
Description of a Novel Fish Pathogen, Plesiomonas shigelloides subsp. oncorhynchi, Isolated from Rainbow Trout (Oncorhynchus mykiss): First Genome Analysis and Comparative Genomics
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacteria Isolation and Phenotypic Characterization
2.2. DNA Extraction, PCR and Sequence Analysis
2.3. Genome Sequencing, Assembly and Identification
2.4. Comparative Genome Analysis
2.5. Antimicrobial Susceptibility and Resistance Gene Characteristics
3. Results
3.1. Phenotypic Characterization
3.2. PCR and Sequence Analysis
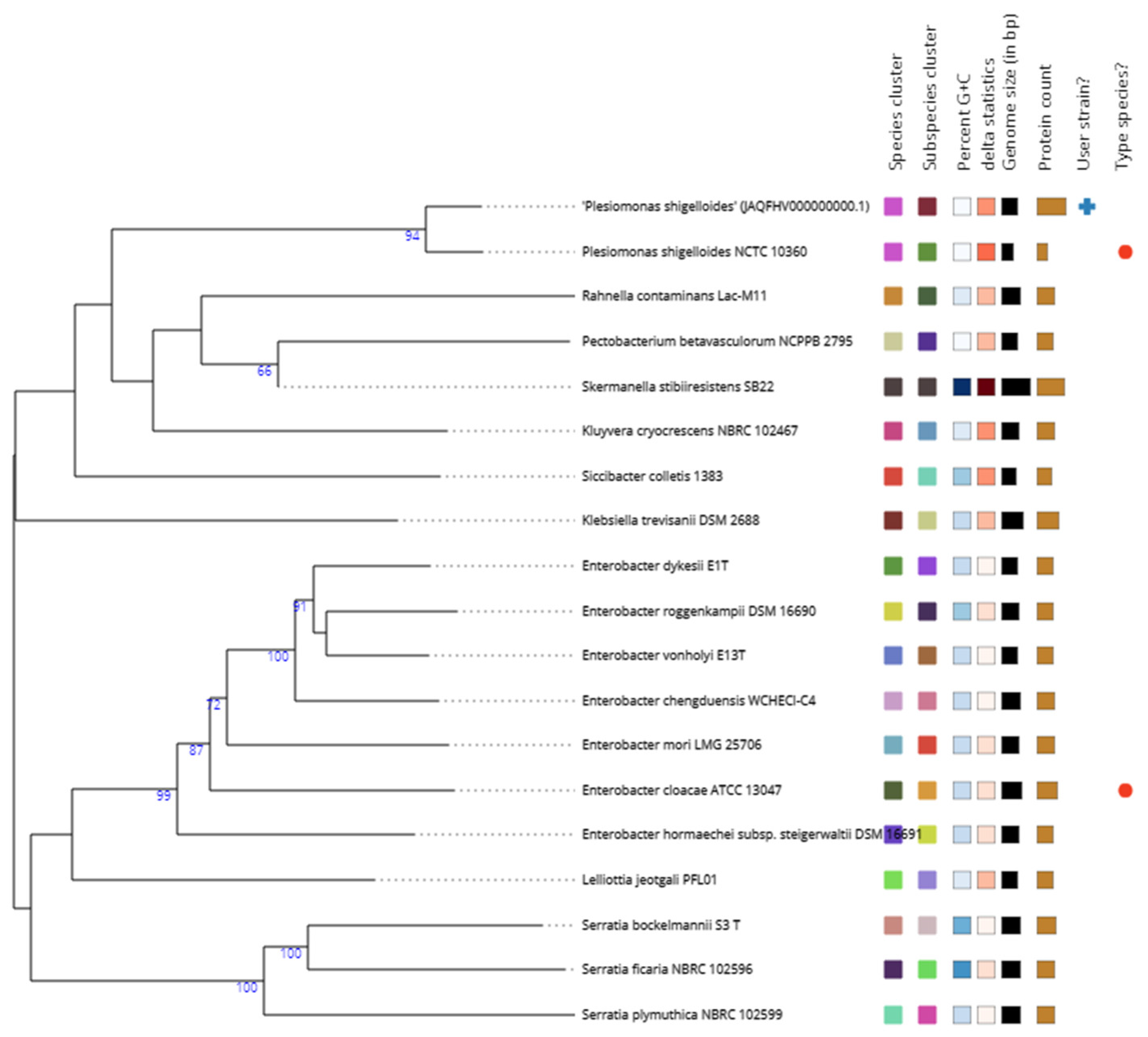
3.3. Phylogenomics
3.4. Functional and Ecological Analyses
3.5. Secondary Metabolites
3.6. Antimicrobial Susceptibility
3.7. Antimicrobial Resistance Genes and Virulence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- Protologue: Emended description of Plesiomonas shigelloides (Bader 1954) Habs and Schubert 1962.
