

Metabonomic Analysis of Macrobrachium rosenbergii with Iron Prawn Syndrome (IPS)
Abstract
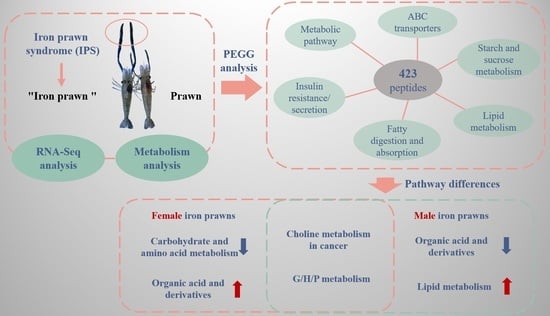
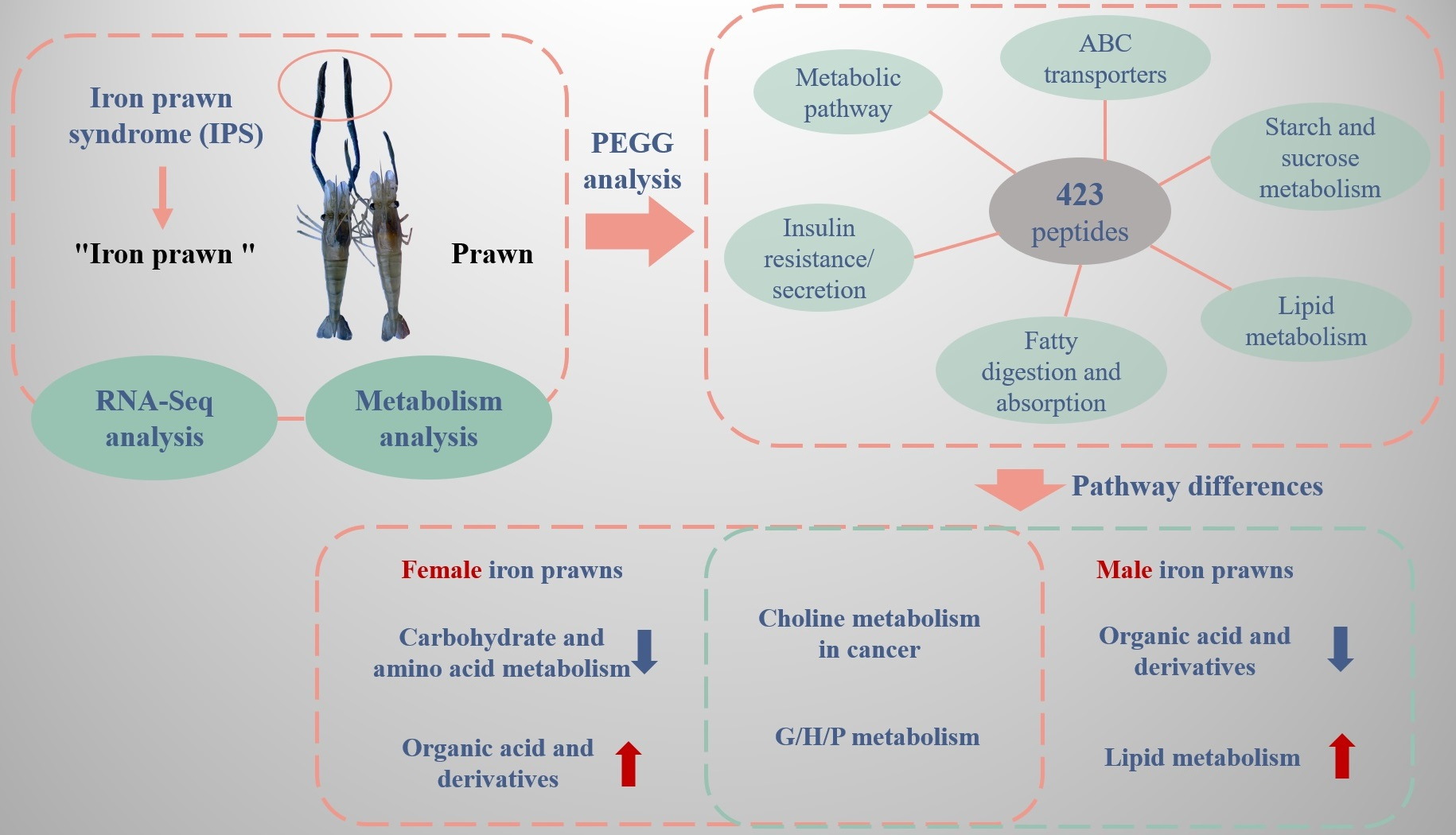
:
1. Introduction
2. Materials and Methods
2.1. Experimental Sample
2.2. Metabonomic Analysis
2.2.1. Total Protein Extraction and Peptide Digestion
2.2.2. Chromatographic Separation and Mass Spectrometry Identification
2.2.3. Non-labeling Quantitation of the Metabonomics
2.3. Transcriptomic Analysis, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Annotation
2.4. The Combined Analysis of Metabonomic and Transcriptomic Data
2.5. Data Analysis
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.L.; Gao, Q.; Shen, P.J.; Zhang, Y.F.; Jiang, W.P.; Huang, Z.Y.; Peng, F.; Gu, Z.M.; Chen, X.F. Proteomic analysis of individual giant freshwater prawn, Macrobrachium rosenbergii, growth retardants. J. Proteom. 2021, 241, 104224. [Google Scholar] [CrossRef]
- Guo, C.; Huang, X.Y.; Yang, M.J.; Wang, S.; Ren, S.T.; Li, H.; Peng, X.X. GC/MS-based metabolomics approach to identify biomarkers differentiating survivals from death in crucian carps infected by Edwardsiella tarda. Fish Shellfish Immunol. 2014, 39, 215–222. [Google Scholar] [CrossRef]
- Young, T.; Alfaro, A.C.; Villas-Bôas, S.G. Metabolic profiling of mussel larvae: Effect of handling and culture conditions. Aquac. Int. 2016, 24, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Du, F.; Li, Y.; Nie, Z.; Xu, P. Integrated application of transcriptomics and metabolomics yields insights into population-asynchronous ovary development in Coilia nasus. Sci. Rep. 2016, 6, 31835. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Chen, Q.; Shen, Z.; Li, D.; Han, T.; Qin, J.; Chen, L.; Du, Z. The metabolomics responses of Chinese mitten-hand crab (Eriocheir sinensis) to different dietary oils. Aquaculture 2017, 479, 188–199. [Google Scholar] [CrossRef]
