
Comparative Analysis of the nrDNA Repeat Unit of Manila Clam Ruditapes philippinarum and Quahog Mercenaria mercenaria

Abstract
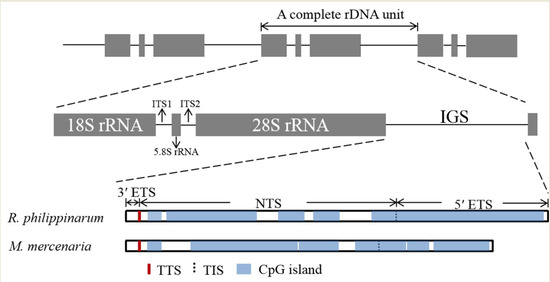
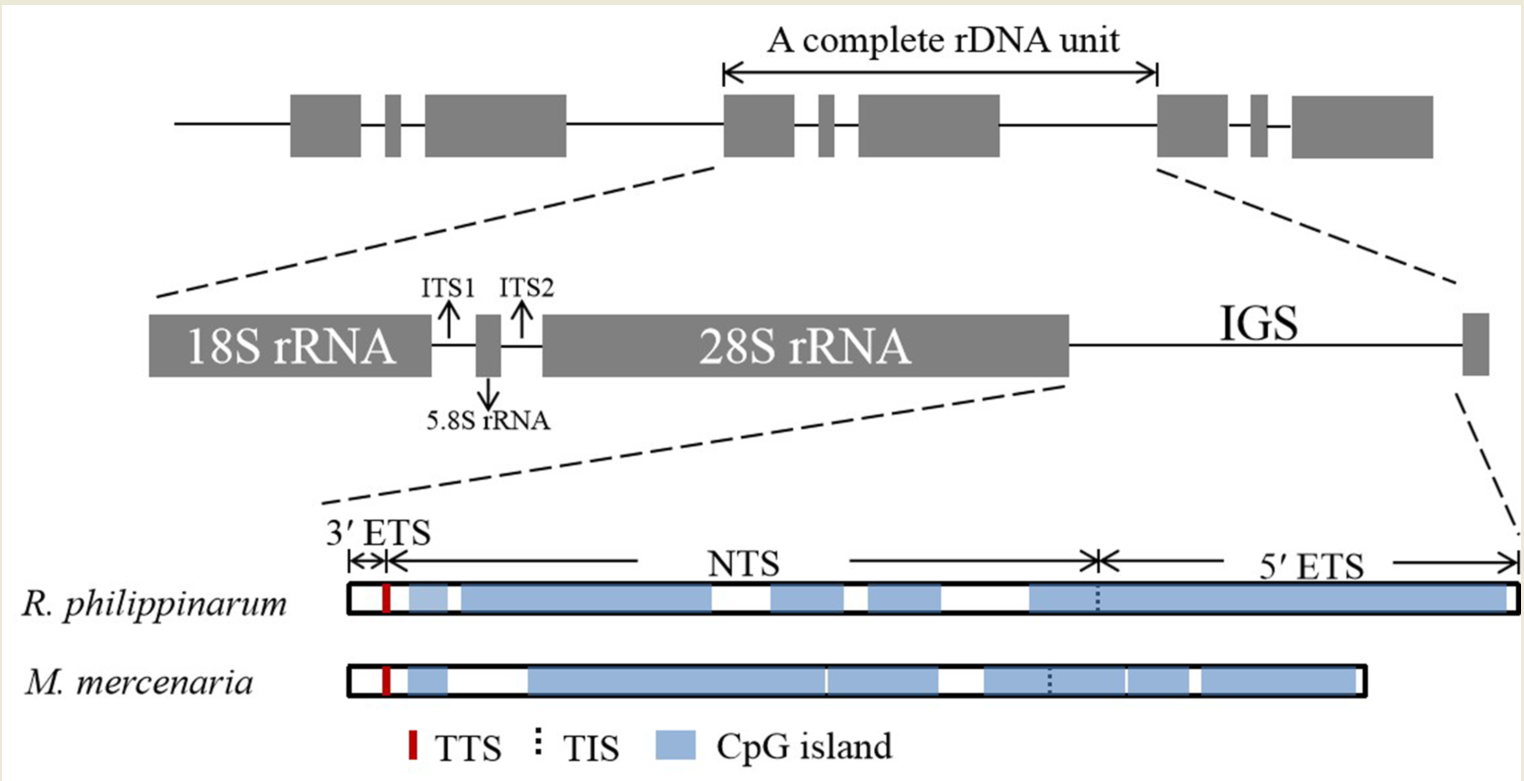
:
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. PCR Amplification, Cloning and Sequencing
2.3. Sequence Analysis
3. Results
3.1. Complete nrDNA Sequences of R. philippinarum and M. mercenaria
3.2. Informative Characteristics of the IGS Region
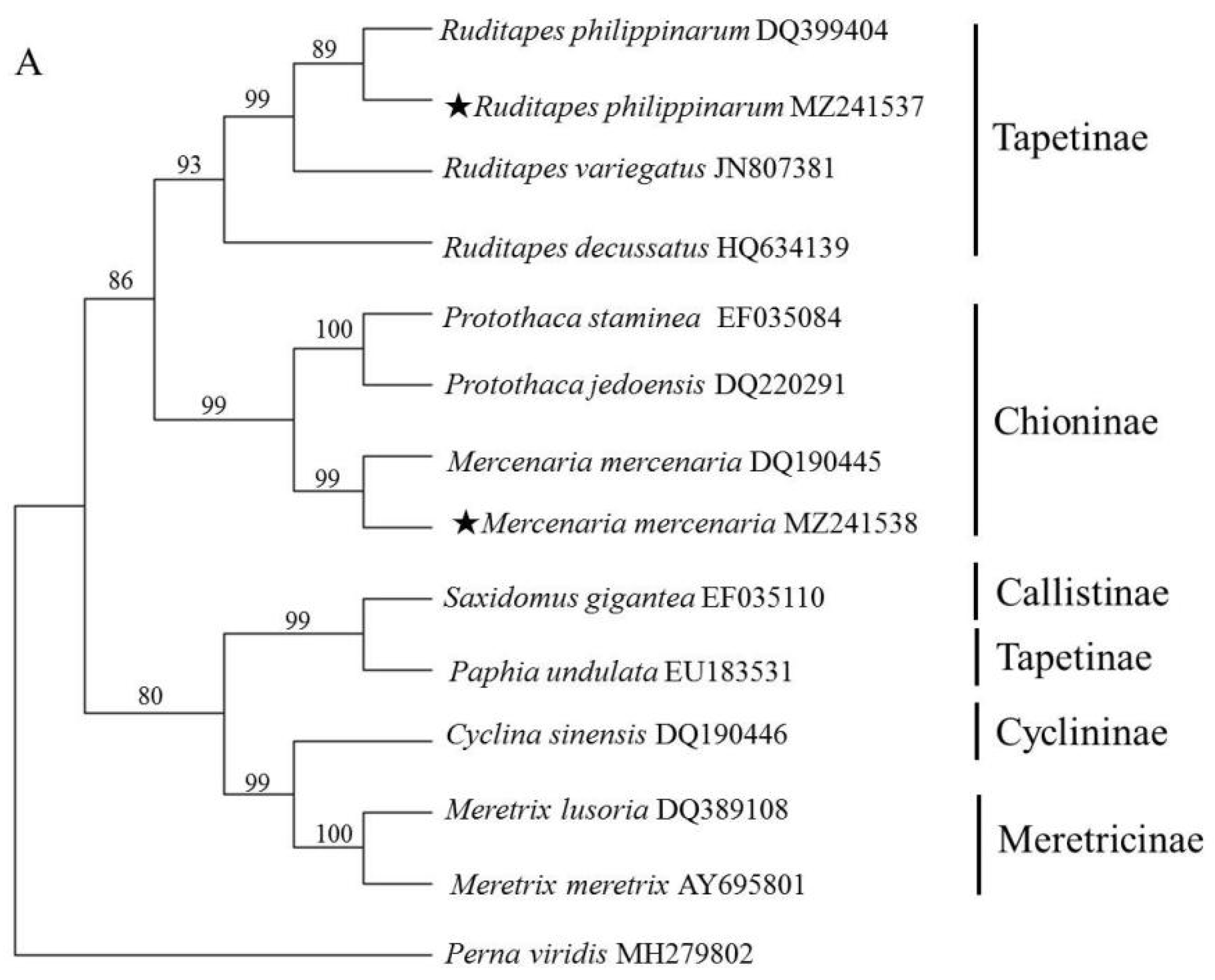
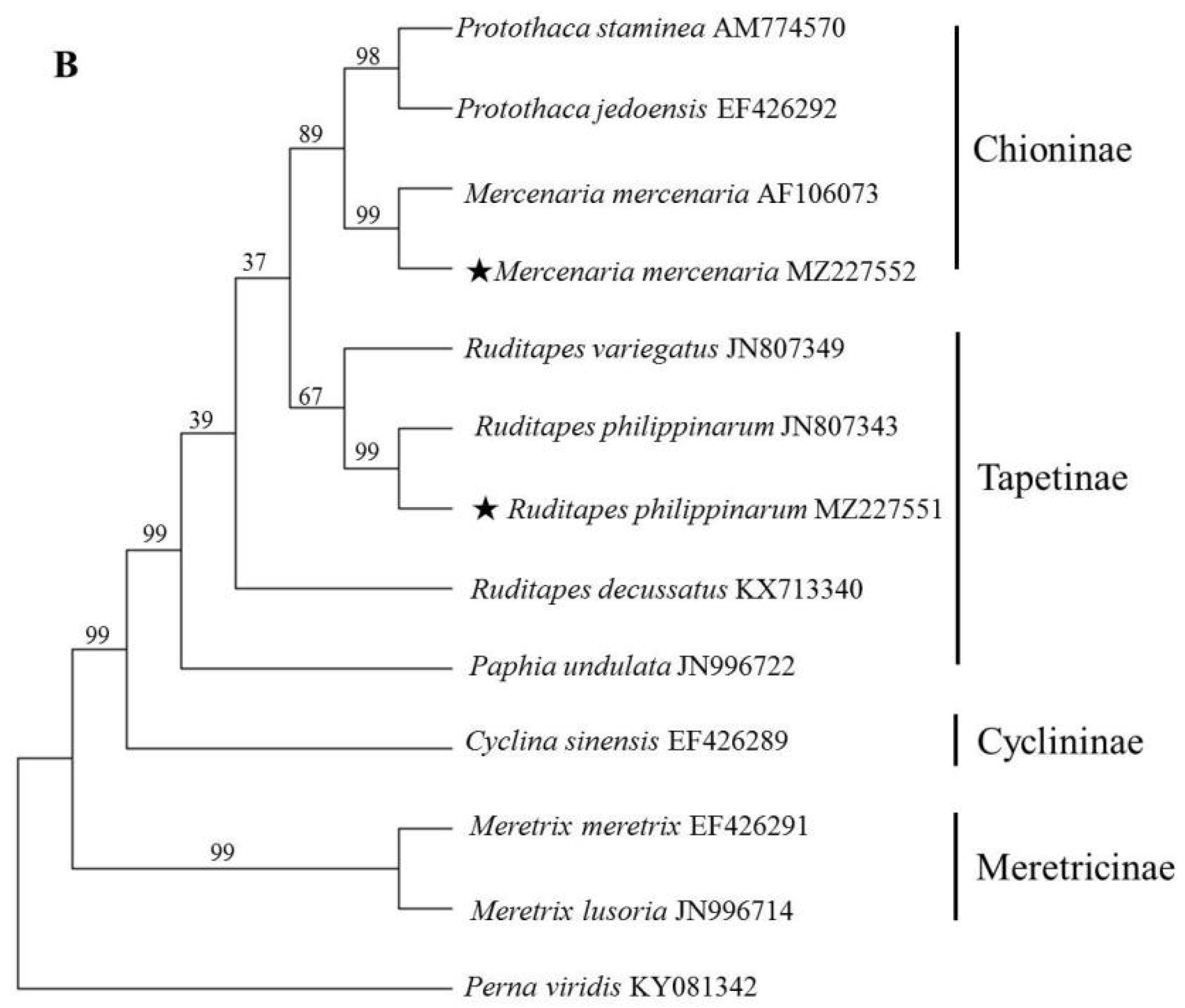
3.3. Phylogenetic Analysis of Veneridae Based on ITS and 18S rRNA Sequence
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Canapa, A.; Schiaparelli, S.; Marota, I.; Barucca, M. Molecular data from the 16S rRNA gene for the phylogeny of Veneridae (Mollusca: Bivalvia). Mar. Biol. 2003, 142, 1125–1130. [Google Scholar] [CrossRef]
- Kappner, I.; Bieler, R. Phylogeny of venus clams (Bivalvia: Venerinae) as inferred from nuclear and mitochondrial gene sequences. Mol. Phylogenet. Evol. 2006, 40, 317–331. [Google Scholar] [CrossRef]
- Chen, J.; Li, Q.; Kong, L.; Zheng, X. Molecular phylogeny of venus clams (Mollusca, Bivalvia, Veneridae) with emphasis on the systematic position of taxa along the coast of mainland China. Zool. Scr. 2011, 40, 260–271. [Google Scholar] [CrossRef]
- Mikkelsen, P.M.; Bieler, R.; Kappner, I.; Rawlings, T.A. Phylogeny of veneroidea (Mollusca: Bivalvia) based on morphology and molecules. Zool. J. Linn. Soc. 2006, 148, 439–521. [Google Scholar] [CrossRef] [Green Version]
- Salvi, D.; Mariottini, P. Molecular phylogenetics in 2D: ITS2 rRNA evolution and sequence-structure barcode from Veneridae to Bivalvia. Mol. Phylogenet. Evol. 2012, 65, 792–798. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Liu, H.Y.; Kong, L.F.; Yu, H.; Liu, S.K.; Li, Q. Phylogeny of Veneridae (Bivalvia) based on mitochondrial genomes. Zool. Scr. 2021, 50, 58–70. [Google Scholar] [CrossRef]
