


Analysis of Processing, Post-Maturation, and By-Products of shRNA in Gene and Cell Therapy Applications


Abstract
1. Introduction
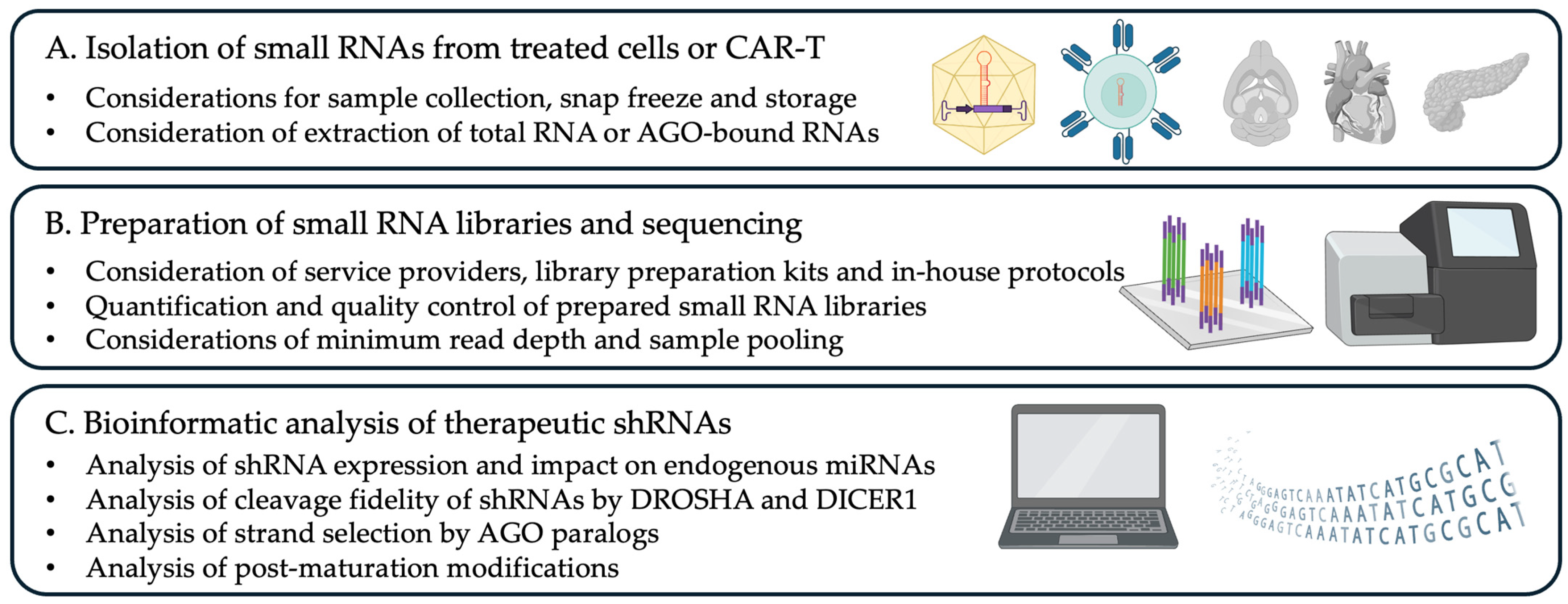
2. Experimental Design
2.1. Materials
- TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA, USA; Cat. no.: 15596026)
- 2.
- QIAseq miRNA Library Kit (Qiagen, Germany, Cat. no.: 331502)
- 3.
- RNA 5′ Adapter (RA5) (7.5 μM)
- 4.
- RNA 3′ Adapter (RA3) (5 μM)
- 5.
- RT Primer
- 6.
- PCR5 Primer
- 7.
- Stop solution (STP) (15 μM)
- 8.
- PCR3 Primer
- 9.
- 10× T4 RNA Ligase 1 buffer (NEB, Ipswich, MA, USA, Cat. no.: M0204L)
- 10.
- RNase Inhibitor (NEB, Ipswich, MA, USA, Cat. no.: M0314L)
- 11.
- T4 RNA Ligase 2, Deletion Mutant (NEB, Ipswich, MA, USA, Cat. no.: M0242S)
- 12.
- Adenosine 5′-Triphosphate (ATP) (NEB, Ipswich, MA, USA, Cat. no.: P0756S)
- 13.
- T4 RNA Ligase (NEB, Ipswich, MA, USA, Cat. no.: M0437M)
- 14.
- SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no.: 18091050)
- 15.
- Phusion High-Fidelity DNA Polymerase (NEB, Ipswich, MA, USA, Cat. no.: M0530L)
- 16.
- RNAlater (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no.: AM7020)
- 17.
- Blue Juice Loading Buffer (10×) (Thermo Fisher, Waltham, MA, USA, Cat. no.: 10816015)
2.2. Equipment
- NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA; Cat. no.: ND-ONE-W)
- Bioanalyzer (Agilent, Santa Clara, CA, USA; Cat. no.: G2939BA)
- Safe Imager 2.0 (Thermo Fisher Scientific, Waltham, MA, USA; Cat. no.: G6600EU)
- ChemiDoc (BioRad, Hercules, CA, USA; Cat. no.: OI91XQ15)
3. Procedure
 CRITICAL STEP: To ensure high quality and reproducibility, all steps during RNA isolation and library preparation must be conducted using sterile, nuclease-free materials. Work with ice-chilled tubes and reagents to prevent RNA degradation and maintain the integrity of small RNA species throughout the process.
CRITICAL STEP: To ensure high quality and reproducibility, all steps during RNA isolation and library preparation must be conducted using sterile, nuclease-free materials. Work with ice-chilled tubes and reagents to prevent RNA degradation and maintain the integrity of small RNA species throughout the process.3.1. Isolation of Small RNAs
CRITICAL STEP: Quality control analysis of the extracted RNA can be performed using a bioanalyzer (Agilent) according to the manufacturer’s protocol. A minimum RNA Integrity Number (RIN) of 8 or higher is expected, indicating high RNA quality. This step also allows for the detection and evaluation of the small RNA fraction present in the sample, ensuring the sample is suitable for downstream applications. CRITICAL STEP: Although small RNAs are generally well preserved, degradation of larger RNA molecules caused by repeated freeze–thaw cycles or nuclease contamination can compromise the overall quality of the library. Ensuring proper handling and storage is critical to maintaining RNA integrity and achieving reliable results. PAUSE STEP: After isolation, the RNA can be stored at −70 °C for extended periods to ensure its stability and integrity.