- Liang, P.; Li, R.; Sun, H.; Zhang, M.; Cheng, W.; Chen, L.; Cheng, X.; Akoh, C.C. Phospholipids composition and molecular species of large yellow croaker (Pseudosciaena crocea) roe. Food Chem. 2018, 245, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Chong, J.; Tang, R.; Li, L.; Xia, J.; Li, D. Metabolomics investigation of dietary effects on flesh quality in grass carp (Ctenopharyngodon idellus). Gigascience 2018, 7, giy111. [Google Scholar] [CrossRef]
- Jiang, W.; Tian, X.; Fang, Z.; Li, L.; Dong, S.; Li, H.; Zhao, K. Metabolic responses in the gills of tongue sole (Cynoglossus semilaevis) exposed to salinity stress using NMR-based metabolomics. Sci. Total Environ. 2019, 653, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, S.; Yu, Y.; Sun, M.; Xiang, J.; Li, F. Effects of ammonia stress on the hemocytes of the Pacific white shrimp Litopenaeus vannamei. Chemosphere 2020, 239, 124759. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.X.; Shi, W.J.; Ma, D.D.; Zhang, J.N.; Ying, G.G.; Zhang, H.; Ong, C.N. Dydrogesterone exposure induces zebrafish ovulation but leads to oocytes over-ripening: An integrated histological and metabolomics study. Environ. Int. 2019, 128, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.T.; Chen, T.Y.; Shiau, C.Y.; Cheng, Y.T.; Chang, Y.W. Study on biochemical divergences of the meat and egg of freshwater prawns (Macrobrachium rosenbergii). Food Sci. Nutr. 2019, 7, 2017–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehnbaum, N.L.; Britz-McKibbin, P. New advances in separation science for metabolomics: Resolving chemical diversity in a post-genomic era. Chem. Rev. 2013, 113, 2437–2468. [Google Scholar] [CrossRef] [PubMed]
- Lulijwa, R.; Alfaro, A.C.; Young, T. Metabolomics in salmonid aquaculture research: Applications and future perspectives. Rev. Aquac. 2021, 14, 547–577. [Google Scholar] [CrossRef]
