
Chromosome-Level Genome Assembly and Comparative Genomic Analysis of the Barbel Chub (Squaliobarbus curriculus) by Integration of PacBio Sequencing and Hi-C Technology


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethic Statement
2.2. Sample DNA and RNA Extraction
2.3. Library Construction and Genome Sequencing
2.4. Genome Size Estimation and Genome Assembly
2.5. Hi-C Analysis and Chromosome-Level Genome Assembly
2.6. Assessment of the Genome Assembly
2.7. Repeat Annotation, Gene Prediction, and Functional Annotation
2.8. Phylogenetic Analysis and Estimation of Divergence Time
2.9. Expansion, Contraction, and Identification of Gene Family
3. Results
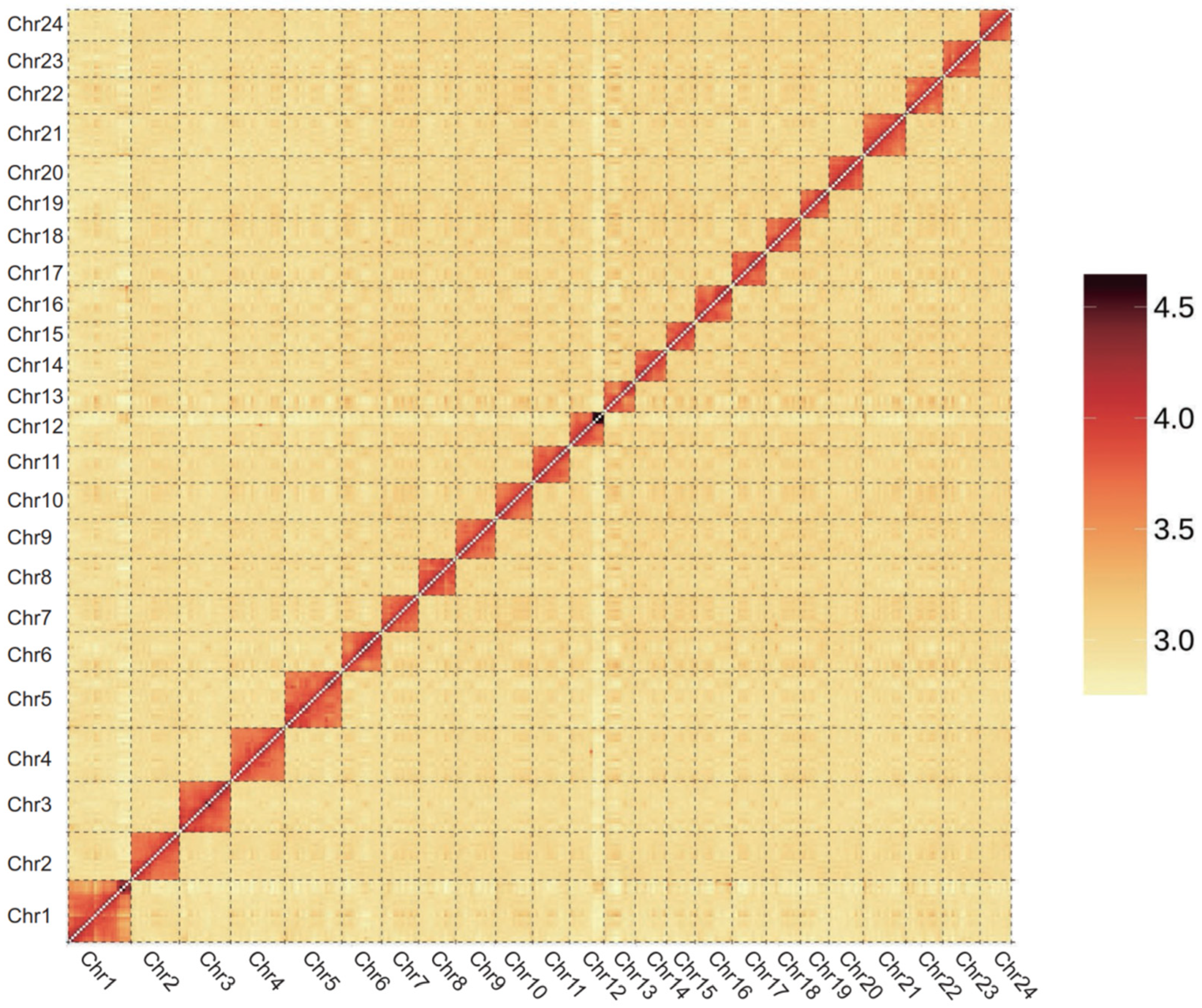
3.1. De Novo Genome Assembly of S. curriculus
3.2. Repeat Annotation, Gene Prediction, and Functional Annotation
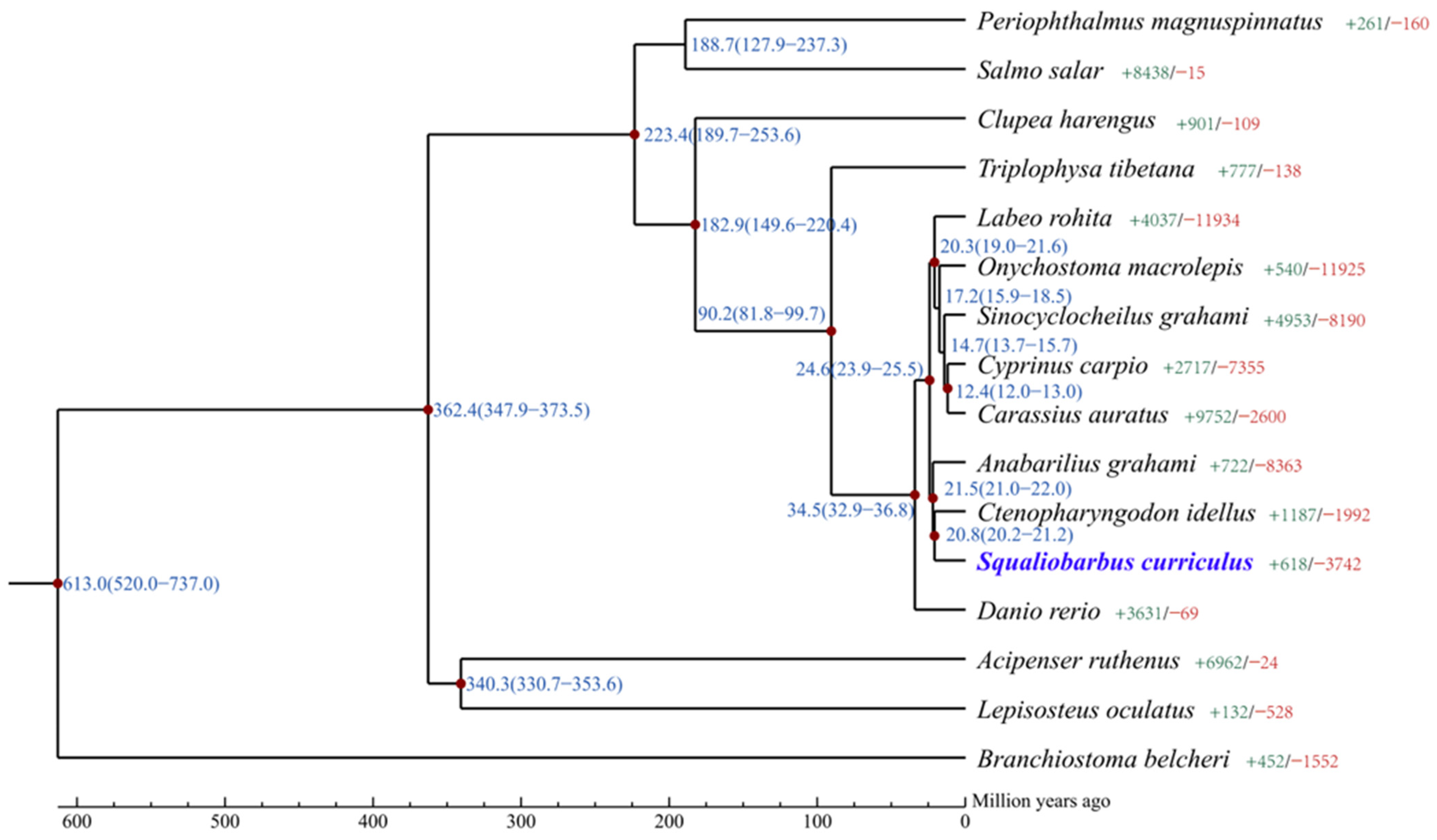
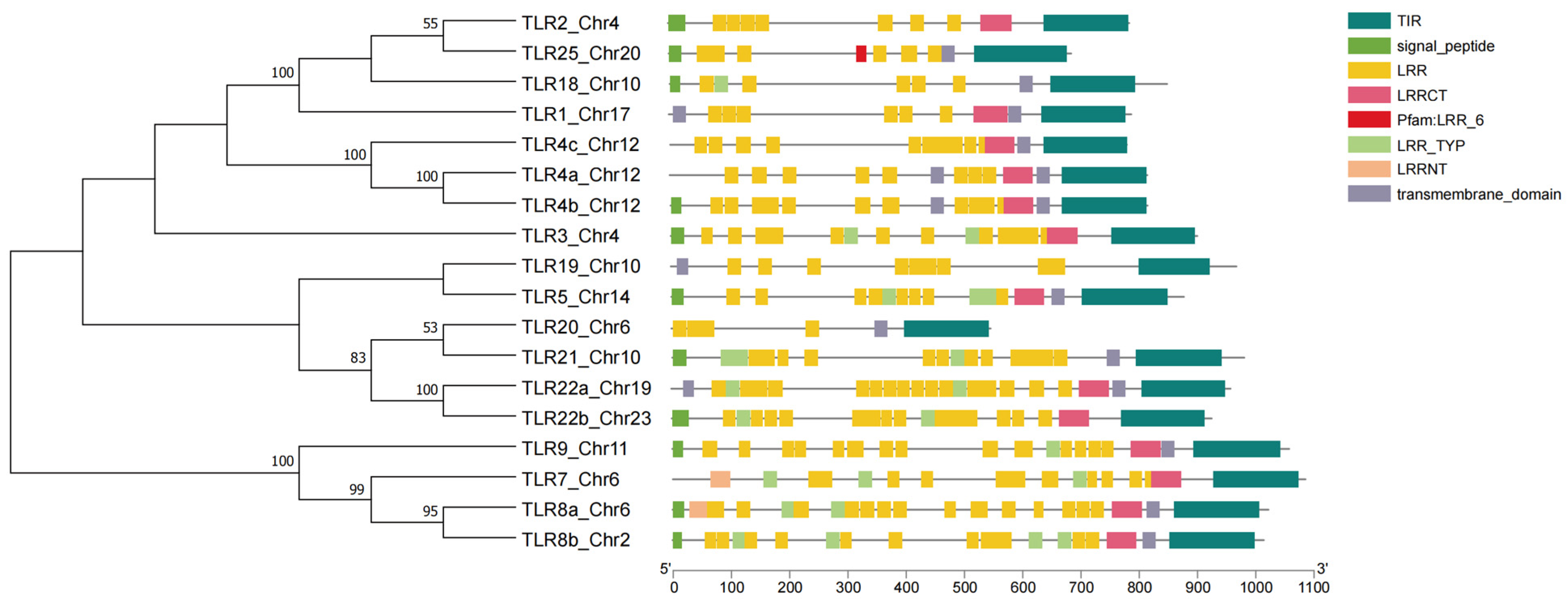
3.3. Comparative Genomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.Z. Fishes of the Yellow River; China Ocean University Press: Qingdao, China, 2017; p. 200. [Google Scholar]
- Luo, Q.S.; Zheng, G.G. A brief introduction to the biology of Squallobarbus curriculus. J. Southwest China Norm. Univ. 1980, 2, 119–122. [Google Scholar]
- Liu, Q.; Xiao, T.; Liu, M.; Zhou, W. Research progress of biology in Squaliobarbus curriculus. Fish. Sci. 2012, 11, 687–691. [Google Scholar]
- Chen, D.Q.; Xiong, F.; Wang, K.; Chang, Y.H. Status of research on Yangtze fish biology and fisheries. Environ. Biol. Fish 2009, 85, 337–357. [Google Scholar] [CrossRef]
- Yi, Y.J.; Yang, Z.F.; Zhang, S.H. Ecological influence of dam construction and river-lake connectivity on migration fish habitat in the Yangtze River basin, China. Procedia Environ. Sci. 2010, 2, 1942–1954. [Google Scholar] [CrossRef]
- Xiang, J.G.; He, F.L. Study on biological characteristics of Squaliobarbus curriculus. Freshw. Fish. 2006, 36, 38–40. [Google Scholar]
- Yang, S.X.; Zheng, T.S. Analysis on flesh rate and muscle nutritional value in Squaliobarbus curriculus Richardson. J. Anhui Agric. Sci. 2010, 38, 11835–11837. [Google Scholar]