- N.L. fem. dim. n. Shigella, a generic name; L. adj. suff. -oides, ressembling, similar; from Gr. neut. adj. suff. -eides, resembling, similar; from Gr. neut. n. eîdos, that which is seen, form, shape, figure; N.L. adj. shigelloides, Shigella-like
- The description is the same as reported by Janda (2015), with the following modifications:
- The genome size of the type strain is 3.4 Mb and genomic G + C content is 52.0%. Type strain: ATCC 14029; CCUG 410; CIP 63.5; DSM 8224; LMG 4242; NCCB 80007; NCTC 10360.
- cvof Plesiomonas shigelloides subsp shigelloides subsp. nov.
- N.L. fem. dim. n. Shigella, a generic name; L. adj. suff. -oides, ressembling, similar; from Gr. neut. adj. suff. -eides, resembling, similar; from Gr. neut. n. eîdos, that which is seen, form, shape, figure; N.L. adj. shigelloides, Shigella-like
- The description is as given by Janda (2015), with the following modifications: The genome size of the type strain is 3.4 Mb and genomic G + C content is 52.0%. Genome size of strains in the subspecies ranges from 3.0 to 4.08. Genomic GC content ranges from 50.9 to 52.4%. Type strain: ATCC 14029; CCUG 410; CIP 63.5; DSM 8224; LMG 4242; NCCB 80007; NCTC 10360.
- Description of Plesiomonas shigelloides subsp. oncorhynchi subsp. nov.
- on.co.rhyn’chi. N.L. gen. masc. n. oncorhynchi, of Oncorhynchus, named after the rainbow trout, Oncorhynchus mykiss, from which the type strain was isolated
- Gram-negative, short bacilli, motile, oxidase and catalase positive, facultatively aerobic, glucose fermentative, non-hemolytic on sheep blood, grown on Mac Conkey agar but not on thiosulfate-citrate-bile salts-sucrose agar (TCBS). Able to tolerate up to 1.5% NaCl and grow at a temperature range of 4–45 °C. Negative for gelatin, Tween 20, and Tween 80 hydrolysis. Susceptible to Vibriostatic agent at 10 µg and 150 µg. The isolate V-63 was collected from rainbow trout weighing 0.5–1 g from farms located in Kütahya (latitude: 39°46′16.5″ N, longitude: 29°38′32.5″ E), and the sampling date was 01.04.2018. The isolate V78 was collected from rainbow trout weighing 200 g from farms located in Kayseri (Latitude: 39°00′15.1″ N, Longitude: 36°38′32.5″ E), and the sampling date was 01.09.2013. AF160 was isolated from goldfish kept in an aquarium producer in Istanbul (latitude: 40°58′41.4″ N, longitude: 28°47′15.1″ E), Turkey. The sampling date for AF160 was 2021.
- The genome size of the type strain was 4.4 Mb, and the genomic G + C content was 51.1%. The GenBank accession number for the whole genome is JAQFHV000000000. The GenBank accession numbers for the 16S rRNA genes are OQ683920 and OQ683923 for strain V-63 and V78, respectively.