- Takahashi, H.; Kai, K.; Shinbo, Y.; Tanaka, K.; Ohta, D.; Oshima, T.; Altaf-Ul-Amin, M.; Kurokawa, K.; Ogasawara, N.; Kanaya, S. Metabolomics approach for determining growth-specific metabolites based on Fourier transform ion cyclotron resonance mass spectrometry. Anal. Bioanal. Chem. 2008, 391, 2769–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rise, M.L.; Martyniuk, C.J.; Chen, M. Comparative physiology and aquaculture: Toward Omics-enabled improvement of aquatic animal health and sustainable production. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 31, 100603. [Google Scholar] [CrossRef]
- Li, F.; Fu, C.; Xie, Y.; Wang, A.; Li, J.; Gao, J.; Cui, X. Transcriptional responses to starvation stress in the hepatopancreas of oriental river prawn Macrobrachium nipponense. Environ. Pollut. 2019, 252 Pt A, 14–20. [Google Scholar] [CrossRef]
- Jiang, Q.; Qian, L.; Gu, S.; Guo, X.; Zhang, X.; Sun, L. Investigation of growth retardation in Macrobrachium rosenbergii based on genetic/epigenetic variation and molt performance. Comp. Biochem. Physiol. Part D: Genom. Proteom. 2020, 35, 100683. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A Novel Integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography-mass spectrometry, XCMS, and chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and opls statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- He, L.; Zhu, D.; Liang, X.; Li, Y.; Liao, L.; Yang, C.; Huang, R.; Zhu, Z.; Wang, Y. Multi-omics sequencing provides insights into age-dependent susceptibility of grass carp (Ctenopharyngodon idellus) to reovirus. Front. Immunol. 2021, 12, 694965. [Google Scholar] [CrossRef]
- Ebert, T.; Painer, J.; Bergman, P.; Qureshi, A.R.; Giroud, S.; Stalder, G.; Kublickiene, K.; Göritz, F.; Vetter, S.; Bieber, C.; et al. Insights in the regulation of trimetylamine N-oxide production using a comparative biomimetic approach suggest a metabolic switch in hibernating bears. Sci. Rep. 2020, 10, 20323. [Google Scholar] [CrossRef]
