The Making of Transgenic Drosophila guttifera


Abstract
:
1. Introduction
2. Experimental Design
2.1. Required Materials and Equipment
2.1.1. Materials
- Plexiglas egg-laying cage measuring 300 mm × 200 mm × 200 mm
- Lamp
- Instant yeast (Instaferm)
- Sponges
- Medium-sized Petri dishes (100 mm × 15 mm)
- 1-L plastic beakers for wet chambers
- 2-L plastic jar
- 5-L beaker
- Egg collection filter with Corning Netwell insert (fine-filter, New York, NY, USA)
- Egg collection filter with Corning Netwell insert (coarse-filter, New York, NY, USA)
- Squeeze bottle
- Micro cover glass (18 mm × 18 mm)
- Microslide (25 mm × 75 mm, 1 mm thick)
- Small brush (to move egg masses)
- Very fine brush (to line up individual eggs)
- Halocarbon oil 700 (Sigma-Aldrich, cat. no. H8898, Albany, NY, USA)
- Halocarbon oil 27 (Sigma-Aldrich, cat. no. H8773, Albany, NY, USA)
- 10-mL syringe
- 50-mL Falcon tube
- FHC capillary tube (Borosil 1.0 mm × 0.75 mm ID/Fiber with Omega dot fiber)
- Nitrogen gas
- CO2 gas
- Fly food vials
- Cornmeal-sucrose-yeast medium (fly food) [10]
- Aluminum foil
- Two pairs of forceps
- Spatula
- Cotton plugs
- Moist chamber
- Anti-fungus paper (see Section 5.1)
2.1.2. Equipment
- Flaming/Brown micropipette puller Model P-97 (Sutter Instruments, Novato, CS, USA)
- Microinjector (Narishige IM 300, Amityville, NY, USA)
- Needle-holder (Narishige, Amityville, NY, USA)
- Micromanipulator (Narishige, Amityville, NY, USA)
- Inverted microscope (Olympus CKX31, Center Valley, PA, USA)
- Dissecting microscope
- Water bath (Thermo Scientific, HAAK S3, Waltham, MA, USA)
- Mercury burner (Olympus U-RFL-T, Center Valley, PA, USA)
- Imaging microscope (Olympus SZX16, Center Valley, PA, USA) with fluorescence filters for GFP and DsRed
3. Methods
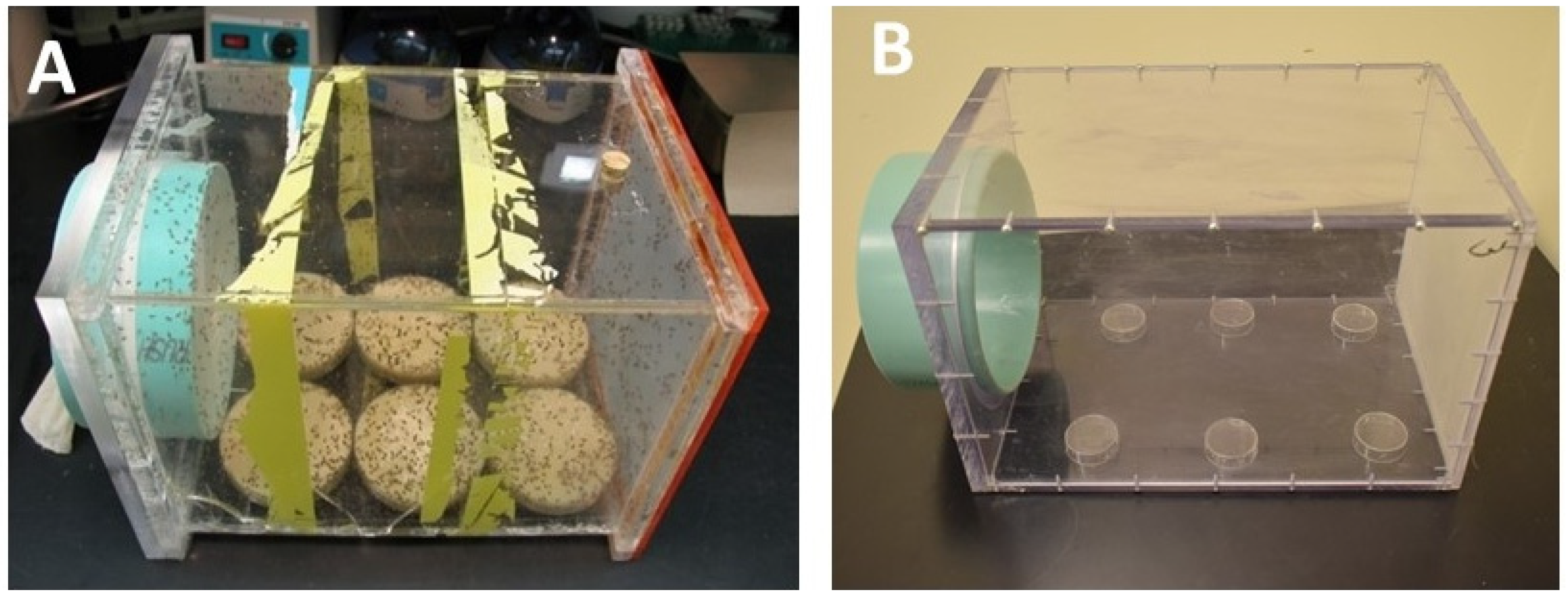
3.1. The Egg-Laying Cage
- (1)
- Knock the plate gently against the round Petri dish “table” to remove any flies before taking out a plate. Use the thumb and middle finger to hold the dish, while holding the index finger in place to prevent the food from falling out.