- Jin, X.L.; Tian, X.C.; Zeng, G.Q.; Wang, M.L. Preliminary study on crossbreeding and seedling cultivation of Squaliobarbus curriculus. Inland Fish. 1997, 12, 6–7. [Google Scholar]
- Long, G.H.; Hu, D.S.; Liu, J.H.; Lu, W.Z. Research on artificial propagation of Squaliobarbus curriculus. Freshw. Fish. 2005, 35, 44–46. [Google Scholar]
- Long, G.H.; Hu, D.S.; Liu, J.H.; Lu, W.Z. Preliminary study on seedling cultivation technology of Squaliobarbus curriculus. Reserv. Fish. 2005, 25, 41. [Google Scholar]
- Xiong, W.Z.; Li, J.; Zhou, P.; Zhang, J.; Chen, C.C.; Wu, D.C. The technical research about the artificial fecundation and rearing of fingerling of the Squaliobarbus curriculus. J. Aquacult. 2005, 26, 12–15. [Google Scholar]
- Chen, Y.C.; Lin, G. Growth characteristics and breeding technology of Squaliobarbus curriculus. Guangxi Agric. Sci. 2007, 38, 97–100. [Google Scholar]
- Mi, G.Q.; Shen, T.S.; Xu, G.X. Studies on artificial propagation techniques of Squaliobarbus curriculus. J. Zhejiang Ocean Univ. (Nat. Sci.) 2007, 26, 272–276. [Google Scholar]
- Guo, S.R.; Feng, X.Y.; Xie, N.; Liu, X.Y.; Wang, Y.X. Culturing experiment of Squaliobarbus curriculus in ponds. J. Hydroecol. 2009, 2, 142–144. [Google Scholar]
- Yang, K.; Gao, Y.A.; Wang, Q.Y.; Xia, R.L.; Zeng, K.W.; Zhu, S.H.; Li, B.; Deng, G.Q.; Cheng, Y.H.; Zheng, C.H. Study on the introduction, domestication and artificial propagation of Squaliobarbus curriculus. Hubei Agric. Sci. 2014, 53, 1367–1369. [Google Scholar]
- Liu, Q.L.; Liu, M.; Xiao, T.Y.; Xu, B.H.; Su, J.M. Mitochondrial DNA sequence of the hybrid of Squaliobarbus curriculus (♀) × Ctenopharyngodon idella (♂). Mitochondrial DNA 2013, 24, 394–396. [Google Scholar] [CrossRef]
- Jin, S.Z.; Zhao, X.; Wang, H.Q.; Su, J.M.; Wang, J.A.; Ding, C.H.; Li, Y.G.; Xiao, T.Y. Molecular characterization and expression of TLR7 and TLR8 in barbel chub (Squaliobarbus curriculus): Responses to stimulation of grass carp reovirus and lipopolysaccharide. Fish Shellfish Immunol. 2018, 83, 292–307. [Google Scholar] [CrossRef]