- Akimov, M.G.; Kudryavtsev, D.S.; Kryukova, E.V.; Fomina-Ageeva, E.V.; Zakharov, S.S.; Gretskaya, N.M.; Zinchenko, G.N.; Serkov, I.V.; Makhaeva, G.F.; Boltneva, N.P.; et al. Arachidonoylcholine and other unsaturated long-chain acylcholines are endogenous modulators of the acetylcholine signaling system. Biomolecules 2020, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Xuan, F.; Fu, H.; Ge, X.; Zhu, J.; Qiao, H.; Jin, S.; Zhang, W. Molecular characterization and mRNA expression of hypoxia inducible factor-1 and cognate inhibiting factor in Macrobrachium nipponense in response to hypoxia. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2016, 196–197, 48–56. [Google Scholar] [CrossRef]
- Lu, Z.B.; Li, Y.D.; Jiang, S.G.; Yang, Q.B.; Jiang, S.; Huang, J.H.; Yang, L.S.; Chen, X.; Zhou, F.L. Transcriptome analysis of hepatopancreas in penaeus monodon under acute low pH stress. Fish Shellfish Immunol. 2022, 131, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Feng, J.; Xie, N.; Ling, F.; Wang, Z.; Ma, K.; Hua, X.; Li, J. RNA-seq provides novel insights into response to acute salinity stress in oriental river prawn Macrobrachium nipponense. Mar. Biotechnol. 2022, 24, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Li, J.; Lv, J.; Liu, P.; Li, J.; Li, S. Full-length transcriptome sequences of ridgetail white prawn Exopalaemon carinicauda provide insight into gene expression dynamics during thermal stress. Sci. Total Environ. 2020, 747, 141238. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Liu, Q.; Kan, D.; Zhao, W.; Guo, H.; Lv, L. Effects of ammonia-N exposure on the growth, metabolizing enzymes, and metabolome of Macrobrachium rosenbergii. Ecotoxicol. Environ. Saf. 2020, 189, 110046. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Kong, Y.; Shao, X.; Zhang, Y.; Ren, C.; Zhao, X.; Yu, W.; Jiang, T.; Ye, J. Growth, antioxidant capacity, intestinal morphology, and metabolomic responses of juvenile Oriental river prawn (Macrobrachium nipponense) to chronic lead exposure. Chemosphere 2019, 217, 289–297. [Google Scholar] [CrossRef]