PAUSE STEP: After isolation, the RNA can be stored at −70 °C for extended periods to ensure its stability and integrity.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Advantages | Disadvantages |
|---|---|---|
| Total RNA (TRIzol) | High yield of RNA | Potential carryover contaminants |
| Total RNA (Spin Column) | High yield of RNA | Potential sequence biases * |
| Reduced carryover of contaminants | ||
| AGO immunoprecipitation | Directly addresses functional AGO-bound small RNAs | Low yield of RNA |
| Increased PCR duplicates | ||
| TraPR (Separation Column) | Retention of AGO-RISC complexes | Retention of other RNA- |
| Easy to use | binding protein complexes |
3.2. Preparation of Small RNA Libraries
CRITICAL STEP: There are multiple commercial kits and customized protocols for the preparation of small RNA libraries for NGS. Recent studies have benchmarked biases and strengths of each method (see [21,25]). In addition, many commercial providers and genomic core facilities provide small RNA sequencing as standard service.3.2.1. Ligation of 3′ Adapter
- Mix 1 μL of 5 μM RNA 3′ Adapter (RA3) with 5 μg of total RNA (starting material can range between 5 ng and 1 μg) in a final volume of 7 μL of nuclease-free water.
- Mix the solution well and incubate the tube at 70 °C for 2 min, and then immediately place the tube on ice.
- Prepare a mix containing:
- 1 μL of 10× T4 RNA ligation buffer,
- 1 μL of RNase Inhibitor,
- 1 μL of T4 RNA Ligase 2 Deletion Mutant,
- In a final volume of 3 μL per reaction.
- Mix the solution well and add 3 μL of the previous mix to the reaction tube containing the RNA and the RA3. Mix the solution by gently pipetting up and down multiple times. The final volume of each reaction is 10 μL.
- Incubate the tube at 28 °C on a preheated block for 1 h.
- 6.
- Add 1 μL Stop Solution (STP, 15 μM). Mix the solution by gently pipetting up and down multiple times.
- 7.
- Incubate the tube at 28 °C for 15 minutes and then place the tube on ice.
3.2.2. Ligation of 5′ Adapter
- Prepare a mix containing:
- 1 μL of 7.5 μM RNA 5′ Adapter (RA5),
- 1 μL of 10 mM ATP,
- 0.6 μL of T4 RNA Ligase 1 (30 U/μL),
- 0.4 μL of 10× T4 RNA ligase buffer,
- In a final volume of 3 μL per reaction.
- Mix the solution well and add 3 μL of the previous mix to the reaction tube containing the small RNA–RA3 ligate. Mix the solution by gently pipetting up and down multiple times. The final volume of each reaction is 14 μL.
- Incubate the tube at 28 °C for 15 minutes and then place the tube on ice.
3.2.3. Reverse Transcription
- Mix 6 μL of RA5–small RNA–RA3 ligate with 1 μL of RT Primer (10 μM). Mix the solution by gently pipetting up and down multiple times.
- Incubate the reaction on a block pre-heated at 70 °C for 2 minutes and then place the tube on ice.
- Prepare a mix containing 2 μL of 5× First Strand Buffer, 0.5 μL of 12.5 mM dNTP mix, 1 μL of 100 mM DTT, 1 μL of RNase Inhibitor, and 1 μL SuperScript II Reverse Transcriptase in a final volume of 5.5 μL per reaction.
- Mix the solution well and add 5.5 μL of the previous mix to the reaction tube with the RA5–small RNA–RA3 and RT Primer. Mix the solution by gently pipetting up and down multiple times. The final volume of each reaction is 12.5 μL.
- Incubate the tube at 50 °C for 1 h, and then place the tube on ice.
PAUSE STEP: After stopping the reverse transcription, the library cDNA can be stored at −20 °C.3.2.4. PCR Amplification and Sample Barcoding
- Prepare a mix containing:
- 21 μL of ultrapure water,
- 10 μL of 5× Phusion HF buffer,
- 1 μL of 10 mM dNTP,
- 2.5 μL of PCR5 primer,
- 2.5 μL of PCR3 primer,
- 0.5 μL Phusion DNA polymerase,
- 12.5 μL library cDNA template.
- The final volume of each reaction is 50 μL.
CRITICAL STEP: Note that each sample will have a different PCR3 primer that contains the Illumina index to facilitate the posterior sample pooling and demultiplexing.- 2.
- Run the PCR amplification on a thermal cycler, with the following steps:
- (Step I) 98 °C for 30 s,
- (Step II) 15 cycles of 98 °C for 10 s,
- 60 °C for 30 s,
- 72 °C for 15 s,
- (Step III) 72 °C for 10 min,
- (Step IV) hold at 4 °C.
PAUSE STEP: After the PCR amplification, the barcoded libraries can be stored at 4 °C for the next few days.3.2.5. Gel Purification
- Prepare a 6% native PAGE gels by mixing:
- 45 mL 6% gel,
- 1 mL 10% APS,
- 20 μL TEMED.
- Pre-warm the gel by running it at 10 wats for 30 min.
- Mix 50 μL of PCR-amplified libraries with 6 μL 10× blue juice.
- Every few wells of the PAGE gel, load 5 μL of 20 bp DNA ladder to allow the alignment of the PCR bands to the corresponding markers at both sides.
- Carefully load the PCR-amplified libraries with blue juice to each well.
- Run the gel at 5 wats for at least 90 min. Allow enough separation of the different dyes on the gel.
- Stain the gel for 3 min with Sybr Gold DNA dye diluted 1:10,000 in 1× TBE.
- Visualize the gel on a blue-light-safe imager.