- (a)
- If the gap between the food and the Petri dish is large due to excessive drying of the food, then bump (knock the flies) towards the large gap. The food will not move much downwards, and no fly will be trapped.
- (b)
- If the gap is narrow, bump towards the side where the food still sticks to the plastic. This prevents most of the flies in the gap from being squeezed to death.
- (2)
- One out, one in! Always take only one plate out at a time and replace immediately with a fresh plate. If there are flies on the food “table” in the cage, they can be gently pushed away with the front edge of the fresh food plate when putting it down. Then repeat this step with the remaining plates.
- (3)
- Freeze and discard the used food as the flies developing from it will be of poor quality and a source of fungus infestations.
- (1)
- Take out all cornmeal-sucrose-yeast medium plates, close them, and continue working at a fly bench with CO2 gas.
- (2)
- Cover an area with unfolded paper towels to your right and put a 5-L beaker on it.
- (3)
- Have a new cage with a fresh entrance ring plus nylon ready.
- (4)
- Put the populated cage to the left of the new cage (not on the towels), the entrance facing upwards to prevent CO2 from leaking out of the cage through the back wall.
- (5)
- Open the CO2 gas, lead it through the nylon to anesthetize the flies, and then close the CO2 gas supply.
- (6)
- Remove the ring with the nylon of the old cage and pour the flies into the 5-L beaker.
- (7)
- Flies that fall on the towels can now be thrown into the beaker as well.
- (8)
- Gas the old cage again to blow loose the remaining living flies and shake the cage over the beaker.
- (9)
- Pour the flies from the beaker into the new cage.
- (10)
- Close the freshly filled cage, bring it to its designed place in the lab and supply the flies with food.
- (11)
- Clean the old cage and the nylon with tap water. Do not use soap or bleach. Rinse with distilled water.