- Guasch-Ferré, M.; Bhupathiraju, S.N.; Hu, F.B. Use of metabolomics in improving assessment of dietary intake. Clin. Chem. 2018, 64, 82–98. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wang, Y.; Xiong, D.; Zhang, J. RNA-seq revealed the signatures of immunity and metabolism in the Litopenaeus vannamei intestine in response to dietary succinate. Fish Shellfish Immunol. 2019, 95, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Driedzic, W.R.; Ewart, K.V. Control of glycerol production by rainbow smelt (Osmerus mordax) to provide freeze resistance and allow foraging at low winter temperatures. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004, 139, 347–357. [Google Scholar] [CrossRef]
- Stalin, A.; Suganthi, P.; Mathivani, S.; Broos, K.V.; Gokula, V.; Sadiq Bukhari, A.; Syed Mohamed, H.E.; Singhal, R.K.; Venu-Babu, P. Effect of cobalt-60 gamma radiation on total hemocyte content and biochemical parameters in Macrobrachium rosenbergii (De Man, 1879). Int. J. Radiat. Biol. 2019, 95, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, W.; Jin, S.; Jiang, S.; Xiong, Y.; Chen, T.; Gong, Y.; Qiao, H.; Fu, H. Transcriptome analysis provides novel insights into the immune mechanisms of Macrobrachium nipponense during molting. Fish Shellfish Immunol. 2022, 131, 454–469. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, J.X.; Zhang, L.; Wu, D.; Tian, J.; Yu, L.J.; He, L.; Zhong, S.; Du, H.; Deng, D.F.; et al. Comprehensive understanding the impacts of dietary exposure to polyethylene microplastics on genetically improved farmed tilapia (Oreochromis niloticus): Tracking from growth, microbiota, metabolism to gene expressions. Sci. Total Environ. 2022, 841, 156571. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddani, S.E.; Houwing-Duistermaat, J.; Salo, P.; Perola, M.; Jongbloed, G.; Uh, H.W. Evaluation of O2PLS in Omics data integration. BMC Bioinform. 2016, 17 (Suppl. S2). [Google Scholar] [CrossRef] [Green Version]
- Fuady, A.M.; El Bouhaddani, S.; Uh, H.W.; Houwing-Duistermaat, J. Estimation of the effect of surrogate multi-omic biomarkers. Theor. Biol. Forum. 2021, 114, 59–73. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