- Capt, C.; Bouvet, K.; Guerra, D.; Robicheau, B.M.; Stewart, D.T.; Pante, E.; Breton, S. Unorthodox features in two venerid bivalves with doubly uniparental inheritance of mitochondria. Sci. Rep. 2020, 10, 1087. [Google Scholar] [CrossRef]
- Chacón, G.M.; Arias-Pérez, A.; Freire, R.; Martínez, L.; Nóvoa, S.; Naveira, H.; Insua, A. Evidence of doubly uniparental inheritance of the mitochondrial DNA in Polititapes rhomboides (Bivalvia, Veneridae): Evolutionary and population genetic analysis of F and M mitotypes. J. Zool. Syst. Evol. Res. 2020, 58, 541–560. [Google Scholar] [CrossRef]
- Passamonti, M.; Plazzi, F. Doubly Uniparental Inheritance and beyond: The contribution of the Manila clam Ruditapes philippinarum. J. Zool. Syst. Evol. Res. 2020, 58, 529–540. [Google Scholar] [CrossRef]
- Dyomin, A.; Galkina, S.; Fillon, V.; Cauet, S.; Lopez-Roques, C.; Rodde, N.; Klopp, C.; Vignal, A.; Sokolovskaya, A.; Saiftdinova, A.; et al. Structure of the intergenic spacers in chicken ribosomal DNA. Genet. Sel. Evol. 2019, 51, 59. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Han, L.; Liang, Z.; Hou, X. Comparative analysis of the ribosomal DNA repeat unit (rDNA) of Perna viridis (Linnaeus, 1758) and Perna canaliculus (Gmelin, 1791). PeerJ 2019, 7, e7644. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, J.; He, Y.; Shen, S.; Zhu, J.; Shen, Z. The complete nuclear ribosomal DNA (nrDNA) cistron sequence of Pyropia yezoensis (Bangiales, Rhodophyta). J. Appl. Phycol. 2016, 28, 663–669. [Google Scholar] [CrossRef]