- Identify the band between 160 and 180 bp corresponding to the size of the mature shRNA ligated with the adapters and PCR barcodes. Amplicons containing no small RNAs, shRNA products, or miRNAs are expected to appear with a size of 144 bp.
- Extract the band by carefully cutting the window between 160 and 180 bp by aligning a clean blade with ladder markers.
- Place the excised bands in gel breaker inserts on 2 mL tubes.
- Centrifuge the bands placed on the 2 mL tubes with the gel breakers at 20,000× g in a benchtop centrifuge for 2 min.
- Discard the gel breaker and add 300 μL ultrapure water to the gel debris.
- Elute the library from the gel by rotating the tubes for at least 2 h at room temperature.
- Add a volume of 1000 μL to each tube with the eluted library.
PAUSE STEP: The elution can be extended to overnight at 4 °C.- 15.
- Transfer the solution and gel debris into an inset containing a 5 μm filter.
- 16.
- Centrifuge the filter for 20 s at 600× g and discard the insert with the debris.
- 17.
- Prepare a mix containing:
- 3 μL Glycoblue,
- 30 μL 3M NaOAc,
- 975 μL of pre-chilled 100% ethanol (−20 °C).
- 18.
- Incubate at −80 °C for 20–30 min to facilitate the subsequent precipitation.
- 19.
- Centrifuge at 20,000× g for 20 min on a benchtop centrifuge pre-cooled to 4 °C.
- 20.
- Identify the blue pellet and carefully remove the supernatant by aspirating with a 1 mL pipette from the top of the solution meniscus.
CRITICAL STEP: If the pellet detaches from the bottom of the tube, spin it again at 20,000× g for 2 min.- 21.
- Wash the blue pellet with 500 μL of 70% ethanol at room temperature.
- 22.
- Centrifuge at 20,000× g at room temperature for 2 min.
- 23.
- Identify the blue pellet and carefully remove the supernatant by aspirating with a 1 mL pipette from the top of the solution meniscus.
- 24.
- Spin the tube and remove the remaining solution with a 10 μL pipette.
- 25.
- Dry the pellet by placing the tube on a 37 °C heat block with open lid for 5–10 min (or until dry).
- 26.
- Resuspend the pellet in 10 μL of 10 mM Tris-HCI, pH 8.5, supplemented with 0.1% Tween 20.
PAUSE STEP: The suspended libraries can be stored at 4 °C for a day or −20 °C for longer periods.3.2.6. Library Quality Control and Sequencing
CRITICAL STEP: Inaccurate library concentration measurements can lead to uneven sequencing coverage, where some samples receive excessive reads, exceeding the required depth for quantification, while others suffer from insufficient coverage, potentially compromising data quality and downstream analyses. CRITICAL STEP: Before loading the pooled libraries into the Illumina sequencing cartridge, denaturation with NaOH is required to ensure proper single-stranded DNA formation for sequencing. It is essential to perform this step using freshly prepared NaOH working solutions to maintain reaction efficiency and prevent degradation of the libraries.3.3. Analysis of shRNA Processing and Endogenous miRNAs on Small RNA Datasets
3.3.1. Adapter Removal with Cutadapt
- Upload the FASTQ files to a project in the cloud computing platform.
- Copy Cutadapt to the project.
- Add the adapters used to generate the library in the corresponding boxes (Figure 3).
- Select discard reads where the adapter is not found.
- Select retain reads where the adapter is found.
- Select to retain only reads that have a minimum length of 15 nucleotides after the adapter removal. Shorter reads are challenging to map and may result from artifacts introduced during the library cloning process. Biologically, AGO-bound small RNAs are typically at least 20–25 nucleotides in length, with shorter sequences being rare and often indicative of degradation [27,28,29].
- 7.
- Inspect the report files. It is expected that on a FASTQ file, 75% of the reads will contain one adapter, and 74% will also fulfill the other filtering criteria previously defined.
3.3.2. Interpretation of Cutadapt Reports
- Check the percentage of reads that contain a recognizable adapter and additional filtering criteria set up for the run (Figure 4).
CRITICAL STEP: Other biases could involve an incomplete adapter sequence.- 3.
- Evaluate the distribution of reads and maximum number of errors allowed in each case. By default, Cutadapt allows a 0.1 error rate, thus allowing 1 error in a subsequence matching the adapter with 10 nucleotides.
3.3.3. Mapping and Analysis of Small RNAs with QuagmiR
- Edit the motif list file (e.g., motif_list_mmu_mirbase22.fa) with a plain text editor to include the guide and passenger strand of your shRNA.
- In the first line of the file add a “>descriptive name” followed by a space and a unique 13-mer motif contained in the middle of all the small RNAs deriving that shRNA arm. In the second line, we provide our intended mature sequence for that shRNA (21–22 nucleotides). For example:
- >shRNA-guide1 GATACAGATACAT
- TCAGGATACAGATACATAACTT
- Repeat the same steps to include also a unique motif and the reference sequence for the passenger strand of your shRNA of interest.
- >shRNA-passenger1 GTAGTAGGTTGTA
- TGAGGTAGTAGGTTGTATAGAA
- 4.
- Save the motif file, edited to contain the guide and passenger strands, along with all the other miRNA sequences expressed endogenously by the treated cells.
CRITICAL STEP: Notice that the miRNA sequences of different organisms can differ. To this end, the QuagmiR repository contains reference files for a large list of organisms including humans, mice, rats, zebrafish, and other model organisms. https://github.com/Bofill-De-Ros-Lab/QuagmiR/tree/master/Motifs (accessed on 1 December 2024).- 5.