- Wang, R.H.; Li, W.; Fan, Y.D.; Liu, Q.L.; Zeng, L.B.; Xiao, T.Y. Tlr22 structure and expression characteristic of barbel chub, Squaliobarbus curriculus provides insights into antiviral immunity against infection with grass carp reovirus. Fish Shellfish Immun. 2017, 66, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, R.H.; Li, Y.G.; Xiao, T.Y. Molecular cloning and functional characterization of interferon regulatory factor 7 of the barbel chub, Squaliobarbus curriculus. Fish Shellfish Immunol. 2017, 69, 185–194. [Google Scholar] [CrossRef]
- Wang, R.H.; Li, Y.G.; Zhou, Z.Y.; Liu, Q.L.; Zeng, L.B.; Xiao, T.Y. Involvement of interferon regulatory factor 3 from the barbel chub Squaliobarbus curriculus in the immune response against grass carp reovirus. Gene 2018, 648, 5–11. [Google Scholar] [CrossRef]
- Li, Y.G.; Jin, S.Z.; Zhao, X.; Luo, H.; Li, R.; Li, D.F.; Xiao, T.Y. Sequence and expression analysis of the cytoplasmic pattern recognition receptor melanoma differentiation-associated gene 5 from the barbel chub Squaliobarbus curriculus. Fish Shellfish Immunol. 2019, 94, 485–496. [Google Scholar] [CrossRef]
- Zhao, X.; Xiao, T.Y.; Jin, S.Z.; Wang, J.A.; Wang, J.Y.; Luo, H.; Li, R.; Sun, T.; Zou, J.; Li, Y.G. Characterization and immune function of the interferon-β promoter stimulator-1 in the barbel chub, Squaliobarbus curriculus. Dev. Comp. Immunol. 2020, 104, 103571. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, X.; Lv, Z.; Hu, X.; Xu, B.; Yang, H.; Xiao, T.; Liu, Q. Comparative transcriptomics analysis reveals unique immune response to Grass Carp Reovirus infection in barbel chub (Squaliobarbus curriculus). Biology 2024, 13, 214. [Google Scholar] [CrossRef]
- Kong, B.; Lin, G.; Li, X.Z.; Hu, X.N. Adaptive domestication of barbel chub (Squaliobarbus curriculus) before artificial releasing in the river. Guangxi Agric. Sci. 2008, 39, 541–543. [Google Scholar]
- Xia, X.L.; Zhai, L.T. Several economic fishes extinct in the wild state of Baiyangdian Lake and their causes. Hebei Fish. 2014, 5, 63–64. [Google Scholar]
- Liu, Q.L.; Xu, B.H.; Xiao, T.Y.; Su, J.M.; Yao, Y.B.; Liu, Y.J. Complete mitochondrial genome of the Xiangjiang barbel chub Squaliobarbus curriculus: Comparative analysis of the genetic variation associated with geographical population. Mitochondrial DNA 2013, 24, 654–656. [Google Scholar] [CrossRef]