- Chiesa, S.; Lucentini, L.; Freitas, R.; Marzano, F.N.; Minello, F.; Ferrari, C.; Filonzi, L.; Figueira, E.; Breda, S.; Baccarani, G.; et al. Genetic diversity of introduced Manila clam Ruditapes philippinarum populations inferred by 16S rDNA. Biochem. Syst. Ecol. 2014, 57, 52–59. [Google Scholar] [CrossRef]
- Hu, Z.; Song, H.; Zhou, C.; Yu, Z.L.; Yang, M.J.; Zhang, T. Complete mitochondrial genome of the hard clam (Mercenaria mercenaria). Mitochondrial DNA B 2019, 4, 3738–3739. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.Y.; Li, J.P.; Gao SQTuerxun, A.; Chang, X.C.; Hu, W.R.; Chen, G.; Huang, Q.S. SsPsaH, a H subunit of the photosystem I reaction center of Suaeda salsa, confers the capacity of osmotic adjustment in tobacco. Genes Genom. 2020, 42, 1455–1465. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Han, L.; Ding, Y.; Hou, X.; Liang, Z. Molecular characterization of the complete nuclear ribosomal DNA sequence of the blacklip abalone Haliotis rubra. N. Z. J. Mar. Fresh. 2018, 52, 430–443. [Google Scholar] [CrossRef]
- Guo, Z.; Hou, X.; Han, L. Complete nuclear ribosomal DNA sequence analyses of the black-footed abalone Haliotis iris. N. Z. J. Mar. Fresh. 2018, 52, 232–246. [Google Scholar] [CrossRef]
- Cheng, H.L.; Peng, Y.X.; Wang, F.; Meng, X.P.; Yan, B.L.; Dong, Z.G. Sequence analysis of 18S rRNA gene of six Veneridae clams (Mollusca: Bivalvia). J. Fish. Sci. Chin. 2008, 15, 559–567, (In Chinese with English abstract). [Google Scholar]
- Chae, J.; Seo, Y.; Yu, W.B.; Yoon, W.D.; Lee, H.E.; Chang, S.J.; Ki, J.S. Comprehensive analysis of the jellyfish Chrysaora pacifca (Goette, 1886) (Semaeostomeae: Pelagiidae) with description of the complete rDNA sequence. Zool. Stud. 2018, 57, 51. [Google Scholar]