References
- Steinberg, J.P.; Lutgring, J.D.; Burd, E.M. Other Gram-Negative and Gram-Variable Bacilli—Clinical Key. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 9, pp. 2847–2864. [Google Scholar]
- Janda, J.M. Plesiomonas. In Bergey’s Manual of Systematics of Archaea and Bacteria; Whitman, W.B., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 324, pp. 1–11. ISBN 9781118960608. [Google Scholar]
- Huber, I.; Spanggaard, B.; Appel, K.F.; Rossen, L.; Nielsen, T.; Gram, L. Phylogenetic Analysis and in Situ Identification of the Intestinal Microbial Community of Rainbow Trout (Oncorhynchus Mykiss, Walbaum). J. Appl. Microbiol. 2004, 96, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Miranda, C.; Palomares, E.; Jurado, M.; Marín, A.; Vega, F.; Soriano-Vargas, E. Isolation and Distribution of Bacterial Flora in Farmed Rainbow Trout from Mexico. J. Aquat. Anim. Health 2010, 22, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Lin, Q.; Shi, C.; Fu, X.; Li, N.; Liu, L.; Wu, S. Isolation and Identification of a Pathogenic Plesiomonas shigelloides from Diseased Grass Carp. Wei Sheng Wu Xue Bao Acta Microbiol. Sin. 2014, 54, 229–235. [Google Scholar]
- Pakingking, R.; Palma, P.; Usero, R. Quantitative and Qualitative Analyses of the Bacterial Microbiota of Tilapia (Oreochromis niloticus) Cultured in Earthen Ponds in the Philippines. World J. Microbiol. Biotechnol. 2015, 31, 265–275. [Google Scholar] [CrossRef]
- Nadirah, M.; Ruhil, H.H.; Jalal, K.C.A.; Najiah, M. Occurrence of Plesiomonas shigelloides in Cultured Red Hybrid Tilapia (Oreochromis niloticus) from Tropical Rivers, East Coast Malaysia. Pakistan J. Biol. Sci. 2012, 15, 600–603. [Google Scholar] [CrossRef]
- Liu, Z.; Ke, X.; Lu, M.; Gao, F.; Cao, J.; Zhu, H.; Wang, M. Identification and Pathological Observation of a Pathogenic Plesiomonas shigelloides Strain Isolated from Cultured Tilapia (Oreochromis niloticus). Wei Sheng Wu Xue Bao Acta Microbiol. Sin. 2015, 55, 96–106. [Google Scholar]
- Michael Janda, J.; Abbott, S.L.; McIver, C.J. Plesiomonas shigelloides Revisited. Clin. Microbiol. Rev. 2016, 29, 349–374. [Google Scholar] [CrossRef]
- Cruz, J.M.; Saraiva, A.; Eiras, J.C.; Branco, R.; Sousa, J.C. An Outbreak of Plesiomonas shigelloides in Farmed Rainbow Trout, Salmo Gairdneri Richardson, in Portugal. Bull. Eur. Assoc. Fish Pathol. 1986, 6, 20. [Google Scholar]
- Vladik, P.; Vitovec, J. Plesiomonas shigelloides in Rainbow Troup Septicemia. Vet. Med. 1974, 19, 297–301. [Google Scholar]
- Wang, X.; Xu, L.; Cao, H.; Wang, J.; Wang, S. Identification and Drug Sensitivity of a Plesiomonas shigelloides Isolated from Diseased Sturgeons. Wei Sheng Wu Xue Bao Acta Microbiol. Sin. 2013, 53, 723–729. [Google Scholar]