- Zhou, W.; Song, N.; Wang, J.; Gao, T.X. Effects of geological changes and climatic fluctuations on the demographic histories and low genetic diversity of Squaliobarbus curriculus in Yellow River. Gene 2016, 590, 149–158. [Google Scholar] [CrossRef]
- Li, C.J.; Teng, T.; Shen, F.F.; Guo, J.Q.; Chen, Y.N.; Zhu, C.K.; Ling, Q.F. Transcriptome characterization and SSR discovery in Squaliobarbus curriculus. J. Oceanol. Limnol. 2019, 37, 235–244. [Google Scholar] [CrossRef]
- Wang, Y.P.; Lu, Y.; Zhang, Y.; Ning, Z.M.; Li, Y.; Zhao, Q.; Lu, H.Y.; Huang, R.; Xia, X.Q.; Feng, Q.; et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat. Genet. 2015, 47, 962. [Google Scholar] [CrossRef]
- Wu, C.S.; Ma, Z.Y.; Zheng, G.D.; Zou, S.M.; Zhang, X.J.; Zhang, Y.A. Chromosome-level genome assembly of grass carp (Ctenopharyngodon idella) provides insights into its genome evolution. BMC Genom. 2022, 7, 23–271. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Genomics: Next regeneration sequencing for reference genomes. Nat. Rev. Genet. 2018, 19, 125. [Google Scholar] [CrossRef]
- Pollard, M.O.; Gurdasani, D.; Mentzer, A.J.; Porter, T.; Sandhu, M.S. Long reads: Their purpose and place. Hum. Mol. Genet. 2018, 27, 234–241. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.Y.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Vij, S.; Kuhl, H.; Kuznetsova, I.S.; Komissarov, A.; Yurchenko, A.A.; Van Heusden, P.; Singh, S.; Thevasagayam, N.M.; Prakki, S.R.; Purushothaman, K.; et al. Chromosomal-level assembly of the Asian seabass genome using long sequence reads and multi-layered scaffolding. PLoS Genet. 2016, 12, e1005954. [Google Scholar]
- Gong, G.R.; Dan, C.; Xiao, S.J.; Guo, W.J.; Huang, P.P.; Xiong, Y.; Wu, J.J.; He, Y.; Zhang, J.C.; Li, X.H.; et al. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. Gigascience 2018, 7, giy120. [Google Scholar] [CrossRef]
- Chen, B.H.; Zhou, Z.X.; Ke, Q.Z.; Wu, Y.D.; Bai, H.Q.; Pu, F.; Xu, P. The sequencing and de novo assembly of the Larimichthys crocea genome using PacBio and Hi-C technologies. Sci. Data 2019, 6, 188. [Google Scholar] [CrossRef]