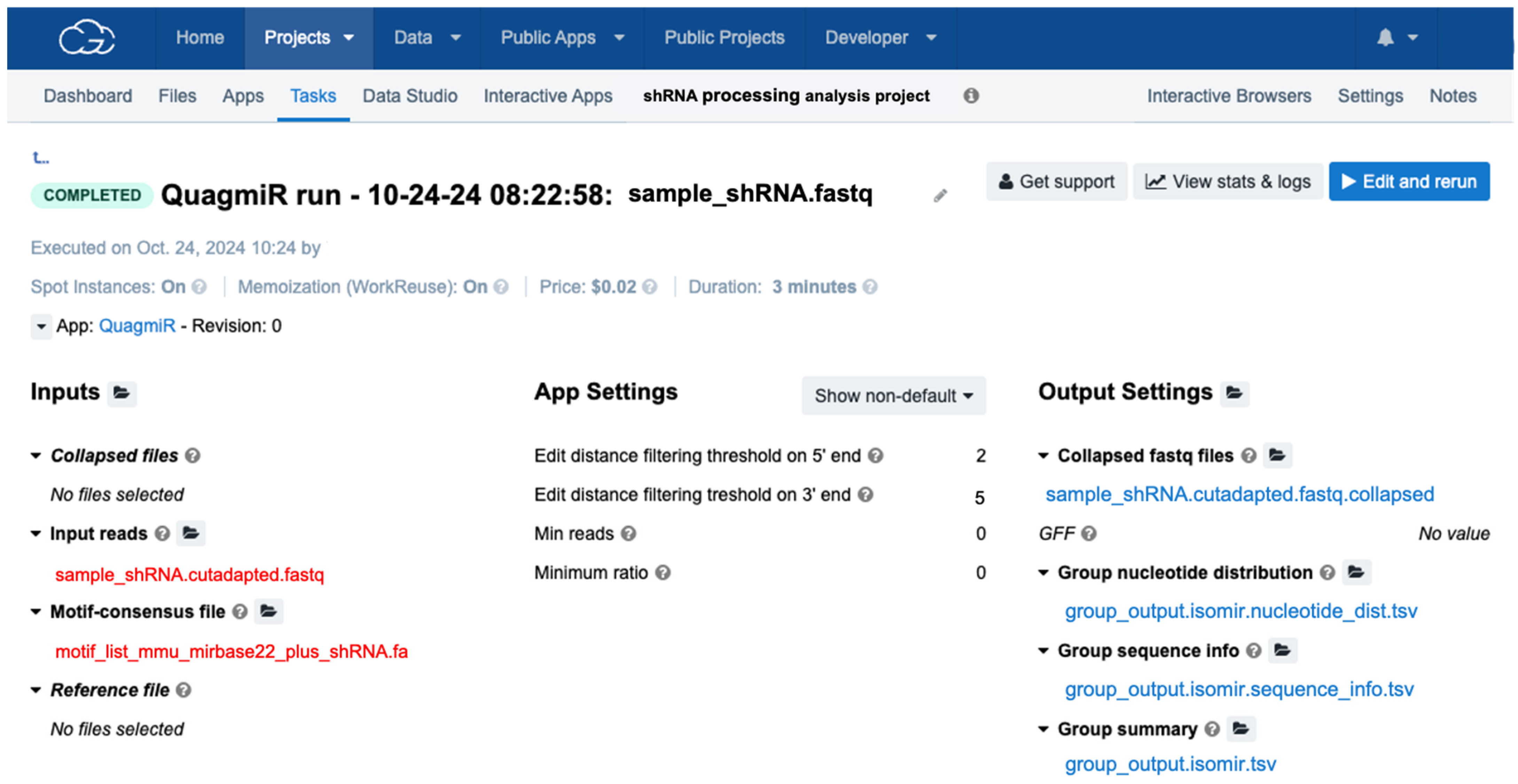
- Upload the edited motif file to the project folder in the cloud computing platform.
- 6.
- Copy QuagmiR to the project.
- 7.
- Select the input cutadapted FASTQ files and motif file (Figure 5).
- 8.
- Select the number of mismatches allowed on the 5′ and 3′ end segments by defining the edit distances (Levenshtein distance). Since modifications on the 3′ end of small reads are more prevalent [27] than 5′ isoforms [26], edit distances of 5 (edit distance 3′ end) and 2 (edit distance 5′ end) are recommended.
3.3.4. Interpretation of QuagmiR Reports
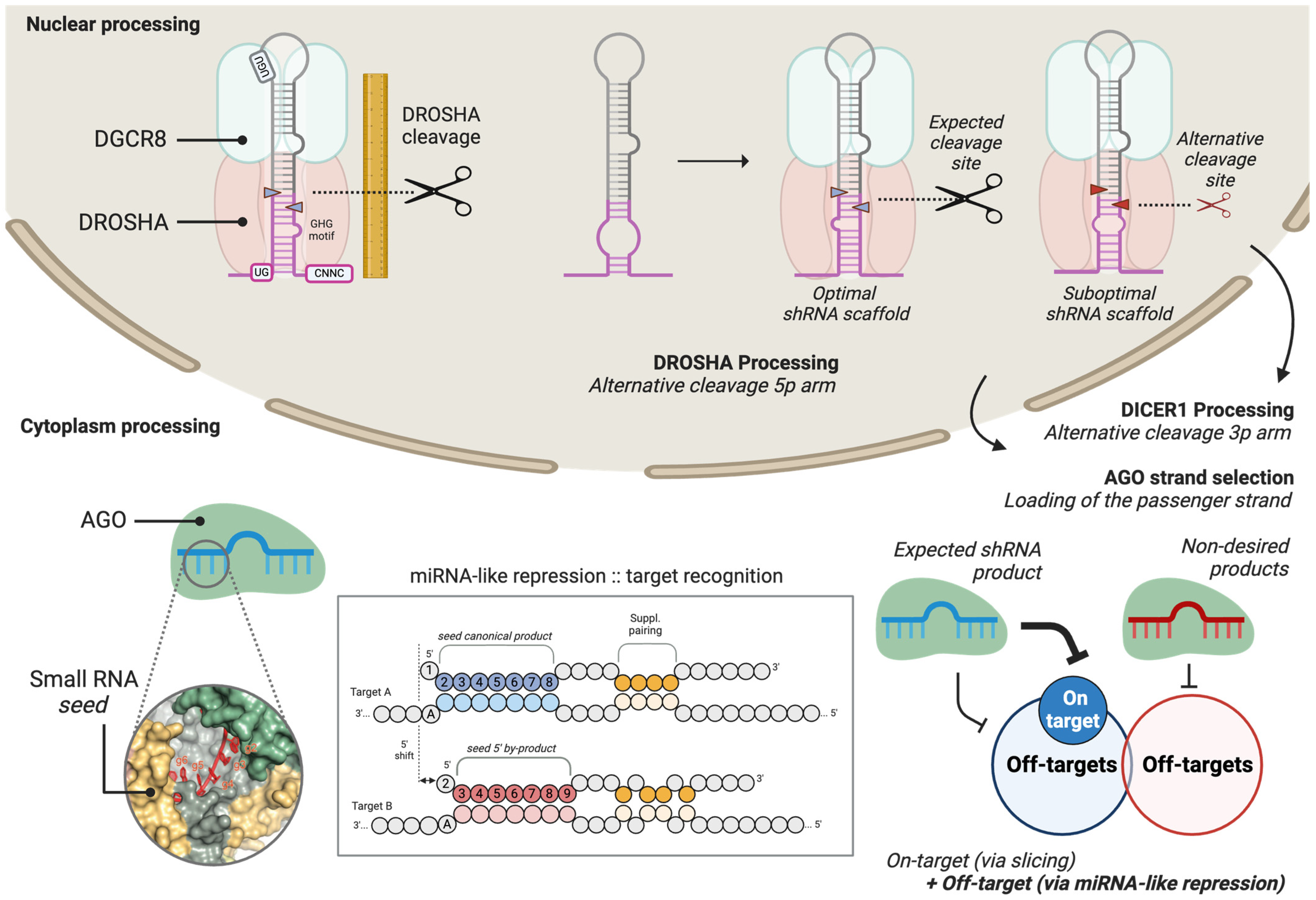
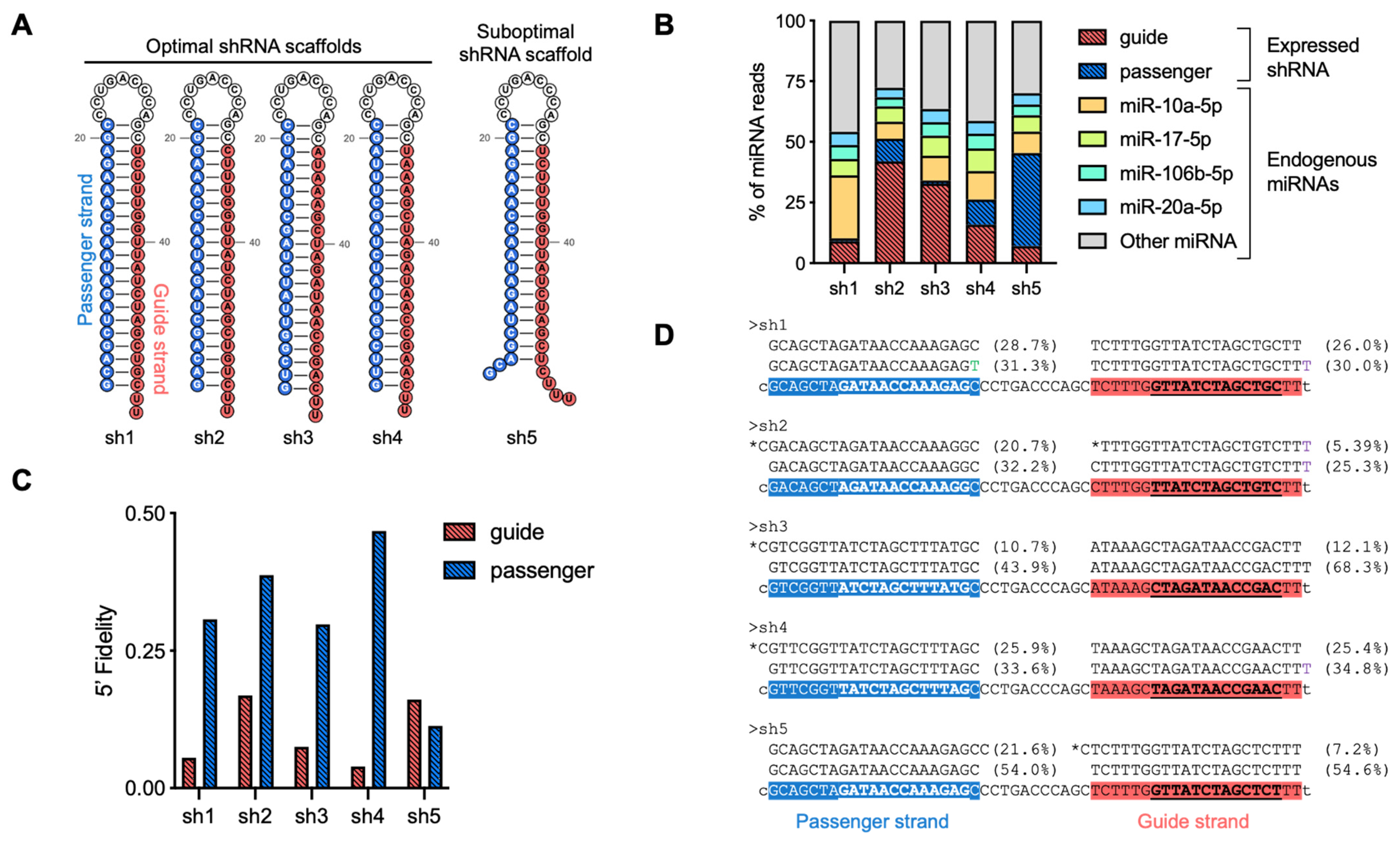
- Examine the relative abundance of the guide and passenger strands of the shRNA in the summary report (Figure 6A). In applications involving transient shRNA expression, both strands may be among the most highly expressed small RNAs, which is a common observation. Monitoring their relative proportions provides insights into strand selection efficiency and potential off-target effects.
- Evaluate 5′ end processing accuracy using the Fidelity_5P metric in the summary report (Figure 6A). This metric quantifies the heterogeneity of 5′ isoforms as a weighted average, typically reflecting variability in cleavage site selection by DROSHA or DICER1. Values range from 0 for highly precise processing to 1 or 2 for inaccurately processed shRNA scaffolds, where increased heterogeneity suggests suboptimal cleavage efficiency.