- Liu, Y.; Rzeszutek, E.; Van Der Voort, M.; Wu, C.H.; Thoen, E.; Skaar, I.; Bulone, V.; Dorrestein, P.C.; Raaijmakers, J.M.; De Bruijn, I. Diversity of Aquatic Pseudomonas Species and Their Activity against the Fish Pathogenic Oomycete Saprolegnia. PLoS ONE 2015, 10, e0136241. [Google Scholar] [CrossRef]
- Nisha, R.G.; Rajathi, V.; Manikandan, R.; Prabhu, N.M. Isolation of Plesiomonas shigelloides from Infected Cichlid Fishes Using 16S RRNA Characterization and Its Control with Probiotic pseudomonas sp. Acta Sci. Vet. 2014, 42, 1–7. [Google Scholar]
- Ruimy, R.; Breittmayer, V.; Elbase, P.; Lafay, B.; Boussemart, O.; Gauthier, M.; Christen, R. Phylogenetic Analysis and Assessment of the Genera Vibrio, Photobacterium, Aeromonas, and Plesiomonas Deduced from Small-Subunit RRNA Sequences. Int. J. Syst. Bacteriol. 1994, 44, 416–426. [Google Scholar] [CrossRef]
- Salerno, A.; Delétoile, A.; Lefevre, M.; Ciznar, I.; Krovacek, K.; Grimont, P.; Brisse, S. Recombining Population Structure of Plesiomonas shigelloides (Enterobacteriaceae) Revealed by Multilocus Sequence Typing. J. Bacteriol. 2007, 189, 7808–7818. [Google Scholar] [CrossRef]
- Abdelhamed, H.; Ozdemir, O.; Tekedar, H.C.; Arick, M.A.; Hsu, C.Y.; Karsi, A.; Lawrence, M.L. Complete Genome Sequence of Multidrug-Resistant Plesiomonas shigelloides Strain MS-17-188. Genome Announc. 2018, 6, e00387-18. [Google Scholar] [CrossRef]
- Piqué, N.; Aquilini, E.; Alioto, T.; Miñana-Galbis, D.; Tomás, J.M. Genome Sequence of Plesiomonas shigelloides Strain 302-73 (Serotype O1). Genome Announc. 2013, 1, e00404-13. [Google Scholar] [CrossRef]
- Behera, B.K.; Bera, A.K.; Paria, P.; Das, A.; Parida, P.K.; Kumari, S.; Bhowmick, S.; Das, B.K. Identification and Pathogenicity of Plesiomonas shigelloides in Silver Carp. Aquaculture 2018, 493, 314–318. [Google Scholar] [CrossRef]
- Herrera, F.C.; Santos, J.A.; Otero, A.; García-López, M.L. Occurrence of Plesiomonas shigelloides in Displayed Portions of Saltwater Fish Determined by a PCR Assay Based on the HugA Gene. Int. J. Food Microbiol. 2006, 108, 233–238. [Google Scholar] [CrossRef]
- Martins, A.F.M.; Fontana, H.; Moreira, B.M.; Bonelli, R.R. Draft Genome Sequence of a Tetracycline-Resistant Plesiomonas shigelloides Strain Isolated from Aquaculture-Reared Tilapia. Microbiol. Resour. Announc. 2018, 7, e00832-18. [Google Scholar] [CrossRef]
- Duman, M.; Buján, N.; Altun, S.; Romalde, J.L.; Saticioglu, I.B. Population Genetic and Evolution Analysis of Vibrio Isolated from Turkish Fish Farms. Aquaculture 2023, 562, 738728. [Google Scholar] [CrossRef]
- PubMLST, P. Databases for Molecular Typing and Microbial Genome Diversity Vibrio spp.|PubMLST. Available online: https://pubmlst.org/organisms/vibrio-spp (accessed on 6 March 2023).
