
Transcriptome Analysis Reveals Cross-Tissue Metabolic Pathway Changes in Female Rana dybowskii during Emergence from Hibernation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Samples
2.2. Total RNA Extraction and RNA Sequencing
2.3. Sequence Alignment and Functional Annotation of RNA-Seq Data
2.4. Functional Enrichment Analysis of Differentially Expressed Genes
2.5. Validation of Gene Expression Data by Real-Time Quantitative RT-PCR
3. Results
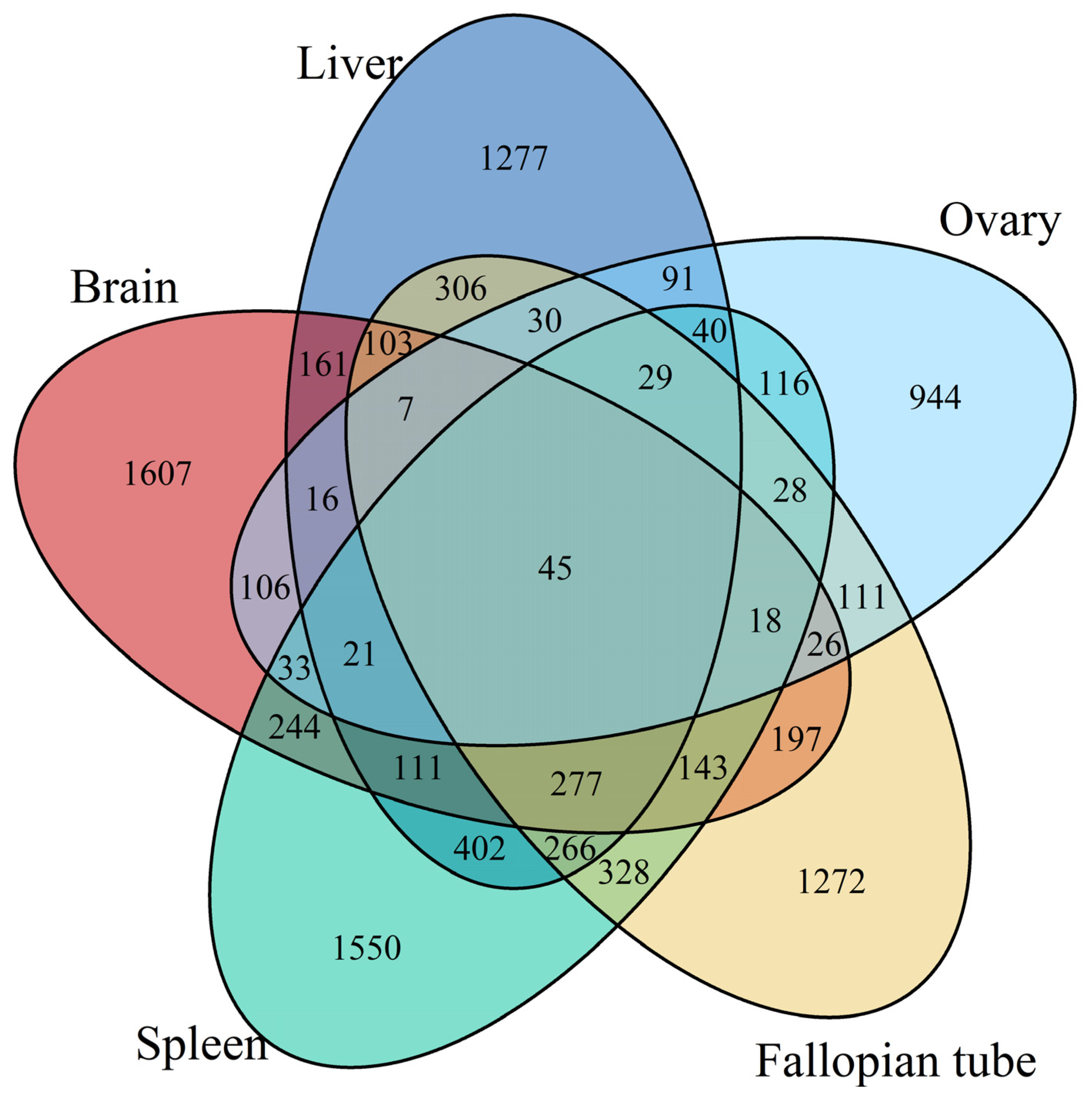
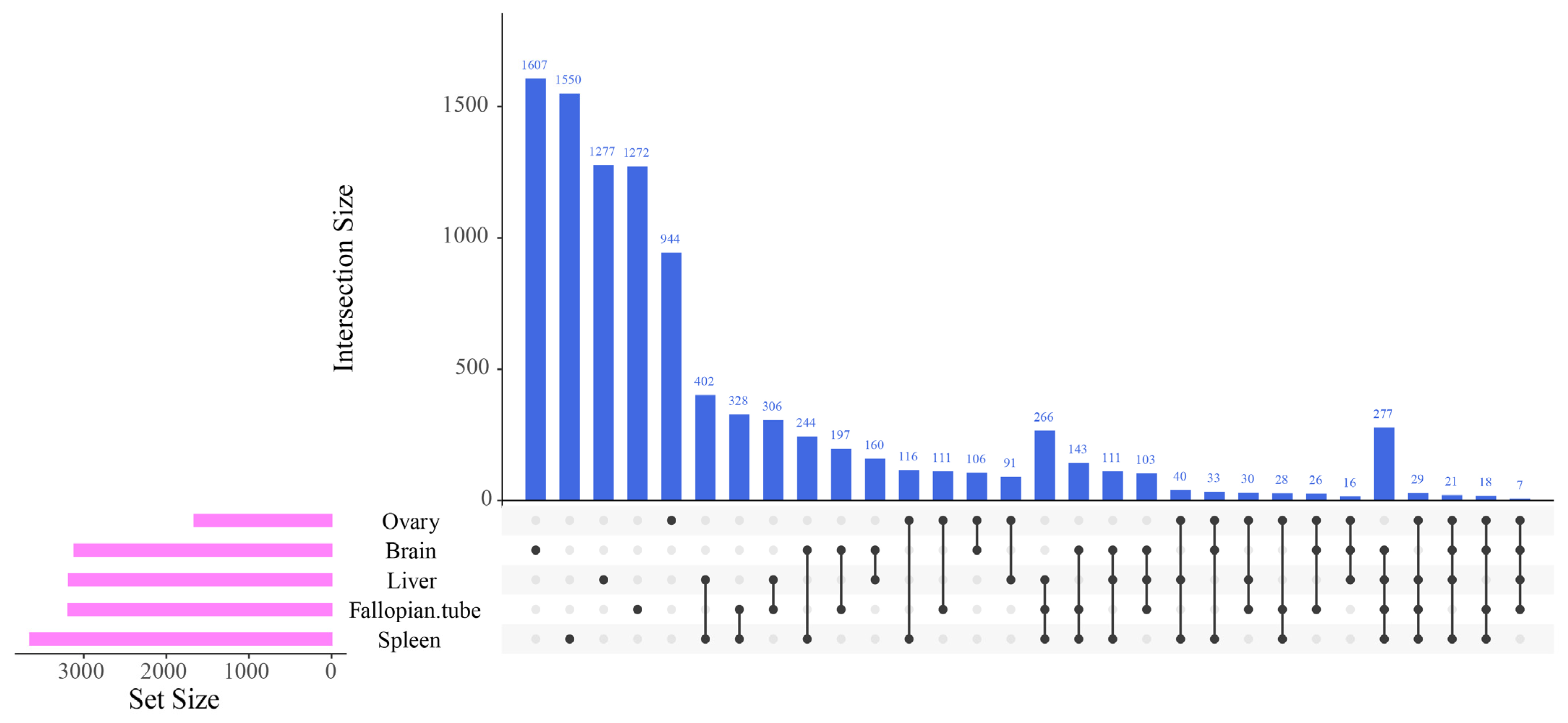
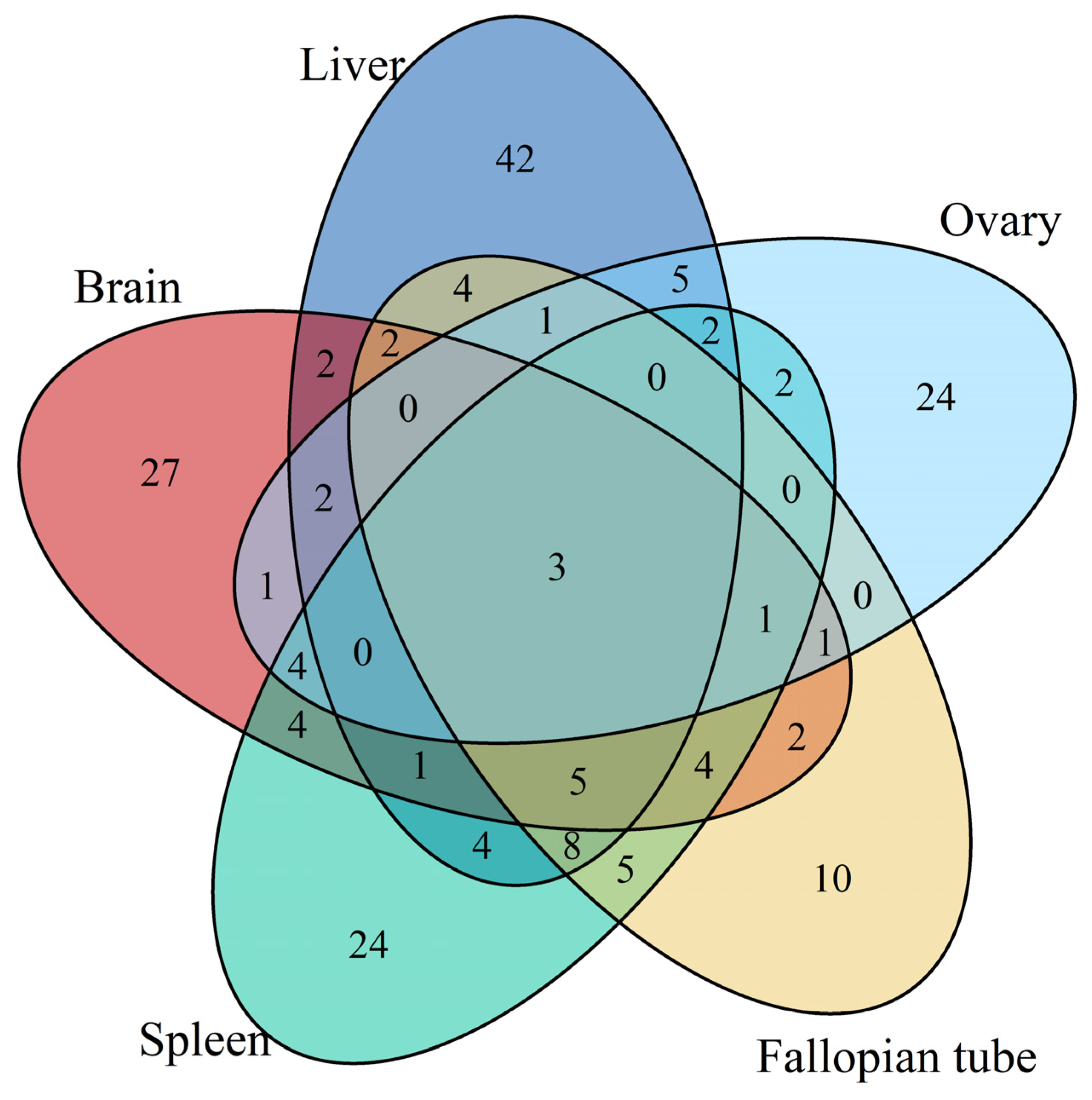
3.1. Differentially Expressed Gene Identification Involved in the Emergence from Hibernation
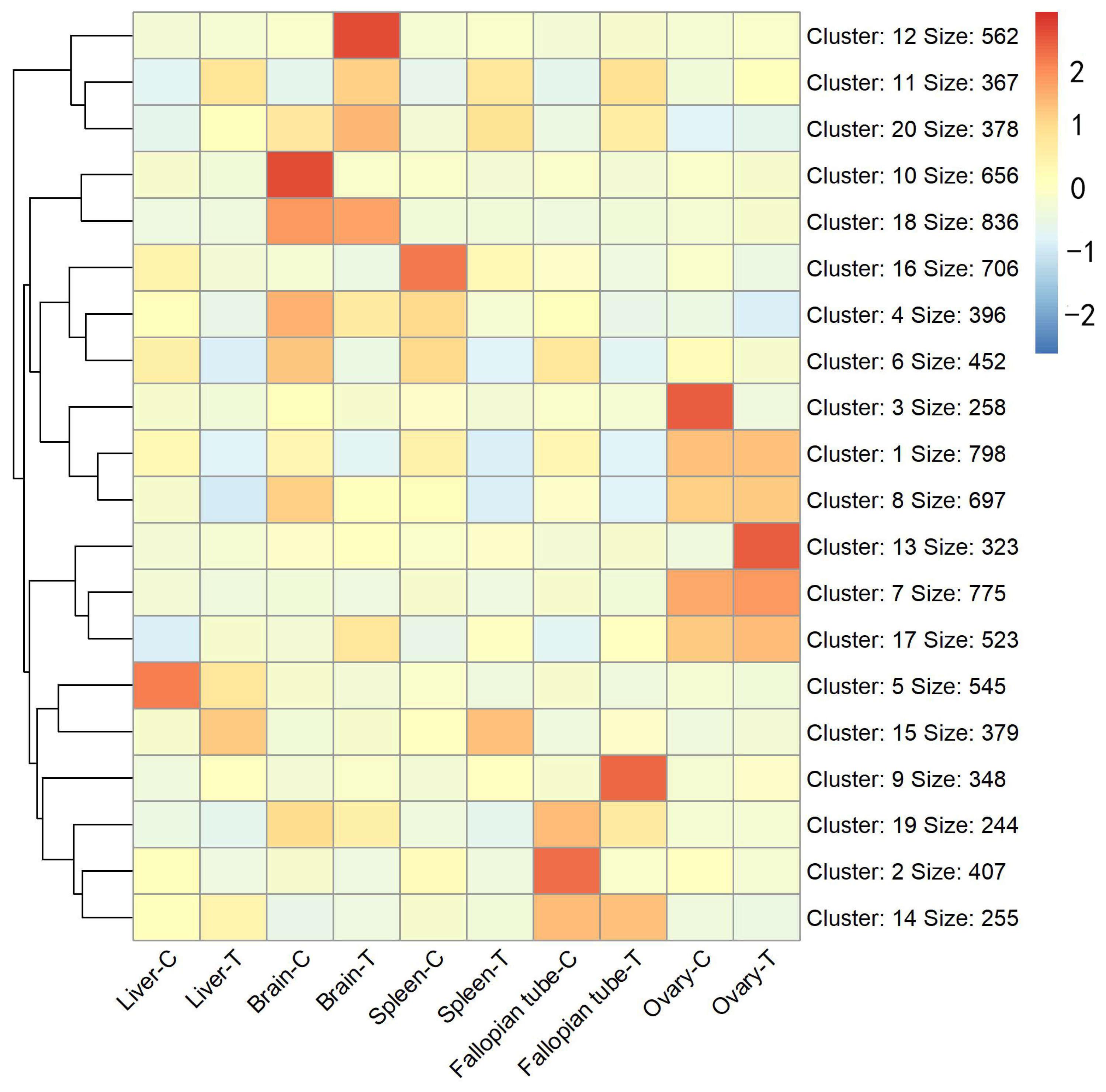
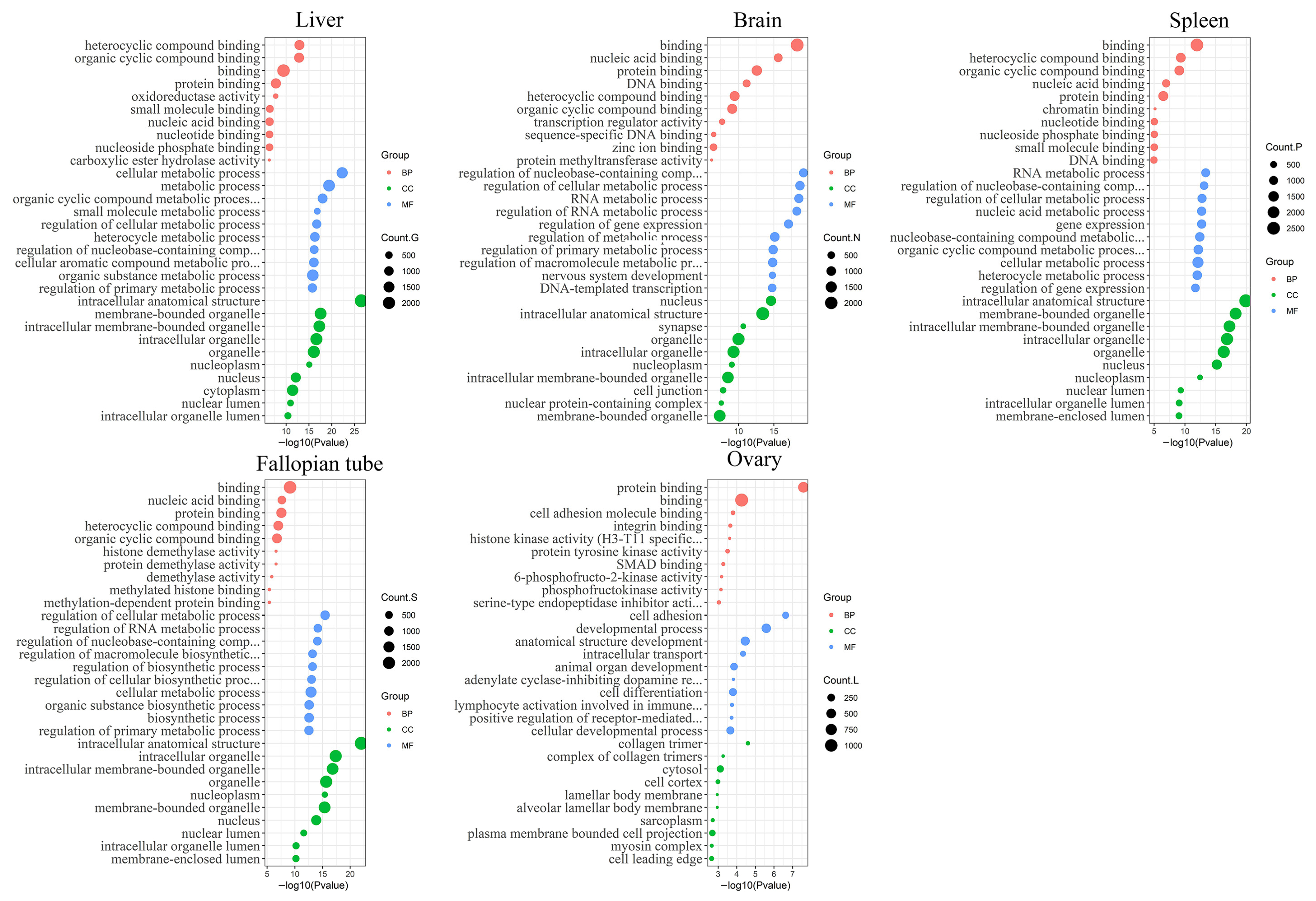
