.; Ausubel, F.M. Virulence of selected phytopathogenic pseudomonads in Arabidopsis thaliana. MPMI 1991, 4, 477–488. [Google Scholar] [CrossRef]
- Cuppels, D.M. Generation and characterization of Tn5 insertion mutations in Pseudomonas syringae pv. tomato. Appl. Environ. Microbiol. 1986, 51, 323–327. [Google Scholar] [PubMed]
- Parte, A.C. LPSN—List of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014, 42, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Jullien, N. AmplifX Version 1.7.0. Available online: http://crn2m.univ-mrs.fr/pub/amplifx-dist (accessed on 25 June 2018).
- Kalendar, R.; Lee, D.; Schulman, A.H. Java web tools for PCR, in silico PCR, and oligonucleotide assembly and analyses. Genomics 2011, 98, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Khassenov, B.; Ramankulov, Y.; Samuilova, O.; Ivanov, K.I. FastPCR: An in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 2017, 109, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Tselykh, T.V.; Khassenov, B.; Ramanculov, E.M. Introduction on Using the FastPCR Software and the Related Java Web Tools for PCR and Oligonucleotide Assembly and Analysis; Springer: New York, NY, USA, 2017; pp. 33–64. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Perez, F. Serial Cloner 2.6.1. Available online: http://serialbasics.free.fr/Serial_Cloner.html (accessed on 25 June 2018).
- Frackman, B.S.; Kobs, G.; Simpson, D.; Storts, D. Betaine and DMSO: Enhancing Agents for PCR. Promega Notes 1998, 65, 9–12. [Google Scholar]
- Wu, M.; Wen, J.; Chang, M.; Yang, G.; Zhou, S. Pseudomonas sihuiensis sp. nov., isolated from a forest soil in South China. Antonie van Leeuwenhoek. Int. J. Gen. Mol. Microbiol. 2014, 105, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.; Hon, G.; Chandonia, J.; Brenner, S. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Frasson, D.; Opoku, M.; Picozzi, T.; Torossi, T.; Balada, S.; Smits, T.H.M.; Hilber, U. Pseudomonas wadenswilerensis sp. nov. and Pseudomonas reidholzensis sp. nov., two new species within the Pseudomonas putida group isolated from forest soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 2853–2861. [Google Scholar] [CrossRef] [PubMed]
- Mulet, M.; Gomila, M.; Scotta, C.; Sánchez, D.; Lalucat, J.; García-Valdés, E. Concordance between whole-cell matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry and multilocus sequence analysis approaches in species discrimination within the genus Pseudomonas. Syst. Appl. Microbiol. 2012, 35, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Peix, A.; Valverde, A.; Rivas, R.; Igual, J.M.; Ramírez-Bahena, M.-H.; Mateos, P.F.; Santa-Regina, I.; Rodríguez-Barrueco, C.; Martínez-Molina, E.; Velázquez, E. Reclassification of Pseudomonas aurantiaca as a synonym of Pseudomonas chlororaphis and proposal of three subspecies, P. chlororaphis subsp. chlororaphis subsp. nov., P. chlororaphis subsp. aureofaciens subsp. nov. Int. J. Syst. Evol. Microbiol. 2007, 57, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Peix, A.; Ramírez-Bahena, M.-H.; Velázquez, E. Historical evolution and current status of the taxonomy of genus Pseudomonas. Infect. Genet. Evol. 2009, 9, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Mulet, M.; Gomila, M.; Gruffaz, C.; Meyer, J.-M.; Palleroni, N.J.; Lalucat, J.; García-Valdés, E. Phylogenetic analysis and siderotyping as useful tools in the taxonomy of Pseudomonas stutzeri: Description of a novel genomovar. Int. J. Syst. Evol. Microbiol. 2008, 58, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Sanz, D.; Meier-Kolthoff, J.P.; Göker, M.; Martín, M.; Rivilla, R.; Redondo-Nieto, M. Genomic and genetic diversity within the Pseudomonas fluoresces complex. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Peix, A.; Ramírez-Bahena, M.H.; Velázquez, E. The current status on the taxonomy of Pseudomonas revisited: An update. Infect. Genet. Evol. 2018, 57, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Cladera, A.M.; Bennasar, A.; Barceló, M.; Lalucat, J.; García-valdés, E.; Cladera, A.M.; Bennasar, A.; Barcelo, M.; Lalucat, J.; García-valde, E. Comparative genetic diversity of Pseudomonas stutzeri genomovars, clonal structure, and phylogeny of the species. J. Bacteriol. 2004, 186, 5239–5248. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Wu, J.; Liu, W.; Zhu, D.; Hu, Y.; Deng, J.; Zhang, X.-E.; Bi, L.; Wang, D.-C. Crystal structure of DNA gyrase B’ domain sheds lights on the mechanism for T-segment navigation. Nucleic Acids Res. 2009, 37, 5908–5916. [Google Scholar] [CrossRef] [PubMed]
- Lagares, A.; Agaras, B.; Bettiol, M.P.; Gatti, B.M.; Valverde, C. A cultivation-independent PCR-RFLP assay targeting oprF gene for detection and identification of Pseudomonas spp. in samples from fibrocystic pediatric patients. J. Microbiol. Methods 2015, 114, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Manceau, C.; Horvais, A. Assessment of genetic diversity among strains of Pseudomonas syringae by PCR-restriction fragment length polymorphism analysis of rRNA operons with special emphasis on P. syringae pv. tomato. Appl. Environ. Microbiol. 1997, 63, 498–505. [Google Scholar] [PubMed]
- Pesaro, M.; Widmer, F. Identification and specific detection of a novel pseudomonadaceae cluster associated with soils from winter wheat plots of a long-term agricultural field experiment. Appl. Environ. Microbiol. 2006, 72, 37–43. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.T.; Raaijmakers, J.M. Polymorphisms within the prnD and pltC genes from pyrrolnitrin and pyoluteorin-producing Pseudomonas and Burkholderia spp. FEMS Microbiol. Ecol. 2003, 43, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Agaras, B.C.; Wall, L.G.; Valverde, C. Influence of agricultural practices and seasons on the abundance and community structure of culturable pseudomonads in soils under no-till management in Argentina. Plant Soil 2014, 382, 117–131. [Google Scholar] [CrossRef]
- Fernández, L.A.; Agaras, B.; Wall, L.G.; Valverde, C. Abundance and ribotypes of phosphate-solubilizing bacteria in Argentinean agricultural soils under no-till management. Ann. Microbiol. 2015, 65. [Google Scholar] [CrossRef]
- Wang, L.T.; Lee, F.L.; Tai, C.J.; Kasai, H. Comparison of gyrB gene sequences, 16S rRNA gene sequences and DNA-DNA hybridization in the Bacillus subtilis group. Int. J. Syst. Evol. Microbiol. 2007, 57, 1846–1850. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).