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A Modular and Extensible Implementation of the RAST Algorithm for Building Custom Annotation Pipelines and Annotating Batches of Genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Mungan, M.D.; Alanjary, M.; Blin, K.; Weber, T.; Medema, M.H.; Ziemert, N. ARTS 2.0: Feature Updates and Expansion of the Antibiotic Resistant Target Seeker for Comparative Genome Mining. Nucleic Acids Res. 2020, 48, W546–W552. [Google Scholar] [CrossRef]
- Bertelli, C.; Gray, K.L.; Woods, N.; Lim, A.C.; Tilley, K.E.; Winsor, G.L.; Hoad, G.R.; Roudgar, A.; Spencer, A.; Peltier, J.; et al. Enabling Genomic Island Prediction and Comparison in Multiple Genomes to Investigate Bacterial Evolution and Outbreaks. Microb. Genom. 2022, 8, 000818. [Google Scholar] [CrossRef]
- Hitch, T.C.A.; Riedel, T.; Oren, A.; Overmann, J.; Lawley, T.D.; Clavel, T. Automated Analysis of Genomic Sequences Facilitates High-Throughput and Comprehensive Description of Bacteria. ISME Commun. 2021, 1, 16. [Google Scholar] [CrossRef]
- CLSI—Clinical and Laboratory Standards Institute. VET03-A Methods for Antimicrobial Disk Susceptibility Testing of Bacteria Isolated From Aquatic Animals; Approved Guideline A Guideline for Global Application Developed through the Clinical and Laboratory Standards Institute Consensus Process; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2006. [Google Scholar]
- Jain, C.; Rodriguez, R.L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Qin, Q.L.; Xie, B.B.; Zhang, X.Y.; Chen, X.L.; Zhou, B.C.; Zhou, J.; Oren, A.; Zhang, Y.Z. A Proposed Genus Boundary for the Prokaryotes Based on Genomic Insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A Comprehensive Open Resource of Processed 16S RRNA Microbial Profiles for Ecology and Diversity Studies. Sci. Rep. 2016, 6, 33721. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef]
- EUCAST-European Committee on Antimicrobial Susceptibility Testing EUCAST: Clinical Breakpoints and Dosing of Antibiotics. Available online: https://www.eucast.org/clinical_breakpoints/ (accessed on 16 March 2023).
- Monteil, H.; Harf-Monteil, C. Plesiomonas Shigelloides: Une Bactérie Exotique. La Lett. L’infectiologue 1997, 12, 255–262. [Google Scholar]
- Levin, R.E. Plesiomonas shigelloides—An Aquatic Food Borne Pathogen: A Review of Its Characteristics, Pathogenicity, Ecology, and Molecular Detection. Food Biotechnol. 2008, 22, 189–202. [Google Scholar] [CrossRef]
- Miller, M.L.; Koburger, J.A. Plesiomonas Shigelloides: An Opportunistic Food and Waterborne Pathogen. J. Food Prot. 1985, 48, 449–457. [Google Scholar] [CrossRef]
- Habs, H.; Schubert, R.H. Uber Die Biochemischen Merkmale Und Die Taxonomische Stellung von Pseudomonas Shigelloides (Bader). Zent. Bakteriol. Parasitenkd. Infekt. Hyg. Abteılung 1962, 186, 316. [Google Scholar]