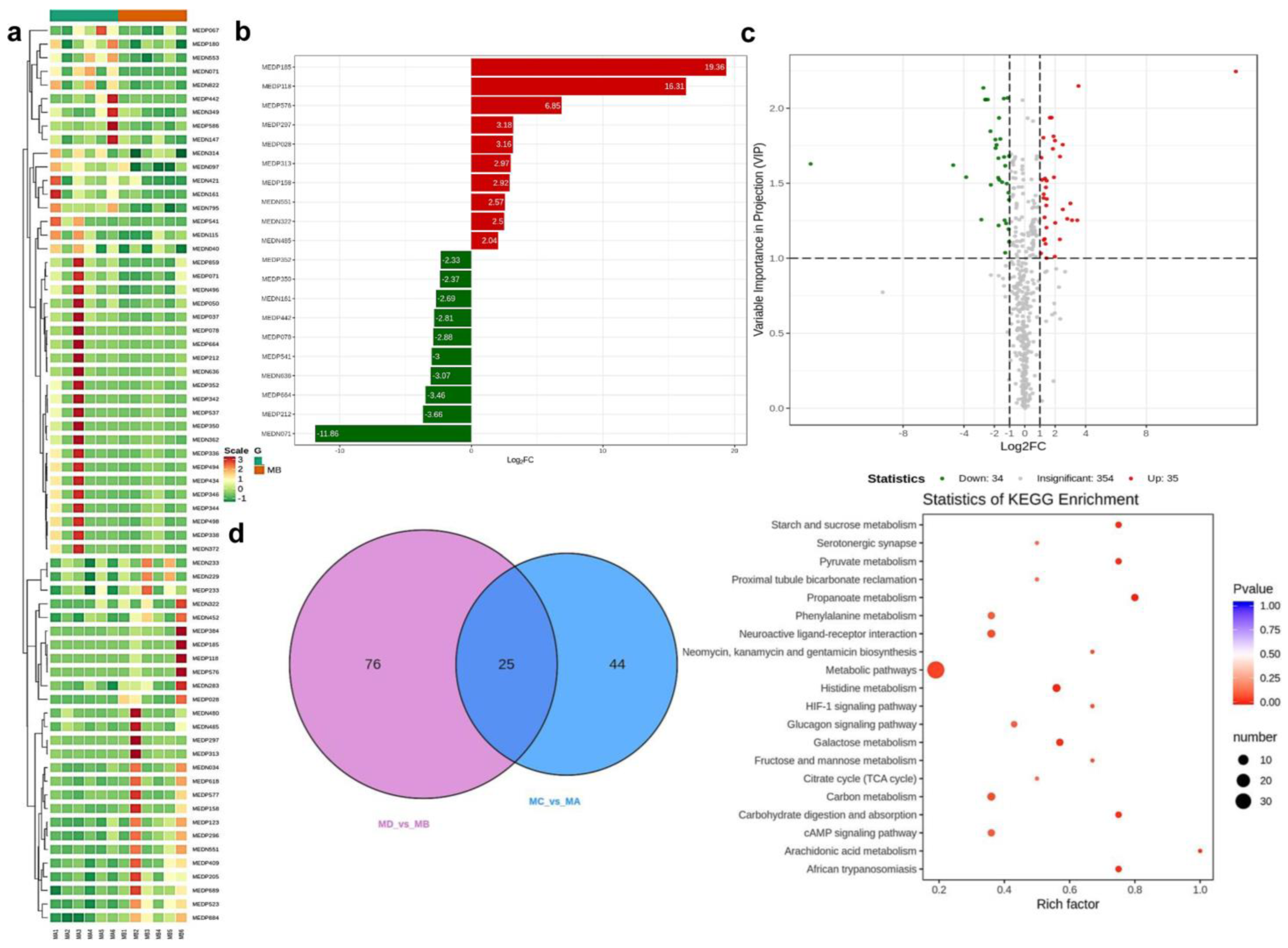
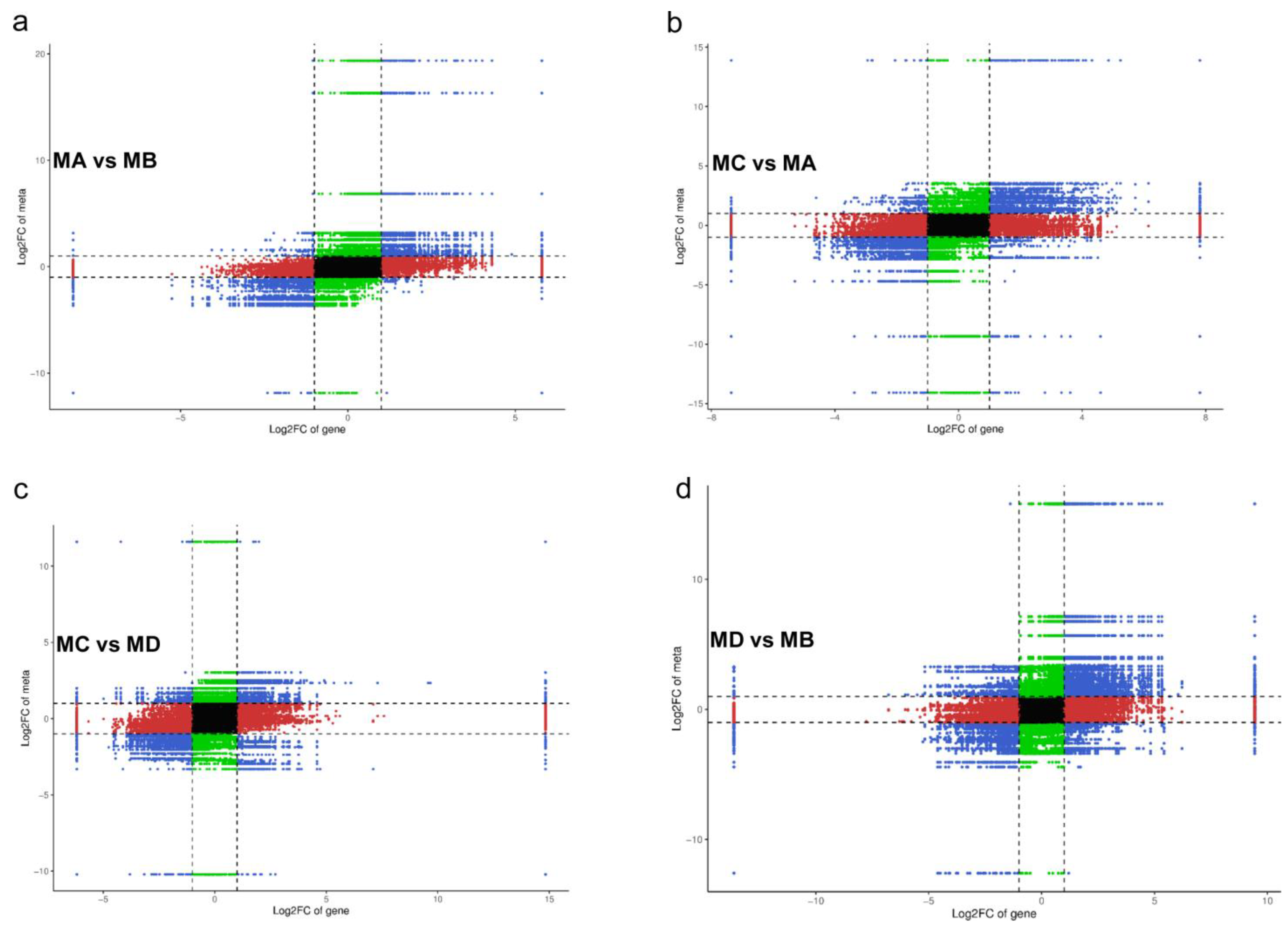
| Group Comparison | Total Sig Metabolites | Down-Regulated | Up-Regulated |
|---|---|---|---|
| MC_vs_MA | 69 | 34 | 35 |
| MD_vs_MB | 101 | 56 | 45 |
| MA_vs_MB | 66 | 39 | 27 |
| MC_vs_MD | 58 | 33 | 25 |
| Index | Compounds | Type | cpd_ID |
|---|---|---|---|
| MEDN049 | L-Saccharopine | up | C00449 |
| MEDN065 | O-Phospho-L-Serine | up | C01005 |
| MEDN097 | P–Hydroxyphenyl Acetic Acid | up | C00642 |
| MEDN009 | L-Aspartic Acid | down | C00049 |
| MEDN011 | L-Glutamic Acid | down | C00025 |
| MEDN070 | Sarcosine | down | C00213 |
| MEDN082 | P-Coumaryl Alcohol | down | C02646 |
| MEDN098 | 2-Picolinic Acid | down | C10164 |
| MEDN120 | Dulcitol | down | C01697 |
| MEDN200 | L-Malic Acid | down | C00149 |
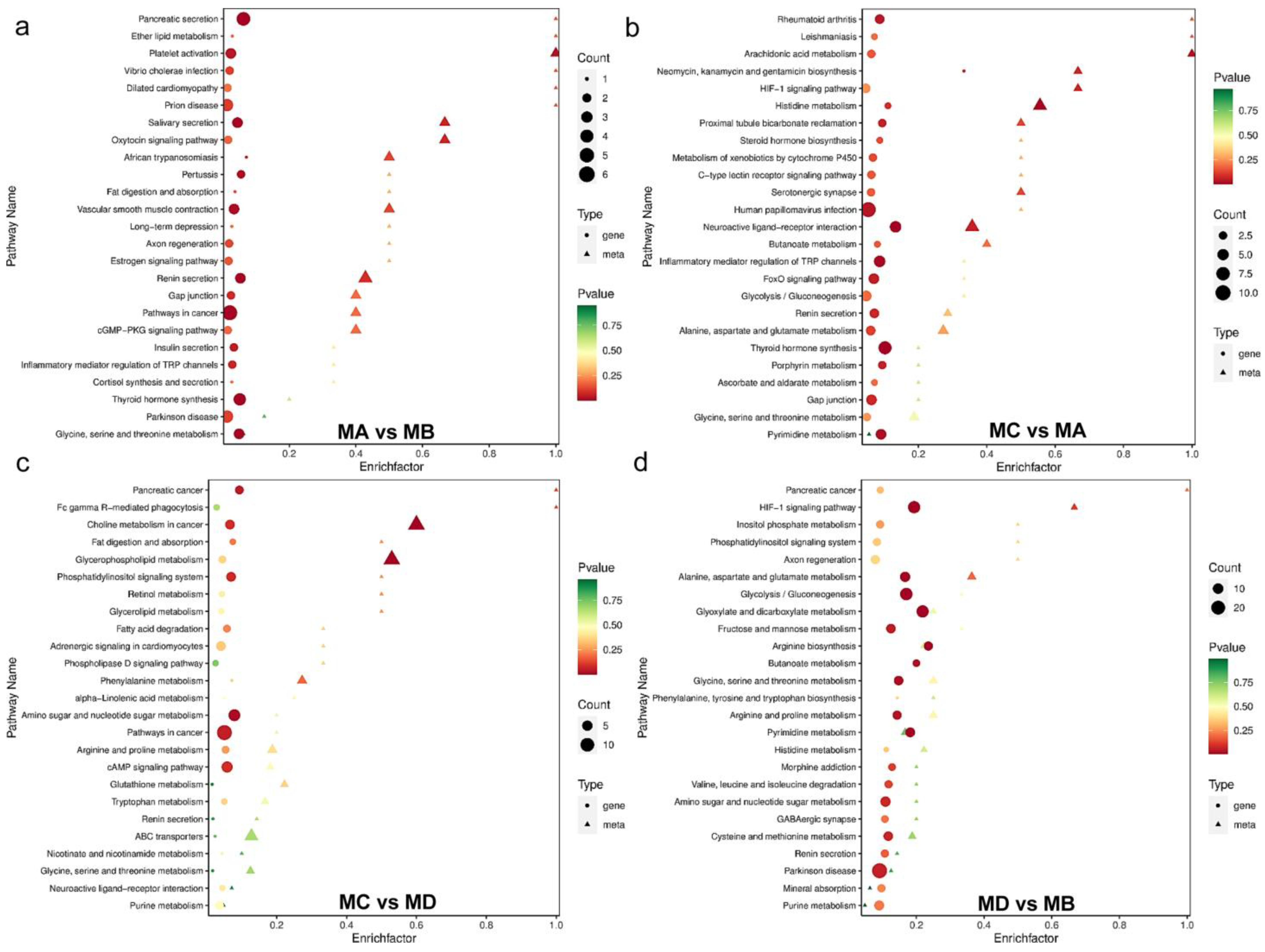
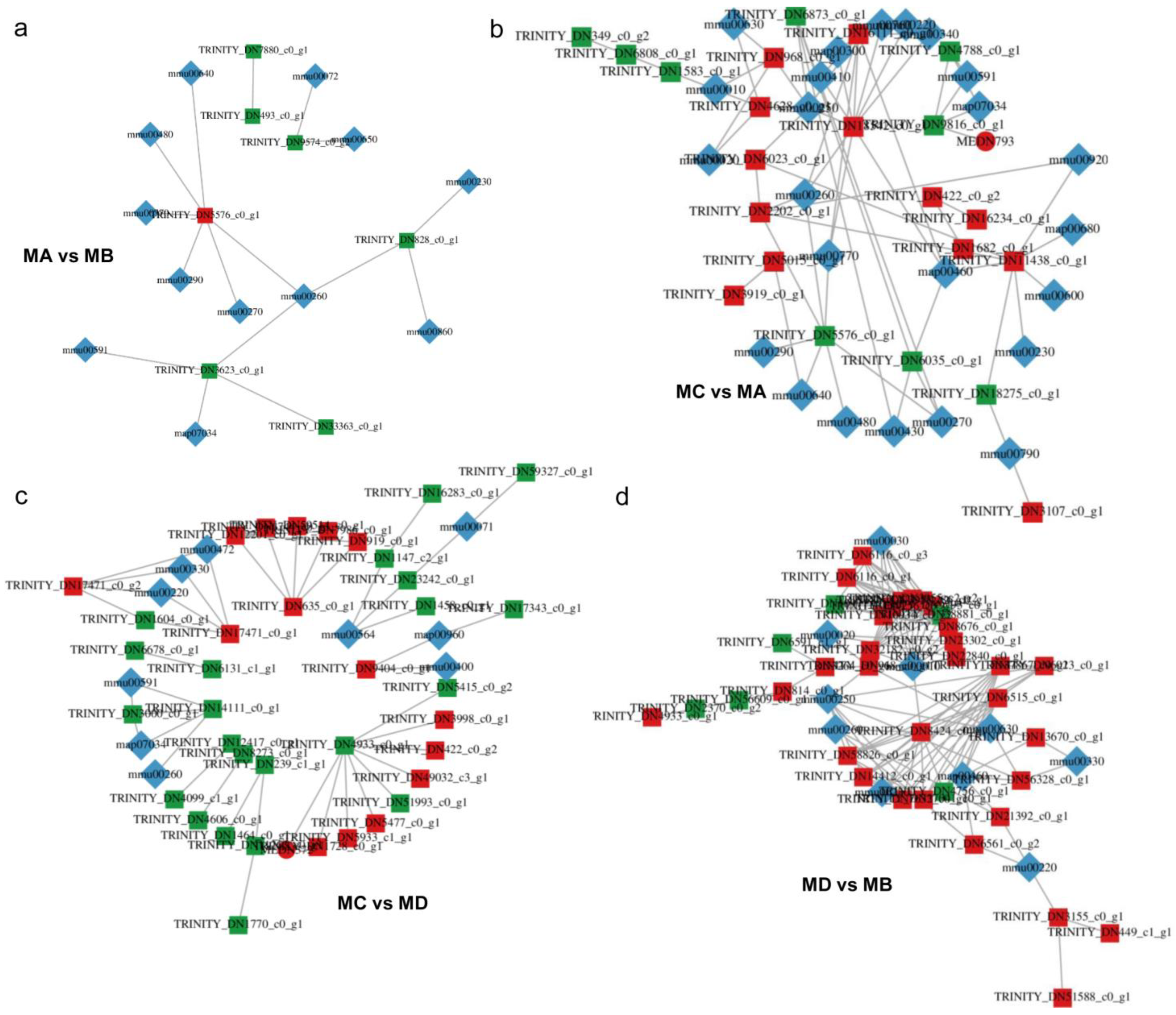
| Comparison Group | Pathway | ko_ID | Unique Compound |
|---|---|---|---|
| MA vs. MB | Metabolic pathways | ko01100 | 28 |
| ⃞ | Choline metabolism in cancer | ko05231 | 11 |
| ⃞ | Glycerophospholipid metabolism | ko00564 | 11 |
| ⃞ | Bile secretion | ko04976 | 4 |
| ⃞ | Cortisol synthesis and secretion | ko04927 | 1 |
| ⃞ | Dopaminergic synapse | ko04728 | 1 |
| ⃞ | Endocrine and other factor-regulated calcium reabsorption | ko04961 | 1 |
| ⃞ | Endocrine resistance | ko01522 | 1 |
| ⃞ | Estrogen-signaling pathway | ko04915 | 1 |
| ⃞ | GnRH-signaling pathway | ko04912 | 2 |
| ⃞ | Oocyte meiosis | ko04114 | 1 |
| ⃞ | Ovarian steroidogenesis | ko04913 | 1 |
| ⃞ | Thyroid hormone-signaling pathway | ko04919 | 1 |
| ⃞ | Thyroid hormone synthesis | ko04918 | 1 |
| ⃞ | Vascular smooth muscle contraction | ko04270 | 2 |
| MC vs. MA | Metabolic pathways | ko01100 | 37 |
| ⃞ | Biosynthesis of secondary metabolites | ko01110 | 11 |
| ⃞ | ABC transporters | ko02010 | 9 |
| ⃞ | Central carbon metabolism in cancer | ko05230 | 6 |
| ⃞ | Histidine metabolism | ko00340 | 5 |