- Guo, Z.; Ding, Y.; Zhang, X.; Hou, X. Complete nuclear ribosomal DNA sequence analysis of Pacific abalone Haliotis discus hannai. Fish. Sci. 2017, 83, 777–784. [Google Scholar] [CrossRef]
- Ki, J.S.; Kim, I.C.; Lee, J.S. Comparative analysis of nuclear ribosomal DNA from the moon jelly Aurelia sp.1 (Cnidaria: Scyphozoa) with characterizations of the 18S, 28S genes, and the intergenic spacer (IGS). Hydrobiologia 2009, 616, 229–239. [Google Scholar] [CrossRef]
- Ki, J.S.; Park, H.G.; Lee, J.S. Extensive analysis of nuclear cistron rDNA sequence of Paracyclopina nana (Cyclopoida: Cyclopettidae). Hydrobiologia 2011, 666, 3–9. [Google Scholar] [CrossRef]
- Gonzalez, I.L.; Sylvester, J.E. Complete sequence of the 43 kb human ribosomal DNA repeat: Analysis of the intergenic spacer. Genomics 1995, 27, 320–328. [Google Scholar] [CrossRef]
- Grozdanov, P.; Georgiev, O.; Karagyozov, L. Complete sequence of the 45-kb mouse ribosomal DNA repeat: Analysis of the intergenic spacer. Genomics 2003, 82, 637–643. [Google Scholar] [CrossRef]
- Yang, M.; Kong, X.Y.; Shi, W.; Gong, L.; Luo, H.R.; Wu, B.S. Remarkable sequence polymorphisms in 18S rDNA of Pleuronichthys cornutus (Pleuronectiformes: Pleuronectidae). Gene 2018, 677, 251–258. [Google Scholar] [CrossRef]
- Gong, L.; Shi, W.; Yang, M.; Si, L.Z.; Kong, X.Y. Non-concerted evolution in ribosomal ITS2 sequence in Cynoglossus zanzibarensis (Pleuronectiformes: Cynoglossidae). Biochem. Syst. Ecol. 2016, 66, 181–187. [Google Scholar] [CrossRef]
- Gong, L.; Shi, W.; Yang, M.; Kong, X.Y. Characterization of 18S-ITS1-5.8S rDNA in eleven species in Soleidae: Implications for phylogenetic analysis. Hydrobiologia 2018, 819, 161–175. [Google Scholar] [CrossRef]