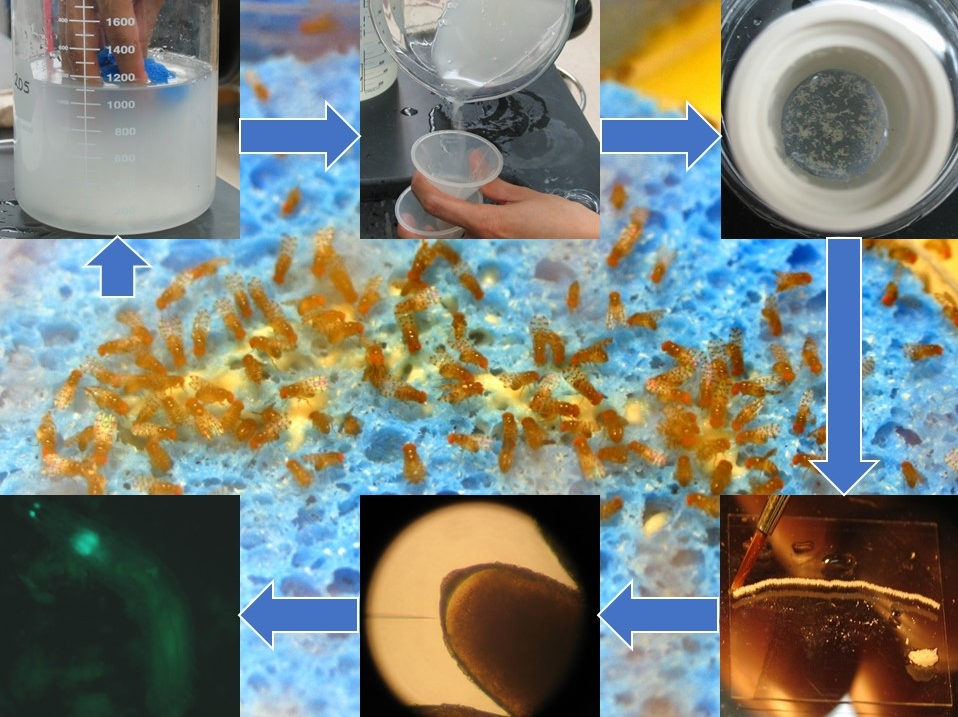
3.2. Egg Collection and Preparation for Microinjections
-
![Mps 03 00031 i001]()
- CRITICAL STEPS: Never wash D. guttifera eggs with ethanol, because it seems to harden the chorion and prevents the injection needles from penetrating it, and never leave the cage without food.

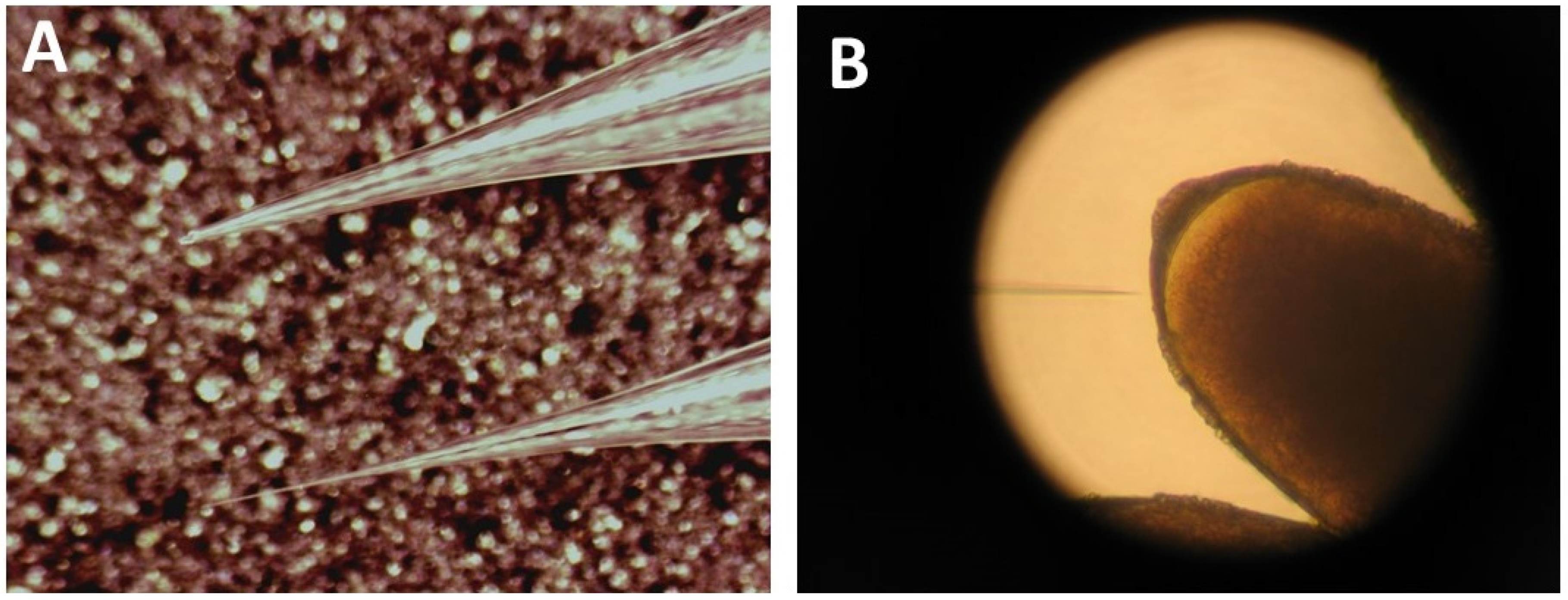
3.3. Lining up the Eggs for Microinjections
-
![Mps 03 00031 i001]()
- CRITICAL STEPS: If the eggs become too desiccated (oil added too late), they die and become deformed when the needle pokes against them. If, however, the oil is put on too early, the eggs will not stick to the glass and move away when the injection needle is poked against them. Only a few seconds lay between both extremes. Do not try to inject a slide with swimming or dead eggs. Instead, save your needles for a better slide.
-
![Mps 03 00031 i001]()
- CRITICAL STEPS: Note the following important timelines: egg-laying in the cage = 1 h, egg collection and lining up = 20 min, injecting one slide containing about 100 eggs = 15 min. It is important to ensure that the eggs are in the syncytial blastoderm stage during injection, which lasts for about 2 h or less at room temperature.
3.4. Preparation of Transgene DNA
- (1)
- Follow the manufacturer’s instructions on the isolation of the plasmid DNA containing the transgene of interest from an overnight Escherichia coli culture. In this described experiment, we use the Invitrogen HiPure Plasmid Midi-Prep Kit by Thermo Fisher Scientific (Waltham, MA, USA).