- Gu, W.; Gonzalez-Rey, C.; Krovacek, K.; Levin, R.E. Genetic Variability among Isolates of Plesiomonas shigelloides from Fish, Human Clinical Sources and Fresh Water, Determined by RAPD Typing. Food Biotechnol. 2006, 20, 1–12. [Google Scholar] [CrossRef]
- Holmberg, S.D.; Wachsmuth, I.K.; Hickman-Brenner, F.W.; Blake, P.A.; Farmer, J.J. Plesiomonas Enteric Infections in the United States. Ann. Intern. Med. 1986, 105, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Kain, K.C.; Kelly, M.T. Clinical Features, Epidemiology, and Treatment of Plesiomonas shigelloides Diarrhea. J. Clin. Microbiol. 1989, 27, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, Y.; Yan, L.; Yan, Q.; Wen, X.; Cao, S.; Huang, Y.; Huang, X.; Ma, X.; Han, X. Identification and Pathogenicity of Plesiomonas shigelloides from Acipenser Dabryanus in China. Aquac. Res. 2021, 52, 2286–2293. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. Expression of Hemolytic Activity by Plesiomonas Shigelloides. J. Clin. Microbiol. 1993, 31, 1206–1208. [Google Scholar] [CrossRef]
- Baratéla, K.C.; Saridakis, H.O.; Gaziri, L.C.J.; Pelayo, J.S. Effects of Medium Composition, Calcium, Iron and Oxygen on Haemolysin Production by Plesiomonas shigelloides Isolated from Water. J. Appl. Microbiol. 2001, 90, 482–487. [Google Scholar] [CrossRef]
- Falcón, R.; Carbonell, G.V.; Figueredo, P.M.S.; Butiao, F.; Saridakis, H.O.; Pelayo, J.S.; Yano, T. Intracellular Vacuolation Induced by Culture Filtrates of Plesiomonas shigelloides Isolated from Environmental Sources. J. Appl. Microbiol. 2003, 95, 273–278. [Google Scholar] [CrossRef]
- Abbott, S.L.; Kokka, R.P.; Janda, J.M. Laboratory Investigations on the Low Pathogenic Potential of Plesiomonas shigelloides. J. Clin. Microbiol. 1991, 29, 148–153. [Google Scholar] [CrossRef]
- Gardner, S.E.; Fowlston, S.E.; George, W.L. In Vitro Production of Cholera Toxin-like Activity by Plesiomonas shigelloides. J. Infect. Dis. 1987, 156, 720–722. [Google Scholar] [CrossRef]
- Kaszowska, M.; Stojkovic, K.; Niedziela, T.; Lugowski, C. The O-Antigen of Plesiomonas shigelloides Serotype O36 Containing Pseudaminic Acid. Carbohydr. Res. 2016, 434, 1–5. [Google Scholar] [CrossRef]
- Hoel, S.; Vadstein, O.; Jakobsen, A.N. Species Distribution and Prevalence of Putative Virulence Factors in Mesophilic Aeromonas Spp. Isolated from Fresh Retail Sushi. Front. Microbiol. 2017, 8, 931. [Google Scholar] [CrossRef]
- Qian, Q.; Chen, Z.; Xu, J.; Zhu, Y.; Xu, W.; Gao, X.; Jiang, Q.; Zhang, X. Pathogenicity of Plesiomonas shigelloides Causing Mass Mortalities of Largemouth Bass (Micropterus salmoides) and Its Induced Host Immune Response. Fish Shellfish. Immunol. 2023, 132, 108487. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Host | Country | Total Genes | Housekeeping Genes | Known Resistance | Virulence Genes |
|---|---|---|---|---|---|---|
| zfcc0051 * | Zebra fish | USA | 3818 | 596 | 31 | 41 |
| V78 * | Rainbow trout | Turkey | 5454 | 297 | 19 | 41 |
| P5462 | Gentoo penguin | Hong Kong | 3287 | 596 | 33 | 40 |