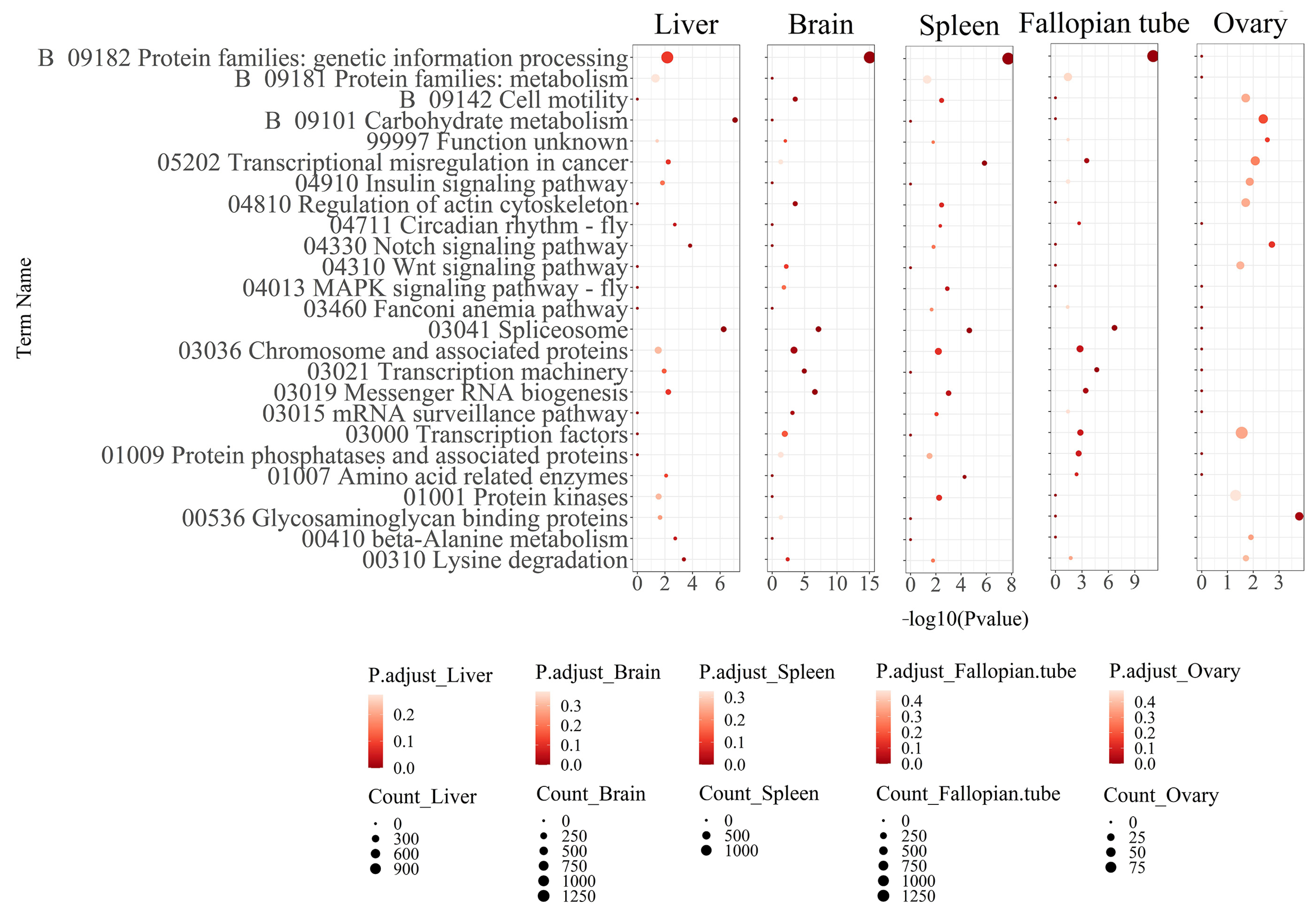
3.2. Functional Annotation Analysis of the Differentially Expressed Transcripts
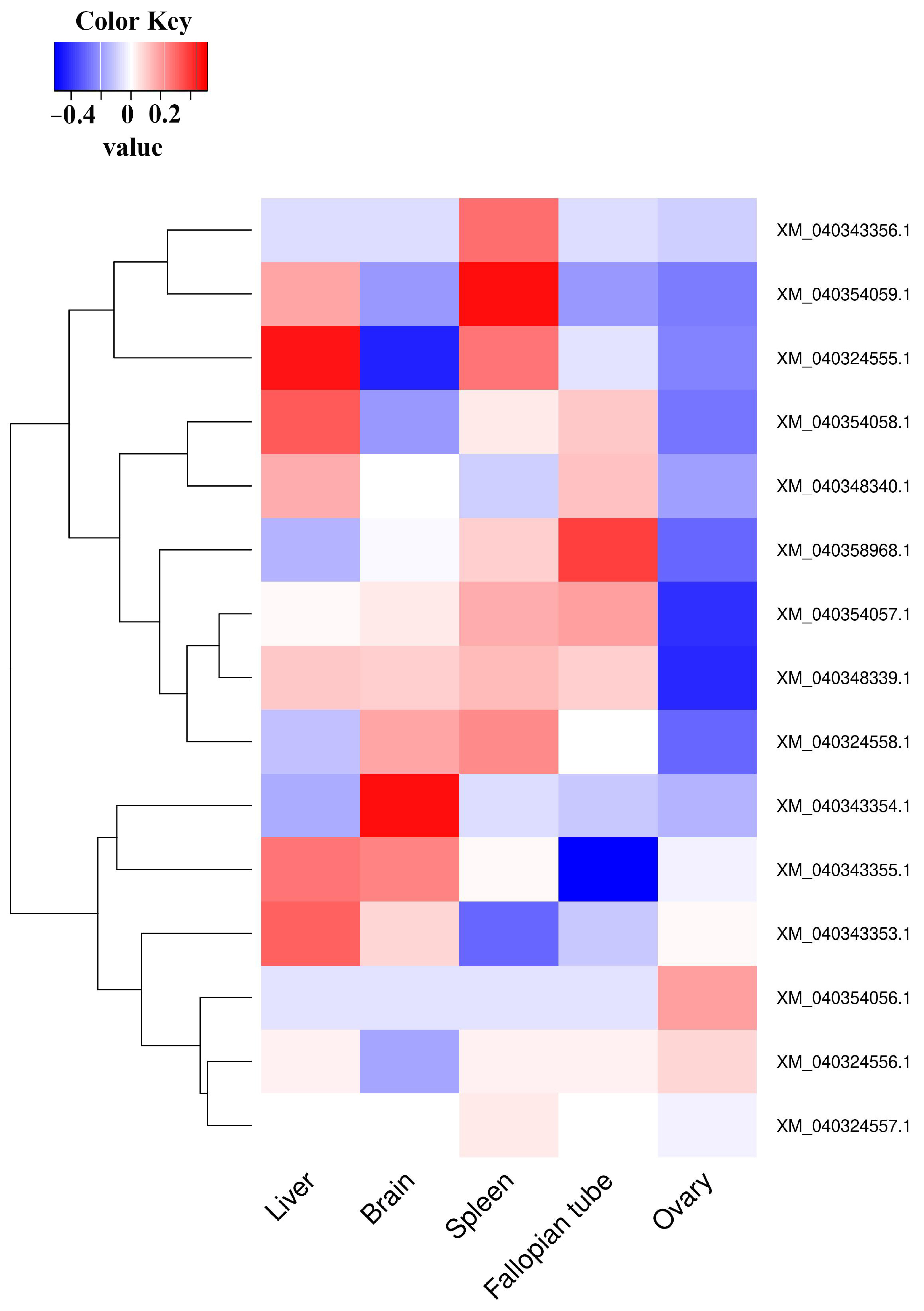
3.3. Validation of Expression Data by Real-Time Quantitative RT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, B.-T.; Zhou, Y.; Min, M.-S.; Matsui, M.; Dong, B.-J.; Li, P.-P.; Fong, J.J. Diversity and phylogeography of Northeast Asian brown frogs allied to Rana dybowskii (Anura, Ranidae). Mol. Phylogenet. Evol. 2017, 112, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, H.; Shang, Y.; Zhu, R.; Hong, X.; Song, Z.; Yang, Z. Development of the general chapters of the Chinese Pharmacopoeia 2020 edition: A review. J. Pharm. Anal. 2021, 11, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gan, Y.; Mao, X.; Kan, H.; Li, N.; Zhang, C.; Wang, Z.; Wang, Y. Antioxidant activity evaluation of Oviductus ranae protein hydrolyzed by different proteases. Molecules 2021, 26, 1625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jia, X.-y.; Ma, Y.-d.; Ma, J.-z. Genetic diversity and differentiation of the Dybowski’s frog (Rana dybowskii) in Northeast China. J. For. Res. 2010, 21, 239–245. [Google Scholar] [CrossRef]
- Staples, J.F. Metabolic flexibility: Hibernation, torpor, and estivation. Compr. Physiol. 2016, 6, 737–771. [Google Scholar]
- Malan, A. Adaptation to poikilothermy in endotherms. J. Therm. Biol. 1983, 8, 79–84. [Google Scholar] [CrossRef]
- Lin, J.-Q.; Yu, J.; Jiang, H.; Zhang, Y.; Wan, Q.-H.; Fang, S.-G. Multi-omics analysis reveals that natural hibernation is crucial for oocyte maturation in the female Chinese alligator. BMC Genom. 2020, 21, 774. [Google Scholar] [CrossRef]
- Santana, F.E.; Swaisgood, R.R.; Lemm, J.M.; Fisher, R.N.; Clark, R.W. Chilled frogs are hot: Hibernation and reproduction of the endangered mountain yellow-legged frog Rana muscosa. Endanger. Species Res. 2015, 27, 43–51. [Google Scholar] [CrossRef]
- Costanzo, J.P.; do Amaral, M.C.F.; Rosendale, A.J.; Lee, R.E., Jr. Hibernation physiology, freezing adaptation and extreme freeze tolerance in a northern population of the wood frog. J. Exp. Biol. 2013, 216, 3461–3473. [Google Scholar] [CrossRef]