- (2)
- Add 3.5 mL of isopropanol to the eluted DNA in a Corex glass vial.
- (3)
- Close the Corex vial with Parafilm, invert 10 times, take away the Parafilm, and wipe off any drops with Kimwipes.
- (4)
- Centrifuge at 11,000 rpm for 30 min at 4 °C.
- (5)
- Mark the area of the DNA pellet with a marker, decant, and invert the vial on a paper towel.
- (6)
- Add 350 µL of sterile MQ water and dissolve the DNA pellet by vortexing.
- (7)
- Collect the plasmid DNA in a 1.5-mL Eppendorf tube.
- (8)
- Add 35 µL of 3 M of sodium acetate (pH 5.5).
- (9)
- Add 875 µL of 200-proof ethanol and invert 10 times.
- (10)
- Spin the DNA at 14,000 rpm for 20 min at 4 °C on a bench-top centrifuge.
- (11)
- Remove supernatant and wash with 300 µL of 70% ethanol by inverting the tube 10 times.
- (12)
- Centrifuge again at 14,000 rpm for 10 min at room temperature.
- (13)
- Remove supernatant and centrifuge for 1 min.
- (14)
- Remove all ethanol and dry the pellet for 5 min.
- (15)
- Dissolve the DNA in 50–100 µL of elution buffer, measure the DNA concentration, and add more elution buffer if necessary, to a final concentration of 1 µg/µL.
3.5. Preparation of the Injection Cocktail
- (1)
- On ice, add 20 µL of transgene-containing piggyBac construct (1 µg/µL) to a 1.5-mL test tube, 5 µL of the piggyBac helper plasmid (phspBac ([1 µg/µL)), and 15 µL of sterile distilled water. Note: We do not add food color, because it is not necessary for the visualization of the injected material.