- Zhang, B.D.; Li, Y.L.; Xue, D.X.; Liu, J.X. Population genomic evidence for high genetic connectivity among populations of small yellow croaker (Larimichthys polyactis) in inshore waters of China. Fish. Res. 2020, 225, 105505. [Google Scholar] [CrossRef]
- Zhang, B.D.; Li, Y.L.; Xue, D.X.; Liu, J.X. Population genomics reveals shallow genetic structure in a connected and ecologically important fish from the northwestern Pacific Ocean. Front. Mar. Sci. 2020, 7, 374. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Liu, B.H.; Shi, Y.J.; Yuan, J.Y.; Hu, X.S.; Zhang, H.; Li, N.; Li, Z.Y.; Chen, Y.X.; Mu, D.S.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563. [Google Scholar] [CrossRef]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Synteny reduction for third-gen diploid genome assemblies. BioRxiv 2018. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.D.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119. [Google Scholar] [CrossRef]
- Shu, H.; Liu, Y.B.; Wei, Q.L.; Liu, L.; Yang, L.D.; Qiang, L.; Hou, L.P. Studies on chromosome karyotype, Ag-NORs and C-banding patterns of wild Ctenopharyngodon idellus and Squaliobarbus curriculus in the Pearl River. J. Guangzhou Univ. 2014, 13, 53–59. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R. RepeatModeler Open-1.0. 2010, 2008–2015. Available online: https://www.repeatmasker.org (accessed on 28 December 2021).
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Guigó, R.; Knudsen, S.; Drake, N.; Smith, T. Prediction of gene structure. J. Mol. Biol. 1992, 226, 141–157. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open-source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. Bmc Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat Manual. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Mulder, N.; Apweiler, R. InterPro and InterProScan: Tools for protein sequence classification and comparison. Methods Mol. Biol. 2007, 396, 59–70. [Google Scholar] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Mackey, A.J.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL-DB: Querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006, 34, D363–D368. [Google Scholar] [CrossRef]