- Van Wjormhoudt, A.; Gaume, B.; Le Bras, Y.; Roussel, V.; Huchette, S. Two different and functional nuclear rDNA genes in the abalone Haliotis tuberculata: Tissue differential expression. Genetica 2011, 139, 1217–1227. [Google Scholar] [CrossRef]
- Gálvez, L.; Clarkson, J.P.; Palmero, D. IGS region polymorphisms are responsible for failure of commonly used species-specific primers in Fusarium proliferatum isolates from diseased garlic. Plant. Pathol. 2020, 69, 713–722. [Google Scholar] [CrossRef]
- Chou, C.H.; Chiang, Y.C.; Chiang, T.Y. Within- and between-individual length heterogeneity of the rDNA IGS in Miscanthus sinensis var. glaber (Poaceae): Phylogenetic analyses. Genome 1999, 42, 1088–1093. [Google Scholar]
- Krawczyk, K.; Nobis, M.; Nowak, A.; Szczecińska, M.; Sawicki, J. Phylogenetic implications of nuclear rRNA IGS variation in Stipa L. (Poaceae). Sci. Rep. 2017, 7, 11506. [Google Scholar] [CrossRef] [Green Version]
- Inacio, V.; Rocheta, M.; Morais-Cecilio, L. Molecular organization of the 25S-18S rDNA IGS of Fagus sylvatica and Quercus suber: A comparative analysis. PLoS ONE 2014, 9, e98678. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yu, F.; Li, X.; Luo, L.; Wu, J.; Yang, Y.; Deng, Z.; Chen, R.; Zhang, M. Comparative genetic analysis of the 45S rDNA intergenic spacers from three Saccharum species. PLoS ONE 2017, 12, e0183447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madani, M.; Ward, L.; Vierstraete, A.; De Boer, S.H.; Moens, M. The ribosomal intergenic spacer (IGS) in the potato and tobacco cyst nematodes, Globodera pallida, G. rostochiensis and G. tabacum. Mol. Cell. Probe. 2019, 48, 101441. [Google Scholar] [CrossRef]