- (2)
- Centrifuge the DNA mixture for 20 min at maximum speed, and transfer the upper 39 µL of it to a fresh tube (do not touch the bottom of the tube with the pipette tip to prevent transferring undissolved material that would clog the needle). Spin the DNA again for 20 min and transfer 38 µL of the injection mix into another fresh tube. This DNA is now clear of debris and can be used to load the needles. The piggyBac system pBac (backbone)/phspBac (piggyBac helper) results in 1 transformation event every 50 D. guttifera eggs with an empty vector and 1 every 500 eggs with an 8-kb insert [18].
3.6. Needle Preparation and Microinjections
-
![Mps 03 00031 i001]()
- CRITICAL STEP: The needles must be changed when the eggs start leaking out a lot of granular cytoplasm, whereas small clear droplets are acceptable and normal.
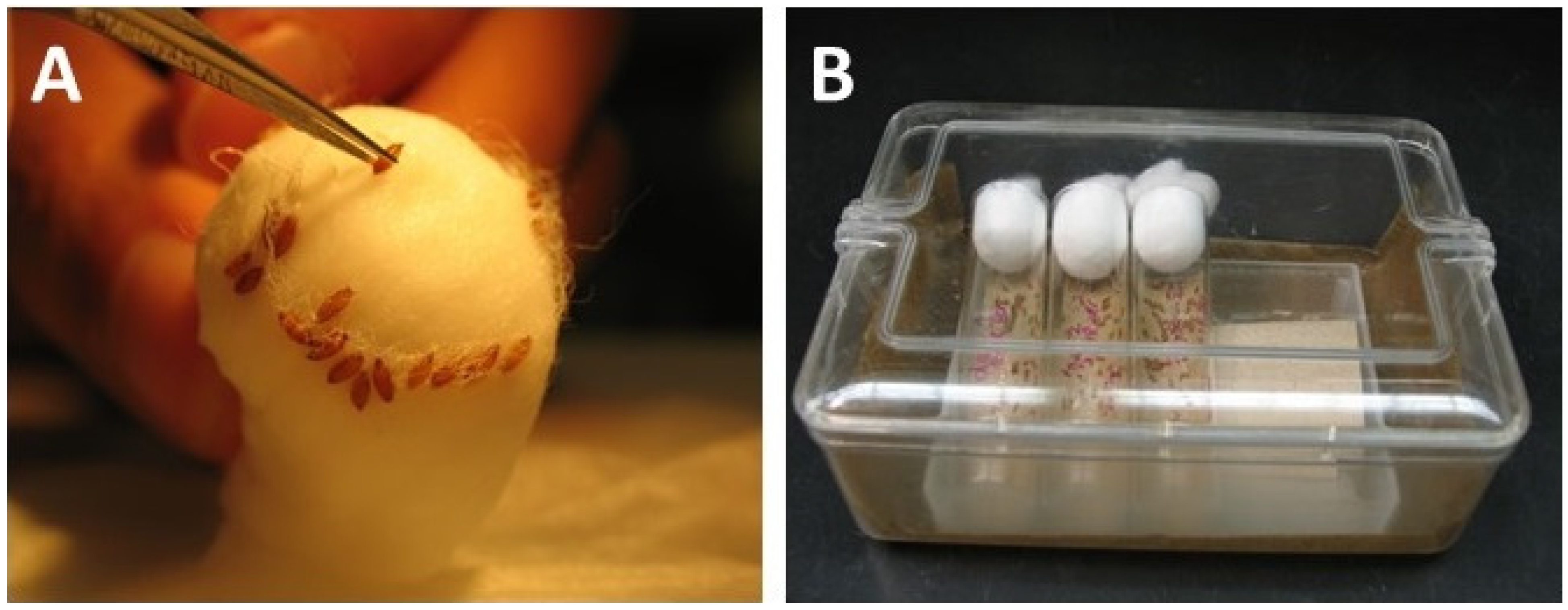
3.7. Post-Injection Treatment and Heat Shock of the Eggs
3.8. After-Care and Fly Crosses
-
![Mps 03 00031 i001]()
- CRITICAL STEP: Moisten the anti-fungus paper inside the glass vials whenever needed, and never let the anti-fungal paper dry out. Check the chamber twice daily.