- Examine 3′ end heterogeneity in the summary report (Figure 6A), which is assessed through multiple parameters, including the number of isoforms as well as the percentage of sequence trimming and tailing. Unusually high values in any of these metrics may indicate suboptimal shRNA processing, such as premature termination or imprecise cleavage, or post-maturation modifications, such as target-directed microRNA degradation (TDMD).
- For a more detailed analysis of the generated isoforms, inspection of sequence-level reports is recommended (Figure 6B). This allows for the sorting of reads and the identification of isoform sequence compositions, particularly those with altered 5′ ends. Additionally, this analysis can help determine whether the presence of 3′ isoforms results from shRNA scaffold misprocessing (templated isoforms) or from endogenous cellular processing mechanisms (non-templated isoforms).
3.4. Calculation of Advanced shRNA Biogenesis Metrics
3.4.1. Analysis of shRNA Strand Selection
TROUBLESHOOTING: Suboptimal strand selection can occur if the 5′ nucleotide of the guide strand is not optimal for Argonaute loading. Structural studies have demonstrated that Argonaute proteins preferentially incorporate guide strands that begin with a uridine (U) or adenine (A) at the 5′ end [38], as well as thermodynamic features of the shRNA duplex [39].3.4.2. Analysis of shRNA 5′ End Isoforms
TROUBLESHOOTING: Suboptimal generation of guide sequences can indicate defects in the shRNA scaffold. The use of reported scaffolds such as pri-miR-22 or pri-miR-16 is encouraged. To ensure proper processing, it is also a good practice to evaluate the folding of the shRNA scaffold with RNAfold [40], Mfold [41], or similar RNA-folding algorithms.4. Expected Results
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corydon, I.J.; Fabian-Jessing, B.K.; Jakobsen, T.S.; Jørgensen, A.C.; Jensen, E.G.; Askou, A.L.; Aagaard, L.; Corydon, T.J. 25 years of maturation: A systematic review of RNAi in the clinic. Mol. Ther. Nucleic Acids 2023, 33, 469–482. [Google Scholar] [CrossRef]
- Chancellor, D.; Barrett, D.; Nguyen-Jatkoe, L.; Millington, S.; Eckhardt, F. The state of cell and gene therapy in 2023. Mol. Ther. 2023, 31, 3376–3388. [Google Scholar] [CrossRef] [PubMed]
- Bofill-De Ros, X.; Gu, S. Guidelines for the optimal design of miRNA-based shRNAs. Methods 2016, 103, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, J.; Zheng, G.; Wang, Q.; Li, X.; Feng, Y.; Shang, F.; He, S.; Jiang, Q.; Shi, B.; et al. Co-Expression of miR155 or LSD1 shRNA Increases the Anti-Tumor Functions of CD19 CAR-T Cells. Front. Immunol. 2021, 12, 811364. [Google Scholar] [CrossRef]
- Zhou, J.-E.; Yu, J.; Wang, Y.; Wang, H.; Wang, J.; Wang, Y.; Yu, L.; Yan, Z. ShRNA-mediated silencing of PD-1 augments the efficacy of chimeric antigen receptor T cells on subcutaneous prostate and leukemia xenograft. Biomed. Pharmacother. 2021, 137, 111339. [Google Scholar] [CrossRef]
- Guo, Q.; Zhang, J.; Parikh, K.; Brinkley, A.; Lin, S.; Zakarian, C.; Pernet, O.; Shimizu, S.; Khamaikawin, W.; Hacke, K.; et al. In vivo selection of anti-HIV-1 gene-modified human hematopoietic stem/progenitor cells to enhance engraftment and HIV-1 inhibition. Mol. Ther. 2024, 32, 384–394. [Google Scholar] [CrossRef]
- Siolas, D.; Lerner, C.; Burchard, J.; Ge, W.; Linsley, P.S.; Paddison, P.J.; Hannon, G.J.; Cleary, M.A. Synthetic shRNAs as potent RNAi triggers. Nat. Biotechnol. 2005, 23, 227–231. [Google Scholar] [CrossRef]
- Rossi, M.; Steklov, M.; Huberty, F.; Nguyen, T.; Marijsse, J.; Jacques-Hespel, C.; Najm, P.; Lonez, C.; Breman, E. Efficient shRNA-based knockdown of multiple target genes for cell therapy using a chimeric miRNA cluster platform. Mol. Ther. Nucleic Acids 2023, 34, 102038. [Google Scholar] [CrossRef]
- Fellmann, C.; Hoffmann, T.; Sridhar, V.; Hopfgartner, B.; Muhar, M.; Roth, M.; Lai, D.Y.; Barbosa, I.A.M.; Kwon, J.S.; Guan, Y.; et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013, 5, 1704–1713. [Google Scholar] [CrossRef]
- Bofill-De Ros, X.; Hong, Z.; Birkenfeld, B.; Alamo-Ortiz, S.; Yang, A.; Dai, L.; Gu, S. Flexible pri-miRNA structures enable tunable production of 5′ isomiRs. RNA Biol. 2022, 19, 279–289. [Google Scholar] [CrossRef]
- Kampmann, M.; Horlbeck, M.A.; Chen, Y.; Tsai, J.C.; Bassik, M.C.; Gilbert, L.A.; Villalta, J.E.; Kwon, S.C.; Chang, H.; Kim, V.N.; et al. Next-generation libraries for robust RNA interference-based genome-wide screens. Proc. Natl. Acad. Sci. USA 2015, 112, E3384–E3391. [Google Scholar] [CrossRef]
- Denise, H.; Moschos, S.A.; Sidders, B.; Burden, F.; Perkins, H.; Carter, N.; Stroud, T.; Kennedy, M.; Fancy, S.-A.; Lapthorn, C.; et al. Deep Sequencing Insights in Therapeutic shRNA Processing and siRNA Target Cleavage Precision. Mol. Ther. Nucleic Acids 2014, 3, e145. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, J.; Cheng, H.; Ke, X.; Sun, L.; Zhang, Q.C.; Wang, H.-W. Cryo-EM Structure of Human Dicer and Its Complexes with a Pre-miRNA Substrate. Cell 2018, 173, 1191–1203.e12. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-Y.; Lee, H.; Kim, H.; Kim, V.N.; Roh, S.-H. Structure of the human DICER-pre-miRNA complex in a dicing state. Nature 2023, 615, 331–338. [Google Scholar] [CrossRef]