- Robert, C.E. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic. Acids. Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Q.; Tang, W.; Huang, Z.; Wang, G.; Wang, Y.; Shi, J.; Xu, H.; Lin, L.; Li, Z.; et al. The evolutionary origin and domestication history of goldfish (Carassius auratus). Proc. Natl. Acad. Sci. USA 2020, 117, 29775–29785. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, X.F.; Wang, X.M.; Li, J.T.; Liu, G.M.; Kuang, Y.Y.; Xu, J.; Zheng, X.H.; Ren, L.F.; Wang, G.L.; et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.X.; Chen, X.L.; Bai, J.; Fang, D.M.; Qiu, Y.; Jiang, W.S.; Yuan, H.; Bian, C.; Lu, J.; He, S.Y.; et al. The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Omori, Y.; Koren, S.; Shirokiya, T.; Kuroda, T.; Miyamoto, A.; Wada, H.; Fujiyama, A.; Toyoda, A.; Zhang, S.Y.; et al. De Novo assembly of the goldfish (Carassius auratus) genome and the evolution of genes after whole genome duplication. Sci. Adv. 2019, 5, eaav0547. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.N.; Gao, T.; Wang, F.L.; Qin, Z.L.; Yan, L.X.; Tao, W.J.; Li, M.H.; Jin, C.B.; Ma, L.; Kocher, T.D.; et al. Chromosome-level genome assembly of a cyprinid fish Onychostoma macrolepis by integration of nanopore sequencing, Bionano and Hi-C technology. Mol. Ecol. Resour. 2020, 20, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 505, 248. [Google Scholar] [CrossRef]
- Yang, L.; Sado, T.; Hirt, M.V.; Pasco-Viel, E.; Arunachalam, M.; Li, J.B.; Wang, X.Z.; Freyhof, J.; Saitoh, K.; Simons, A.M.; et al. Phylogeny and polyploidy: Resolving the classification of cyprinine fishes (Teleostei: Cypriniformes). Mol. Phylogenet. Evol. 2015, 85, 97–116. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, H.; Chen, Y.; Kuwahara, M.; Nakajima, T.; Zhong, Y. Molecular phylogenetic relationships of Eastern Asian Cyprinidae (Pisces: Cypriniformes) inferred from cytochrome b sequences. Sci. China Life Sci. 2004, 47, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Secombes, C.J.; Wang, T. The innate and adaptive immune system of fish. Infect. Dis. Aquac. 2012, 14, 3–68. [Google Scholar]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll receptors and pathogen resistance. Cell Microbiol. 