3.9. Screening for Transgenic Larvae
- (1)
- Switch on the UV lamp 10 min prior to screening.
- (2)
- Microscope settings: light filter = “GFP”, bottom filter = “Oblique”, objective = 1× shutter open, camera tract closed, and all visible microscope lights off (check that the bottom light is off as well!).
- (3)
- Repeat the following steps until all vials have been screened:
- (a)
- Remove the larvae from a food vial by squirting distilled water (squeeze bottle) and stirring with a rough brush; then, pour the larva/food soup into a medium-sized Petri dish bottom half.
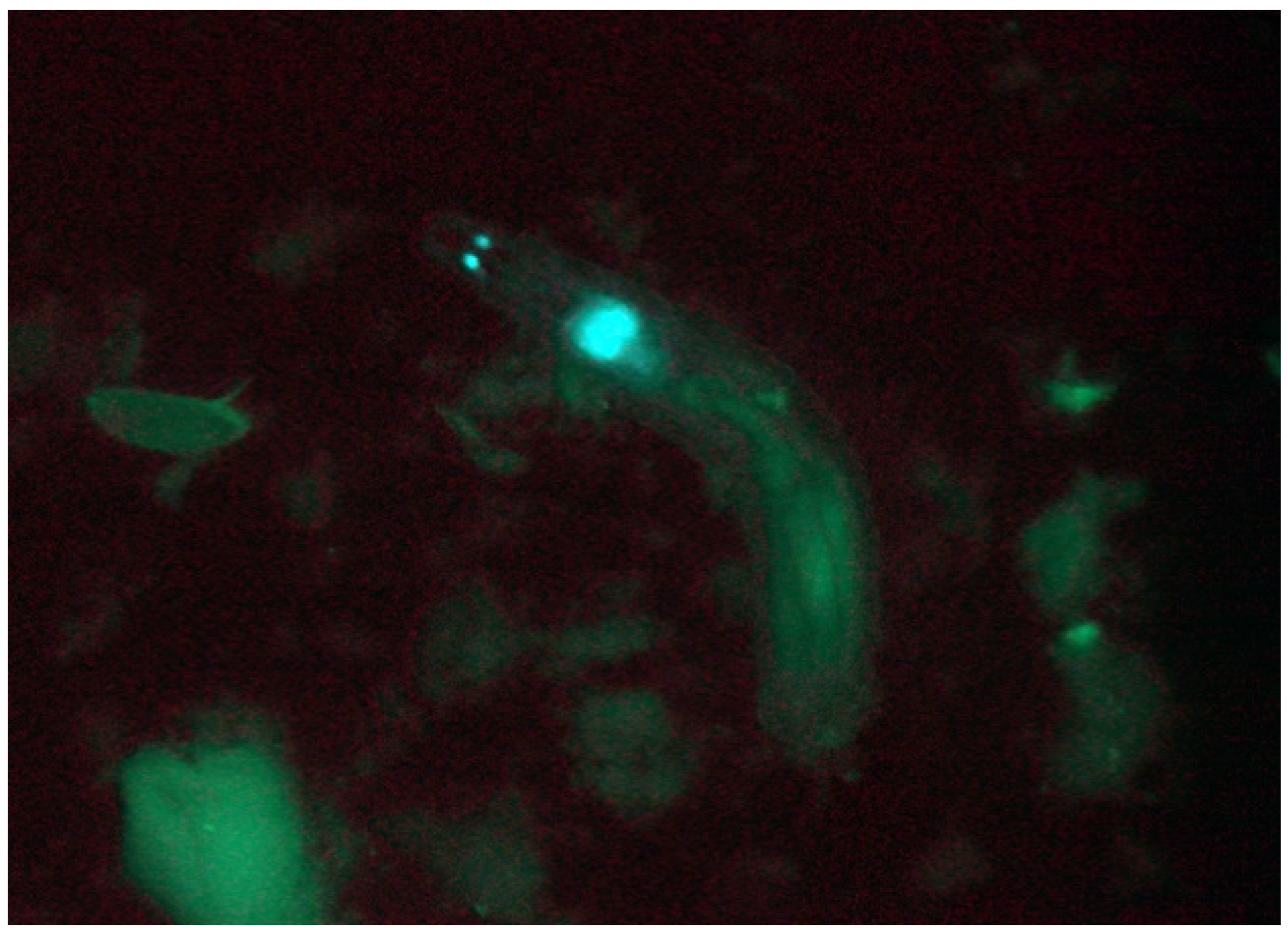
- (b)
- Collect positive larvae (Figure 8) with a pair of forceps into 2-mL tubes filled with 1-mL cornmeal-sucrose-yeast medium (briefly angle the food by centrifugation, allowing the larvae to be removed more easily from the collection forceps). Wash and screen each vial 3 times for transgenic larvae.