- Jouravleva, K.; Golovenko, D.; Demo, G.; Dutcher, R.C.; Hall, T.M.T.; Zamore, P.D.; Korostelev, A.A. Structural basis of microRNA biogenesis by Dicer-1 and its partner protein Loqs-PB. Mol. Cell 2022, 82, 4049–4063.e6. [Google Scholar] [CrossRef] [PubMed]
- Zapletal, D.; Taborska, E.; Pasulka, J.; Malik, R.; Kubicek, K.; Zanova, M.; Much, C.; Sebesta, M.; Buccheri, V.; Horvat, F.; et al. Structural and functional basis of mammalian microRNA biogenesis by Dicer. Mol. Cell 2022, 82, 4064–4079.e13. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Trinh, T.A.; Bao, S.; Nguyen, T.A. Secondary structure RNA elements control the cleavage activity of DICER. Nat. Commun. 2022, 13, 2138. [Google Scholar] [CrossRef]
- Lee, Y.-Y.; Kim, H.; Kim, V.N. Sequence determinant of small RNA production by DICER. Nature 2023, 615, 323–330. [Google Scholar] [CrossRef]
- Shibata, A.; Shirohzu, H.; Iwakami, Y.; Abe, T.; Emura, C.; Aoki, E.; Ohgi, T. Terminal bridging of siRNA duplex at the ribose 2′ position controls strand bias and target sequence preference. Mol. Ther. Nucleic Acids 2023, 32, 468–477. [Google Scholar] [CrossRef]
- Bofill-De Ros, X.; Chen, K.; Chen, S.; Tesic, N.; Randjelovic, D.; Skundric, N.; Nesic, S.; Varjacic, V.; Williams, E.H.; Malhotra, R.; et al. QuagmiR: A cloud-based application for isomiR big data analytics. Bioinformatics 2019, 35, 1576–1578. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.; Kim, K.; Chang, H.; You, K.; Kim, V.N. Bias-minimized quantification of microRNA reveals widespread alternative processing and 3′ end modification. Nucleic Acids Res. 2019, 47, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Bofill-De Ros, X.; Stanton, R.; Shao, T.-J.; Villanueva, P.; Gu, S. TENT2, TUT4, and TUT7 selectively regulate miRNA sequence and abundance. Nat. Commun. 2022, 13, 5260. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Yeo, J.; Kim, B.; Ha, M.; Kim, V.N. Short Structured RNAs with Low GC Content Are Selectively Lost During Extraction from a Small Number of Cells. Mol. Cell 2012, 46, 893–895. [Google Scholar] [CrossRef]
- Gómez-Martín, C.; Aparicio-Puerta, E.; van Eijndhoven, M.A.J.; Medina, J.M.; Hackenberg, M.; Pegtel, D.M. Reassessment of miRNA variant (isomiRs) composition by small RNA sequencing. Cell Rep. Methods 2023, 3, 100480. [Google Scholar] [CrossRef]
- Bofill-De Ros, X.; Kasprzak, W.K.; Bhandari, Y.; Fan, L.; Cavanaugh, Q.; Jiang, M.; Dai, L.; Yang, A.; Shao, T.-J.; Shapiro, B.A.; et al. Structural Differences between Pri-miRNA Paralogs Promote Alternative Drosha Cleavage and Expand Target Repertoires. Cell Rep. 2019, 26, 447–459.e4. [Google Scholar] [CrossRef]
- Yang, A.; Shao, T.-J.; Bofill-De Ros, X.; Lian, C.; Villanueva, P.; Dai, L.; Gu, S. AGO-bound mature miRNAs are oligouridylated by TUTs and subsequently degraded by DIS3L2. Nat. Commun. 2020, 11, 2765. [Google Scholar] [CrossRef]
- Sim, G.; Kehling, A.C.; Park, M.S.; Secor, J.; Divoky, C.; Zhang, H.; Malhotra, N.; Bhagdikar, D.; Abd El-Wahab, E.W.; Nakanishi, K. Manganese-dependent microRNA trimming by 3′→5′ exonucleases generates 14-nucleotide or shorter tiny RNAs. Proc. Natl. Acad. Sci. USA 2022, 119, e2214335119. [Google Scholar] [CrossRef]
- Sim, G.; Kehling, A.C.; Park, M.S.; Divoky, C.; Zhang, H.; Malhotra, N.; Secor, J.; Nakanishi, K. Determining the defining lengths between mature microRNAs/small interfering RNAs and tinyRNAs. Sci. Rep. 2023, 13, 19761. [Google Scholar] [CrossRef]
- Mansur, F.; Ivshina, M.; Gu, W.; Schaevitz, L.; Stackpole, E.; Gujja, S.; Edwards, Y.J.K.; Richter, J.D. Gld2-catalyzed 3′ monoadenylation of miRNAs in the hippocampus has no detectable effect on their stability or on animal behavior. RNA 2016, 22, 1492–1499. [Google Scholar] [CrossRef]
- Morgan, M.; Much, C.; DiGiacomo, M.; Azzi, C.; Ivanova, I.; Vitsios, D.M.; Pistolic, J.; Collier, P.; Moreira, P.N.; Benes, V.; et al. mRNA 3′ uridylation and poly(A) tail length sculpt the mammalian maternal transcriptome. Nature 2017, 548, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Wardaszka, P.; Kuzniewska, B.; Guminska, N.; Hojka-Osinska, A.; Puchalska, M.; Milek, J.; Stawikowska, A.; Krawczyk, P.; Pauzin, F.P.; Wojtowicz, T.; et al. Terminal nucleotidyltransferase Tent2 microRNA tailing regulates excitatory/inhibitory balance in the hippocampus. BioRxiv 2024. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Emde, A.-K.; Grunert, M.; Weese, D.; Reinert, K.; Sperling, S.R. MicroRazerS: Rapid alignment of small RNA reads. Bioinformatics 2010, 26, 123–124. [Google Scholar] [CrossRef]
- Weese, D.; Holtgrewe, M.; Reinert, K. RazerS 3: Faster, fully sensitive read mapping. Bioinformatics 2012, 28, 2592–2599. [Google Scholar] [CrossRef] [PubMed]