- Kakou, B.; Angers, B.; Glémet, H. Extensive length variation in the ribosomal DNA intergenic spacer of yellow perch (Perca flavescens). Genome 2015, 59, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Shen, S.D.; Shen, Z. Cloning and Application of the Complete Nuclear Ribosomal DNA (nrDNA) Cistron Sequence of Pyropia haitanensis (Bangiales, Rhodophyta). Bot. Mar. 2017, 60, 1515–1526. [Google Scholar] [CrossRef]
- Qiu, Y.Y.; Gao, Y.; Li, Y.; Ma, X.X.; Lv, Q.B.; Hu, Y.; Qiu, H.Y.; Chang, Q.C.; Wang, C.R. Comparative analyses of complete ribosomal DNA sequences of Clonorchis sinensis and Metorchis orientalis: IGS sequences may provide a novel genetic marker for intraspecific variation. Infect. Genet. Evol. 2020, 78, 104125. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Genus | Species | ITS | 18S | 28S | |||
|---|---|---|---|---|---|---|---|---|
| GenBank Accession Number | Length (bp) | GenBank Accession Number | Length (bp) | GenBank Accession Number | Length (bp) | |||
| Meretricinae | Meretrix | Meretrix lusoria | DQ389108 | 1567 | JN996714 | 1899 | MW221234 | 739 |
| Meretricinae | Meretrix | Meretrix meretrix | AY695801 | 1480 | EF426291 | 1900 | / | / |
| Tapetinae | Ruditapes | Ruditapes philippinarum | DQ399404 | 1303 | JN807343 | 1833 | AM779742 | 1476 |
| Tapetinae | Ruditapes | Ruditapes variegatus | JN807381 | 1265 | JN807349 | 1832 | DQ343858 | 1388 |
| Tapetinae | Ruditapes | Ruditapes decussatus | HQ634139 | 1259 | KX713340 | 1779 | KX713427 | 2078 |
| Tapetinae | Paratapes | Paphia undulata | EU183531 | 1236 | JN996722 | 1831 | JQ277802 | 743 |
| Chioninae | Leukoma | Protothaca staminea | EF035084 | 1379 | AM774570 | 1779 | AM779744 | 1478 |
| Chioninae | Leukoma | Protothaca jedoensis | DQ220291 | 1437 | EF426292 | 1831 | DQ343856 | 1390 |
| Chioninae | Mercenaria | Mercenaria mercenaria | DQ190445 | 1332 | AF106073 | 1804 | KX713401 | 2085 |