- (c)
- Poke two holes into each lid (not too big that the larvae can escape) after putting larvae into the 2-mL food tubes (only add up to 10 larvae per vial). Label each vial with the construct and line information.
- (d)
- Discard screening soup with negative larvae and food debris into an empty 5-L beaker. Rinse the Petri dish with distilled water after screening each vial.
4. Expected Results
5. Reagents
5.1. Anti-Fungus Paper
5.2. Halocarbon Oil Mixture
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rubin, G.M.; Spradling, A.C. Genetic transformation of Drosophila with transposable element vectors. Science 1982, 218, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaria, P.; Roberts, D. Drosophila: A Practical Approach; IRL Press: Oxford, UK, 1986. [Google Scholar]
- Handler, A.M.; James, A.A. Insect Transgenesis: Methods and Applications; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Venken, K.J.; He, Y.; Hoskins, R.A.; Bellen, H.J. P[acman]: A BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 2006, 314, 1747–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, M.P.; Groth, A.C.; Calos, M.P.; Nusse, R. Creating transgenic Drosophila by microinjecting the site-specific φC31 integrase mRNA and a transgene-containing donor plasmid. Nat. Protoc. 2007, 2, 2325. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.J.; Bellen, H.J. Transgenesis upgrades for Drosophila melanogaster. Development 2007, 134, 3571–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, A.C.; Fish, M.; Nusse, R.; Calos, M.P. Construction of transgenic Drosophila by using the site-specific integrase from phage φC31. Genetics 2004, 166, 1775–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, B.G.; Weeks, D.L. Transgenic Xenopus laevis embryos can be generated using φC31 integrase. Nat. Methods 2005, 2, 975–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmo, D.; Alphey, L.; Meredith, J.; Eggleston, P. High efficiency site-specific genetic engineering of the mosquito genome. Insect Mol. Boil. 2006, 15, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, T.; Steenwinkel, T.; Jaenike, J. Drosophilids of the Midwest and Northeast; Michigan Technological University: Houghton, MI, USA, 2018. [Google Scholar]
- Koshikawa, S.; Fukutomi, Y.; Matsumoto, K. Drosophila guttifera as a model system for unraveling color pattern formation. In Diversity and Evolution of Butterfly Wing Patterns; Springer: Berlin/Heidelberg, Germany, 2017; pp. 287–301. [Google Scholar]
- Werner, T.; Koshikawa, S.; Williams, T.M.; Carroll, S.B. Generation of a novel wing colour pattern by the Wingless morphogen. Nature 2010, 464, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, S. Evolution of wing pigmentation in Drosophila: Diversity, physiological regulation, and cis-regulatory evolution. Dev. Growth Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, S.; Giorgianni, M.W.; Vaccaro, K.; Kassner, V.A.; Yoder, J.H.; Werner, T.; Carroll, S.B. Gain of cis-regulatory activities underlies novel domains of wingless gene expression in Drosophila. Proc. Natl. Acad. Sci. USA 2015, 112, 7524–7529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukutomi, Y.; Matsumoto, K.; Agata, K.; Funayama, N.; Koshikawa, S. Pupal development and pigmentation process of a polka-dotted fruit fly, Drosophila guttifera(Insecta, Diptera). Dev. Genes Evol. 2017, 227, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Mollinari, C.; González, C. Microinjection of Drosophila Eggs. In Microinjection and Transgenesis; Springer: Berlin/Heidelberg, Germany, 1998; pp. 587–603. [Google Scholar]