| ⃞ | Carbon metabolism | ko01200 | 5 |
| ⃞ | Neuroactive ligand-–receptor interaction | ko04080 | 5 |
| ⃞ | Bile secretion | ko04976 | 1 |
| ⃞ | Glucagon-signaling pathway | ko04922 | 3 |
| ⃞ | Glutamatergic synapse | ko04724 | 1 |
| ⃞ | Glutathione metabolism | ko00480 | 3 |
| ⃞ | Glycerolipid metabolism | ko00561 | 1 |
| ⃞ | Glycerophospholipid metabolism | ko00564 | 1 |
| ⃞ | Glycine, serine and threonine metabolism | ko00260 | 3 |
| ⃞ | Glycolysis / Gluconeogenesis | ko00010 | 1 |
| ⃞ | Glyoxylate and dicarboxylate metabolism | ko00630 | 3 |
| ⃞ | Insulin resistance | ko04931 | 1 |
| ⃞ | Insulin secretion | ko04911 | 1 |
| ⃞ | Starch and sucrose metabolism | ko00500 | 3 |
| ⃞ | Vitamin digestion and absorption | ko04977 | 1 |
| MC vs. MD | Metabolic pathways | ko01100 | 21 |
| ⃞ | Glycerophospholipid metabolism | ko00564 | 9 |
| ⃞ | Choline metabolism in cancer | ko05231 | 9 |
| ⃞ | ABC transporters | ko02010 | 6 |
| ⃞ | Biosynthesis of secondary metabolites | ko01110 | 5 |
| ⃞ | cAMP-signaling pathway | ko04024 | 2 |
| ⃞ | Fat digestion and absorption | ko04975 | 1 |
| ⃞ | Fatty acid degradation | ko00071 | 1 |
| ⃞ | GnRH-signaling pathway | ko04912 | 1 |
| ⃞ | Regulation of lipolysis in adipocytes | ko04923 | 1 |
| MD vs. MB | Metabolic pathways | ko01100 | 35 |
| ⃞ | Biosynthesis of secondary metabolites | ko01110 | 13 |
| ⃞ | Glycerophospholipid metabolism | ko00564 | 13 |
| ⃞ | Choline metabolism in cancer | ko05231 | 13 |
| ⃞ | Central carbon metabolism in cancer | ko05230 | 6 |
| ⃞ | Biosynthesis of amino acids | ko01230 | 5 |
| ⃞ | ABC transporters | ko02010 | 5 |
| ⃞ | Glycine, serine and threonine metabolism | ko00260 | 4 |
| ⃞ | Biosynthesis of unsaturated fatty acids | ko01040 | 1 |
| ⃞ | cAMP-signaling pathway | ko04024 | 4 |
| ⃞ | Carbohydrate digestion and absorption | ko04973 | 1 |
| ⃞ | Carbon metabolism | ko01200 | 3 |
| ⃞ | Fat digestion and absorption | ko04975 | 1 |
| ⃞ | Glucagon-signaling pathway | ko04922 | 3 |
| ⃞ | Glucosinolate biosynthesis | ko00966 | 1 |