| NCTC1036 | Human | Unknown | 3098 | 584 | 32 | 42 |
| NCTC10363 | Human | Unknown | 3264 | 599 | 30 | 40 |
| NCTC10360 | Dog | Unknown | 2886 | 583 | 28 | 40 |
| MS-17-188 * | Catfish | USA | 3426 | 595 | 32 | 42 |
| MD22D9 | Macrobdella decora | USA | 3190 | 595 | 32 | 40 |
| LS1 * | Percocypris pingi | China | 3484 | 590 | 33 | 43 |
| GT4 * | Oreochromis niloticus | Brazil | 3134 | 600 | 34 | 43 |
| GN7 | Water | Malaysia | 3434 | 596 | 33 | 42 |
| FM82 * | Fish | Brazil | 3233 | 593 | 32 | 41 |
| FDAARGOS_725 | Human | USA | 3250 | 597 | 32 | 40 |
| Colony20 | Food | Thailand | 2598 | 576 | 28 | 43 |
| Colony13 | Human | Thailand | 2648 | 580 | 28 | 39 |
| CAPA010 * | Oreochromis niloticus | Peru | 3176 | 599 | 33 | 42 |
| 7A | River | Sweden | 3298 | 597 | 32 | 41 |
| 302-73 | Human | Japan | 3398 | 592 | 32 | 40 |
| CAPA003 * | Oreochromis niloticus | Peru | 3185 | 596 | 33 | 41 |
| Strain | Type Strain | POCP (%) |
|---|---|---|
| V78 | Plesiomonas shigelloides | 68.1 |
| Yersinia entomophaga | 43.2 | |
| Obesumbacterium proteus | 42.4 | |
| Hafnia paralvei | 42.1 | |
| Buttiauxella agrestis | 41.8 | |
| Citrobacter werkmanii | 41.8 | |
| Buttiauxella noackiae | 41.8 | |
| Citrobacter pasteurii | 41.7 | |
| Salmonella enterica | 41.7 | |
| Buttiauxella brennerae | 41.6 | |
| Citrobacter freundii | 41.5 | |
| Enterobacter soli | 41.2 | |
| Serratia marcescens | 40.9 | |
| Serratia grimesii | 40.8 | |
| Pectobacterium carotovorum | 40.8 | |
| Serratia proteamaculans | 40.6 | |
| Buttiauxella ferragutiae | 40.6 | |
| Serratia nematodiphila | 40.5 | |
| Citrobacter braakii | 40.5 | |
| Serratia odorifera | 40.3 | |
| Chania multitudinisentens | 40.3 | |
| Serratia quinivorans | 40.2 | |
| Yersinia pestis | 40.2 | |
| Serratia fonticola | 40.2 | |
| Buttiauxella gaviniae | 40.1 | |
| Enterobacter hormaechei | 40.0 | |
| Proteus hauseri | 39.9 | |
| Serratia ficaria | 39.9 | |
| Serratia plymuthica | 39.9 | |
| Cronobacter sakazakii | 39.8 | |
| Citrobacter amalonaticus | 39.7 | |
| Pectobacterium parmentieri | 39.5 | |
| Enterobacter cloacae | 39.4 | |
| Dickeya chrysanthemi | 39.4 | |
| Raoultella planticola | 39.0 | |
| Klebsiella pneumoniae | 38.9 | |
| Raoultella ornithinolytica | 38.8 | |
| Lonsdalea quercina | 38.3 | |
| Pantoea agglomerans | 38.1 | |
| Enterobacter ludwigii | 38.0 | |
| Rouxiella chamberiensis | 38.0 | |
| Pantoea allii | 37.2 | |
| Rouxiella silvae | 37.2 | |
| Rouxiella badensis | 37.0 | |
| Sodalis praecaptivus | 36.3 | |
| Pantoea cypripedii | 35.3 |
| Environment | Detection Ratio (%) | Mean Relative Abundance (%) | Standard Deviation (%) |
|---|---|---|---|
| Rhizosphere | 33.9 | 0.71 | 3.47 |
| Freshwater | 28.7 | 3.29 | 12.49 |
| Insect gut | 25.5 | 4.42 | 16.15 |
| Wastewater | 19.5 | 0.20 | 0.55 |
| Plant | 13.7 | 3.26 | 11.07 |
| Soil | 11.1 | 0.20 | 1.37 |
| Activated sludge | 10.0 | 0.02 | 0.05 |
| Coral | 8.5 | 0.38 | 0.66 |