- Raja, K.K.; Shittu, M.O.; Nouhan, P.M.; Steenwinkel, T.E.; Bachman, E.A.; Kokate, P.P.; McQueeney, A.H.; Mundell, E.A.; Armentrout, A.A.; Peabody, A.M.; et al. The regulation of a pigmentation gene in the formation of complex color patterns in Drosophila abdomens. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Horn, C.; Wimmer, E.A. A versatile vector set for animal transgenesis. Dev. Genes Evol. 2000, 210, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.F.; Holtzman, S.L.; Kaufman, T.C. Customized microinjection glass capillary needles for P-element transformations in Drosophila melanogaster. Biotechniques 2002, 33, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, C.; Jaunich, B.; Wimmer, E.A. Highly sensitive, fluorescent transformation marker for Drosophila transgenesis. Dev. Genes Evol. 2000, 210, 623–629. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Problem | Reason | Solution |
|---|---|---|
| Few larvae hatch. | Bad injection needles. | Make sure you use standard needles. Strictly follow the instructions under Section 3.6. |
| Over-desiccation of eggs during lining up. | Add injection oil within 3–5 s after eggs appear completely dry under the microscope. | |
| Toxic DNA. | Use highly pure DNA (no mini-preps). Additional isopropanol precipitation after the first extraction of DNA is necessary. | |
| Too-high heat shock temperature. | The heat shock temperature for D. guttifera transformation is 40 °C; however, if this temperature is causing strong reduction in the survival rate, reduce the temperature by 1–2 degrees but never go below 37 °C. | |
| If the Halocarbon oil mixture is suffocating and killing the eggs. This does not normally happen, but we have seen this problem in our lab. | Use Halocarbon oil 27 only or GEM extra virgin olive oil. We are using extra virgin olive oil in our lab with high success. | |
| Few surviving adults. | Having only a few injection-surviving larvae causes bacterial growth and/or mold formation in the food. | Ensure a high injection survival rate. At least 10–20 larvae can keep the food mold-free. |
| Desiccation of pupae due to dry chamber and/or dry cotton plugs. | Always keep the chamber and the cotton plugs | |
| No transgenic larvae. | Poor DNA quality. | Always clean the DNA by isopropanol precipitation or prepare new DNA samples. |
| Insufficient heat shock temperature. | Always ensure the heat shock temperature is not below 37 °C. Check the temperature of your water bath with a calibrated thermometer. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shittu, M.; Steenwinkel, T.; Koshikawa, S.; Werner, T. The Making of Transgenic Drosophila guttifera. Methods Protoc. 2020, 3, 31. https://doi.org/10.3390/mps3020031
Shittu M, Steenwinkel T, Koshikawa S, Werner T. The Making of Transgenic Drosophila guttifera. Methods and Protocols. 2020; 3(2):31. https://doi.org/10.3390/mps3020031
Chicago/Turabian StyleShittu, Mujeeb, Tessa Steenwinkel, Shigeyuki Koshikawa, and Thomas Werner. 2020. "The Making of Transgenic Drosophila guttifera" Methods and Protocols 3, no. 2: 31. https://doi.org/10.3390/mps3020031
APA StyleShittu, M., Steenwinkel, T., Koshikawa, S., & Werner, T. (2020). The Making of Transgenic Drosophila guttifera. Methods and Protocols, 3(2), 31. https://doi.org/10.3390/mps3020031