| ⃞ | Glutathione metabolism | ko00480 | 2 |
| ⃞ | Glycerolipid metabolism | ko00561 | 2 |
| ⃞ | Glycolysis / Gluconeogenesis | ko00010 | 1 |
| ⃞ | Glyoxylate and dicarboxylate metabolism | ko00630 | 2 |
| ⃞ | Insulin resistance | ko04931 | 1 |
| ⃞ | Insulin secretion | ko04911 | 1 |
| ⃞ | Protein digestion and absorption | ko04974 | 2 |
| ⃞ | Regulation of lipolysis in adipocytes | ko04923 | 1 |
| ⃞ | Vitamin digestion and absorption | ko04977 | 2 |
| Comparison Group | Kegg_Pathway | ko_id | Cluter_Frequency | Corrected_p-Value |
|---|---|---|---|---|
| MA vs. MB | Choline metabolism in cancer | ko05231 | 11 | 0.0001 |
| Glycerophospholipid metabolism | ko00564 | 11 | 0.0004 | |
| MC vs. MA | Propanoate metabolism | ko00640 | 4 | 0.3173 |
| Histidine metabolism | ko00340 | 5 | 0.8170 | |
| MC vs. MD | Choline metabolism in cancer | ko05231 | 9 | 0.0007 |
| Glycerophospholipid metabolism | ko00564 | 9 | 0.0027 | |
| MD vs. MB | Choline metabolism in cancer | ko05231 | 13 | 0.0000 |
| Glycerophospholipid metabolism | ko00564 | 13 | 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.-L.; Shen, P.-J.; Jiang, W.-P.; Meng, J.-L.; Cheng, H.-H.; Gao, Q. Metabonomic Analysis of Macrobrachium rosenbergii with Iron Prawn Syndrome (IPS). Fishes 2023, 8, 196. https://doi.org/10.3390/fishes8040196
Li X-L, Shen P-J, Jiang W-P, Meng J-L, Cheng H-H, Gao Q. Metabonomic Analysis of Macrobrachium rosenbergii with Iron Prawn Syndrome (IPS). Fishes. 2023; 8(4):196. https://doi.org/10.3390/fishes8040196
Chicago/Turabian StyleLi, Xi-Lian, Pei-Jing Shen, Wen-Ping Jiang, Ji-Lun Meng, Hai-Hua Cheng, and Qiang Gao. 2023. "Metabonomic Analysis of Macrobrachium rosenbergii with Iron Prawn Syndrome (IPS)" Fishes 8, no. 4: 196. https://doi.org/10.3390/fishes8040196
APA StyleLi, X.-L., Shen, P.-J., Jiang, W.-P., Meng, J.-L., Cheng, H.-H., & Gao, Q. (2023). Metabonomic Analysis of Macrobrachium rosenbergii with Iron Prawn Syndrome (IPS). Fishes, 8(4), 196. https://doi.org/10.3390/fishes8040196