| Chicken gut | 8.2 | 0.23 | 1.17 |
| Human gut | 7.9 | 6.17 | 16.59 |
| Human skin | 7.4 | 0.22 | 0.67 |
| Pig gut | 5.7 | 0.04 | 0.08 |
| Human vaginal | 5.0 | 0.02 | 0.04 |
| Marine sediment | 4.9 | 0.10 | 0.21 |
| Mouse gut | 4.8 | 0.15 | 0.33 |
| Human lung | 4.3 | 0.38 | 1.61 |
| Marine | 4.2 | 0.05 | 0.22 |
| Bovine gut | 2.5 | 0.03 | 0.05 |
| Human oral | 0.9 | 0.20 | 0.36 |
| Family | Antibiotics | Disk Content (µg) | A. salmonicida subps. salmonicida ATCC 33658 | E. coli ATCC 25922 | P. shigelloides | ||
|---|---|---|---|---|---|---|---|
| V-63 | V78 | AF160 | |||||
| Sulfonamides | Trimethoprim/Sulfamethoxazole (SXT) | 25 | 23 | 27 | 25 (S) | 25 (S) | 21 (S) |
| Aminopenicillins | Amoxicillin (AML) * | 25 | 32 | 23 | 12 | 13 | 25 |
| Tetracyclines | Doxycycline (DO) * | 30 | 31 | 25 | 22 | 20 | 15 |
| Tetracycline (TE) * | 30 | 35 | 30 | 15 | 13 | 12 | |
| Oxytetracycline (OT) * | 30 | 30 | 29 | 10 | 0 (R) | 0 (R) | |
| Quinolone | Oxolinic Acid (OA) * | 2 | 35 | 28 | 24 | 25 | 10 |
| Fluoroquinolone | Enrofloxacin (ENR) * | 5 | 40 | 37 | 31 | 35 | 25 |
| Ciprofloxacin (CIP) | 5 | 50 | 40 | 33 (S) | 33 (S) | 25 (S) | |
| Flumequine (UB) * | 30 | 40 | 35 | 26 | 30 | 28 | |
| Cephalosporins | Cefalexin (CN) * | 10 | 25 | 25 | 20 (S) ** | 20 (S) ** | 21 (S) ** |
| Penicillins | Ampicillin (AMP) * | 10 | 32 | 20 | 11 (R) ** | 12 (R) ** | 10 (R) ** |
| Macrolides | Erythromycin (E) * | 15 | 22 | 8 | 16 | 11 | 10 |
| Clindamycin | Lincomycin (MY) * | 15 | 0 (R) | 0 (R) | 0 (R) | 0 (R) | 0 (R) |
| Chloramphenicol | Florfenicol (FFC) * | 30 | 40 | 24 | 11 | 12 | 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duman, M.; García Valdés, E.; Ay, H.; Altun, S.; Saticioglu, I.B. Description of a Novel Fish Pathogen, Plesiomonas shigelloides subsp. oncorhynchi, Isolated from Rainbow Trout (Oncorhynchus mykiss): First Genome Analysis and Comparative Genomics. Fishes 2023, 8, 179. https://doi.org/10.3390/fishes8040179
Duman M, García Valdés E, Ay H, Altun S, Saticioglu IB. Description of a Novel Fish Pathogen, Plesiomonas shigelloides subsp. oncorhynchi, Isolated from Rainbow Trout (Oncorhynchus mykiss): First Genome Analysis and Comparative Genomics. Fishes. 2023; 8(4):179. https://doi.org/10.3390/fishes8040179
Chicago/Turabian StyleDuman, Muhammed, Elena García Valdés, Hilal Ay, Soner Altun, and Izzet Burcin Saticioglu. 2023. "Description of a Novel Fish Pathogen, Plesiomonas shigelloides subsp. oncorhynchi, Isolated from Rainbow Trout (Oncorhynchus mykiss): First Genome Analysis and Comparative Genomics" Fishes 8, no. 4: 179. https://doi.org/10.3390/fishes8040179
APA StyleDuman, M., García Valdés, E., Ay, H., Altun, S., & Saticioglu, I. B. (2023). Description of a Novel Fish Pathogen, Plesiomonas shigelloides subsp. oncorhynchi, Isolated from Rainbow Trout (Oncorhynchus mykiss): First Genome Analysis and Comparative Genomics. Fishes, 8(4), 179. https://doi.org/10.3390/fishes8040179