- Gao, X.; Jin, C.; Llusia, D.; Li, Y. Temperature-induced shifts in hibernation behavior in experimental amphibian populations. Sci. Rep. 2015, 5, 11580. [Google Scholar] [CrossRef]
- Tong, Q.; Dong, W.J.; Long, X.Z.; Hu, Z.F.; Luo, Z.W.; Guo, P.; Cui, L.Y. Effects of fine-scale habitat quality on activity, dormancy, habitat use, and survival after reproduction in Rana dybowskii (Chordata, Amphibia). BMC Zool. 2023, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Chen, Q.; Storey, K.B.; Teng, L.; Li, X.; Xu, T.; Zhang, H. Physiological ecology of winter hibernation by the high-altitude frog Nanorana parkeri. Physiol. Biochem. Zool. 2022, 95, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Pimentel, H.; Bray, N.L.; Pachter, L. Gene-level differential analysis at transcript-level resolution. Genome Biol. 2018, 19, 53. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef]
- Ungaro, A.; Pech, N.; Martin, J.-F.; McCairns, R.J.S.; Mévy, J.-P.; Chappaz, R.; Gilles, A. Challenges and advances for transcriptome assembly in non-model species. PLoS ONE 2017, 12, e0185020. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Sun, M.A.; Yan, P. Pathway and network analysis of differentially expressed genes in transcriptomes. Methods Mol. Biol. 2018, 1751, 35–55. [Google Scholar]
- Jin, L.; Zuo, X.-Y.; Su, W.-Y.; Zhao, X.-L.; Yuan, M.-Q.; Han, L.-Z.; Zhao, X.; Chen, Y.-D.; Rao, S.-Q. Pathway-based analysis tools for complex diseases: A review. Genom. Proteom. Bioinform. 2014, 12, 210–220. [Google Scholar] [CrossRef]
- Cirillo, E.; Parnell, L.D.; Evelo, C.T. A review of pathway-based analysis tools that visualize genetic variants. Front. Genet. 2017, 8, 174. [Google Scholar] [CrossRef]
- Streicher, J.W. The genome sequence of the common frog, Rana temporaria Linnaeus 1758. Wellcome Open Res. 2021, 6, 286. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Makowski, D.; Jones, J.W.; Brun, F. (Eds.) Chapter 3—The R programming language and software. In Working with Dynamic Crop Models, 2nd ed.; Academic Press: San Diego, CA, USA, 2014; pp. 71–117. [Google Scholar]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment analysis for gene ontology. R Package 2019, version 2.38.1.