| Callistinae | Saxidomus | Saxidomus gigantea | EF035110 | 1332 | / | / | / | |
| Cyclininae | Cyclina | Cyclina sinensis | DQ190446 | 1263 | EF426289 | 1838 | DQ343849 | 1396 |
| outgroup | Perna viridis | MH279802 | 735 | KY081342 | 1698 | MK419106 | 3679 | |
| Amplified Region | Primers | Sequences (5′–3′) | Annealing Temperature | Extension Time | Length (bp) |
|---|---|---|---|---|---|
| 18S rRNA | 18S-ar | TCAAATGTCTGCCCTATC | 53 °C for R. philippinarum, 55 °C for M. mercenaria | 1 min 30 s | 1501–1573 |
| 18S-br | TTCACCTACGGATACCTTG | ||||
| 18S rRNA–ITS-28S rRNA | ITS-ar | TAACAAGGTATCCGTAGGTG | 53 °C for R. philippinarum, 55 °C for M. mercenaria | 1 min | 1436–1841 |
| ITS-br | CGTGCCAGTATTTAGCC | ||||
| 28S rRNA | 28S1-ar | AGTCGGGTTGTTTGGGAATG | 55 °C | 1 min | 1240–1251 |
| 28S1-br | TTGATTCGGCAGGTGAGTTG | ||||
| 28S2-ar | CTGTGGGATGAACCAAACGC | 55 °C | 1 min 30 s | 1649–1691 | |
| 28S2-br | ACCTTAGGACACCTGCGTTA | ||||
| 28S3-ar | TCACCCACTAATAGGGAACG | 55 °C | 1 min | 691–702 | |
| 28S3-br | AAGCACCTAAACCAAATGTC | ||||
| IGS for R. philippinarum | IGS-ar1 | CCAAATGCCTCGTCATCTAA | 59 °C | 6 min 30 s | 7829 |
| IGS-br1 | CTGCCTTCCTTGGATGTG | ||||
| IGS for M. mercenaria | IGS-ar2 | GAATACAGACCGTGAAAGCG | 56 °C | 5 min 30 s | 6434 |
| IGS-br1 | CTGCCTTCCTTGGATGTG |
| Region | Ruditapes philippinarum | Mercenaria mercenaria | Alignment Length (bp) | Pairwise Identity (%) | Pairwise Distance | Variable Site (V) | ||
|---|---|---|---|---|---|---|---|---|
| Length (bp) | GC Content (%) | Length (bp) | GC Content (%) | |||||
| 18S rRNA | 1832 | 51.85 | 1830 | 51.75 | 1833 | 99.13 | 0.007 | 12 |
| ITS1 | 638 | 64.42 | 679 | 63.04 | 692 | 46.24 | 0.599 | 254 |
| 5.8S rRNA | 157 | 57.96 | 157 | 57.96 | 157 | 100.00 | 0.000 | 0 |