- Pantano, L.; Estivill, X.; Martí, E. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res. 2010, 38, e34. [Google Scholar] [CrossRef]
- Aparicio-Puerta, E.; Gómez-Martín, C.; Giannoukakos, S.; Medina, J.M.; Scheepbouwer, C.; García-Moreno, A.; Carmona-Saez, P.; Fromm, B.; Pegtel, M.; Keller, A.; et al. sRNAbench and sRNAtoolbox 2022 update: Accurate miRNA and sncRNA profiling for model and non-model organisms. Nucleic Acids Res. 2022, 50, W710–W717. [Google Scholar] [CrossRef] [PubMed]
- Frank, F.; Sonenberg, N.; Nagar, B. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 2010, 465, 818–822. [Google Scholar] [CrossRef]
- Gu, S.; Jin, L.; Zhang, F.; Huang, Y.; Grimm, D.; Rossi, J.J.; Kay, M.A. Thermodynamic stability of small hairpin RNAs highly influences the loading process of different mammalian Argonautes. Proc. Natl. Acad. Sci. USA 2011, 108, 9208–9213. [Google Scholar] [CrossRef]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Ma, H.; Wu, Y.; Dang, Y.; Choi, J.-G.; Zhang, J.; Wu, H. Pol III promoters to express small rnas: Delineation of transcription initiation. Mol. Ther. Nucleic Acids 2014, 3, e161. [Google Scholar] [CrossRef] [PubMed]
- Monopoli, K.R.; Korkin, D.; Khvorova, A. Asymmetric trichotomous partitioning overcomes dataset limitations in building machine learning models for predicting siRNA efficacy. Mol. Ther. Nucleic Acids 2023, 33, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhong, H.; Wang, T.; Lu, Z.J. OligoFormer: An accurate and robust prediction method for siRNA design. Bioinformatics 2024, 40, btae577. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.M.; Birmingham, A.; Baskerville, S.; Reynolds, A.; Maksimova, E.; Leake, D.; Fedorov, Y.; Karpilow, J.; Khvorova, A. Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA 2008, 14, 853–861. [Google Scholar] [CrossRef]
- Medley, J.C.; Panzade, G.; Zinovyeva, A.Y. microRNA strand selection: Unwinding the rules. Wiley Interdiscip. Rev. RNA 2021, 12, e1627. [Google Scholar] [CrossRef]
- Blue, S.M.; Yee, B.A.; Pratt, G.A.; Mueller, J.R.; Park, S.S.; Shishkin, A.A.; Starner, A.C.; Van Nostrand, E.L.; Yeo, G.W. Transcriptome-wide identification of RNA-binding protein binding sites using seCLIP-seq. Nat. Protoc. 2022, 17, 1223–1265. [Google Scholar] [CrossRef]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]



| Source | Advantages | Disadvantages |
|---|---|---|
| Synthesized siRNA | Can be chemically modified for enhanced stability and reduced immunogenicity | Effects are transient |
| No biogenesis steps involved | Delivery challenges | |
| Pol-III-driven shRNA (e.g., U6 or H1) | Can be encoded on a viral vector | Increased cellular toxicity |
| High intracellular expression | Unintended off-targets | |
| Long-term silencing in stable cells | Limited number of promoters | |
| Pol-II-driven pri-shRNA (e.g., CMV, Tet-ON/OFF) | Can be encoded on a viral vector | Lower intracellular expression |
| Lower risk of off-targets | Higher biogenesis complexity | |
| Multiple promoter options |
| Best Practice | Rationale |
|---|---|
| Guide Selection | |
| Target Site Selection | Use bioinformatics tools to predict and minimize off-target effects by selecting sequences with high specificity for the target mRNA [43,44]. |
| Seed Region Optimization | Avoid pairing in the seed region (nucleotides 2–8) to unintended transcripts or highly prevalent k-mers in the 3′UTR [45]. |
| Hairpin Design | |
| Optimized Hairpins | Design shRNAs with loop structures that promote efficient DROSHA and DICER1 processing, reducing heterogeneous processing and off-target effects [3]. |
| Strand Selection | Ensure preferential loading of the intended guide strand into RISC by modifying thermodynamic asymmetry or using mismatches in the passenger strand [46]. |
| Evaluate Off-Targets | |
| Off-Target Screening | Use CLIP-based approaches (AGO-CLEAR CLIP, PAR-CLIP) to identify unintended targets transcriptome-wide [47]. |
| Mismatched Controls | Use control shRNAs with single or double mismatches to distinguish specific from off-target effects [48]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Z.; Tesic, N.; Bofill-De Ros, X. Analysis of Processing, Post-Maturation, and By-Products of shRNA in Gene and Cell Therapy Applications. Methods Protoc. 2025, 8, 38. https://doi.org/10.3390/mps8020038
Hong Z, Tesic N, Bofill-De Ros X. Analysis of Processing, Post-Maturation, and By-Products of shRNA in Gene and Cell Therapy Applications. Methods and Protocols. 2025; 8(2):38. https://doi.org/10.3390/mps8020038
Chicago/Turabian StyleHong, Zhenyi, Nikola Tesic, and Xavier Bofill-De Ros. 2025. "Analysis of Processing, Post-Maturation, and By-Products of shRNA in Gene and Cell Therapy Applications" Methods and Protocols 8, no. 2: 38. https://doi.org/10.3390/mps8020038
APA StyleHong, Z., Tesic, N., & Bofill-De Ros, X. (2025). Analysis of Processing, Post-Maturation, and By-Products of shRNA in Gene and Cell Therapy Applications. Methods and Protocols, 8(2), 38. https://doi.org/10.3390/mps8020038