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Geiser, F. Metabolic rate and body temperature reduction during hibernation and daily torpor. Annu. Rev. Physiol. 2004, 66, 239–274. [Google Scholar] [CrossRef]
- Lahiri, A.; Senior, R.; Khattar, R. The role of neurohormonal antagonists in hibernating myocardium. J. Cardiovasc. Pharmacol. 1999, 33, S9–S16. [Google Scholar]
- Sotowska-Brochocka, J.; Martyńska, L.; Licht, P. Dopaminergic inhibition of gonadotropic release in hibernating frogs, Rana temporaria. Gen. Comp. Endocrinol. 1994, 93, 192–196. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Li, M.; Liu, S.; Yu, J.; Yan, Z.; Zhou, H. Traditional uses, bioactive constituents, biological functions, and safety properties of Oviductus ranae as functional foods in China. Oxidative Med. Cell. Longev. 2019, 2019, 4739450. [Google Scholar] [CrossRef]
- Raser, J.M.; O’shea, E.K. Noise in gene expression: Origins, consequences, and control. Science 2005, 309, 2010–2013. [Google Scholar] [CrossRef] [PubMed]
- Pian, C.; He, M.; Chen, Y. Pathway-based personalized analysis of Pan-Cancer transcriptomic data. Biomedicines 2021, 9, 1502. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.E. Review of Lysine metabolism with a focus on humans. J. Nutr. 2020, 150, 2548s–2555s. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.; Kwak, H.; Cheong, E.; Lee, C.J. GABA tone regulation and its cognitive functions in the brain. Nat. Rev. Neurosci. 2023, 24, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, X.; Ma, J.; Xu, G. The fine-tuned crosstalk between lysine acetylation and the circadian rhythm. Biochim. Biophys. Acta Gene Regul. Mech. 2023, 1866, 194958. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef]
- Wong, S.H.D.; Yin, B.; Li, Z.; Yuan, W.; Zhang, Q.; Xie, X.; Tan, Y.; Wong, N.; Zhang, K.; Bian, L. Mechanical manipulation of cancer cell tumorigenicity via heat shock protein signaling. Sci. Adv. 2023, 9, eadg9593. [Google Scholar] [CrossRef]
- Tremblay, M.; Sanchez-Ferras, O.; Bouchard, M. GATA transcription factors in development and disease. Development 2018, 145, dev164384. [Google Scholar] [CrossRef]
- Fujikura, J.; Yamato, E.; Yonemura, S.; Hosoda, K.; Masui, S.; Nakao, K.; Miyazaki, J.-i.; Niwa, H. Differentiation of embryonic stem cells is induced by GATA factors. Genes Dev. 2002, 16, 784–789. [Google Scholar] [CrossRef]
- Shu, J.; Zhang, K.; Zhang, M.; Yao, A.; Shao, S.; Du, F.; Yang, C.; Chen, W.; Wu, C.; Yang, W. GATA family members as inducers for cellular reprogramming to pluripotency. Cell Res. 2015, 25, 169–180. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| XM_040324555.1 | TCAAGGACACAGTCATGGAT | TGCCACACAAGTTGTAGAAG |
| XM_040324558.1 | TACTCTACTGCTCCCTTCCT | CACTGTAGCACCACAATTCA |
| XM_040343354.1 | TAAGGGTATAGGTGGAAGCC | TACACTCCTTGTCTTCCTGG |
| XM_040343355.1 | GAGGAGGTGGATGTACTCTT | GGCTTCCACCTATACCCTTA |
| XM_040343356.1 | TAAGGGTATAGGTGGAAGCC | TACACTCCTTGTCTTCCTGG |
| XM_040348339.1 | AGAACAAGCGATCTGGGATA | CCCAGCCCCTTCATAAGAT |
| XM_040348340.1 | TCTGTTGCTGGAGAAAGTTG | GTTGTAGGCTGAGCTCTCT |
| XM_040354056.1 | TACCAGGGTTGGATCTATGC | ATTGATCTCTGCCATTCACG |
| XM_040354057.1 | TACCATTACTCTCCCAGTCC | GAAGACTATGCAGCATGGAG |
| XM_040354058.1 | TACACCCATCTCCTACCTTC | ATTGATCTCTGCCATTCACG |
| β-actin | AAGAATGAGGGCTGGAACA | GTGCGTGACATCAAGGAGAAGC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Luan, P.; Li, M.; Zhang, T.; Shu, Y.; Hu, G. Transcriptome Analysis Reveals Cross-Tissue Metabolic Pathway Changes in Female Rana dybowskii during Emergence from Hibernation. Fishes 2023, 8, 569. https://doi.org/10.3390/fishes8120569
Chen F, Luan P, Li M, Zhang T, Shu Y, Hu G. Transcriptome Analysis Reveals Cross-Tissue Metabolic Pathway Changes in Female Rana dybowskii during Emergence from Hibernation. Fishes. 2023; 8(12):569. https://doi.org/10.3390/fishes8120569
Chicago/Turabian StyleChen, Feng, Peixian Luan, Manman Li, Tianxiang Zhang, Yongjun Shu, and Guo Hu. 2023. "Transcriptome Analysis Reveals Cross-Tissue Metabolic Pathway Changes in Female Rana dybowskii during Emergence from Hibernation" Fishes 8, no. 12: 569. https://doi.org/10.3390/fishes8120569