| ITS2 | 434 | 65.67 | 420 | 65.72 | 446 | 61.66 | 0.432 | 133 |
| 28S rRNA | 3633 | 57.68 | 3612 | 57.22 | 3638 | 96.65 | 0.025 | 90 |
| IGS | 6216 | 61.18 | 5402 | 58.33 | 6239 | 40.47 | 0.878 | 2724 |
| Species | Sub-Repeat | Width (bp) | Copy Number | Location | GC Content (%) | Entropy (0–2) |
|---|---|---|---|---|---|---|
| Ruditapes philippinarum | R-S1 | 36 | 3.8 | 592–727 | 55.65 | 1.99 |
| R-S2 | 154 | 3 | 700–1161 | 54.55 | 1.98 | |
| R-S3 | 4 | 29 | 2635–2752 | 75.00 | 1.49 | |
| R-S4 | 62 | 2 | 3164–3284 | 63.64 | 1.90 | |
| R-S5 | 72 | 1.9 | 3320–3454 | 65.75 | 1.89 | |
| R-S6 | 141 | 2 | 4338–4619 | 65.96 | 1.91 | |
| R-S7 | 72 | 1.9 | 4637–4771 | 65.75 | 1.89 | |
| Mercenaria mercenaria | M-S1 | 48 | 3 | 3615–3761 | 66.67 | 1.89 |
| M-S2 | 96 | 1.8 | 3599–3776 | 64.29 | 1.88 | |
| M-S3 | 48 | 3 | 4054–4197 | 62.50 | 1.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Wang, Z.; Hou, X. Comparative Analysis of the nrDNA Repeat Unit of Manila Clam Ruditapes philippinarum and Quahog Mercenaria mercenaria. Fishes 2021, 6, 42. https://doi.org/10.3390/fishes6030042
Guo Z, Wang Z, Hou X. Comparative Analysis of the nrDNA Repeat Unit of Manila Clam Ruditapes philippinarum and Quahog Mercenaria mercenaria. Fishes. 2021; 6(3):42. https://doi.org/10.3390/fishes6030042
Chicago/Turabian StyleGuo, Zhansheng, Zhen Wang, and Xuguang Hou. 2021. "Comparative Analysis of the nrDNA Repeat Unit of Manila Clam Ruditapes philippinarum and Quahog Mercenaria mercenaria" Fishes 6, no. 3: 42. https://doi.org/10.3390/fishes6030042
APA StyleGuo, Z., Wang, Z., & Hou, X. (2021). Comparative Analysis of the nrDNA Repeat Unit of Manila Clam Ruditapes philippinarum and Quahog Mercenaria mercenaria. Fishes, 6(3), 42. https://doi.org/10.3390/fishes6030042