2003, 5, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Su, J.G.; Yang, C.R.; Xiong, F.; Wang, Y.P.; Zhu, Z.Y. Toll-like receptor 4 signaling pathway can be triggered by grass carp reovirus and Aeromonas hydrophila infection in rare minnow Gobio cyprisrarus. Fish Shellfish Immunol. 2009, 27, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.Y.; Huang, R.; Li, Y.M.; Liao, L.J.; Zhu, Z.Y.; Wang, Y.P. Characterizations of four toll-like receptor 4s in grass carp Ctenopharyngodon idellus and their response to grass carp reovirus infection and lipopolysaccharide stimulation. J. Fish Biol. 2015, 86, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.W.; Wan, Q.Y.; Su, H.; Wu, C.S.; Su, J.G. Pattern recognition receptors in grass carp Ctenopharyngodon idella: I. Organization and expression analysis of TLRs and RLRs. Dev. Comp. Immunol. 2017, 76, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.C.; Glusman, G.; Rowen, L.; Kaur, A.; Purcell, M.K.; Smith, K.D.; Hood, L.E.; Aderem, A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 9577–9582. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.; Korytar, T.; Kaczmarzyk, D.; Nipkow, M.; Kuhn, C.; Goldammer, T.; Rebl, A. Toll-like receptors in maraena whitefish: Evolutionary relation- ship among salmonid fishes and patterns of response to Aeromonas salmonicida. Fish Shellfish Immunol. 2016, 54, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.L.; Chen, S.N.; Huo, H.J.; Nie, P. Identification and expression analysis of sixteen Toll-like receptor genes, TLR1, TLR2a, TLR2b, TLR3, TLR5M, TLR5S, TLR7−9, TLR13a−c, TLR14, TLR21−23 in mandarin fish Siniperca chuatsi. Dev. Comp. Immunol. 2021, 121, 104100. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, A.Y.; Kiron, V.; Dopazo, J.; Fernandes, J.M. Diversification of the expanded teleost-specific toll-like receptor family in Atlantic cod, Gadus morhua. BMC Evol. Biol. 2012, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.T.; Fernandes, J.M.O.; Hong, W.S.; Wu, H.X.; Zhang, Y.T.; Huang, S.; Liu, D.T.; Yu, H.; Wang, Q.; You, X.X.; et al. Paralogues from the expanded Tlr11 gene family in mudskipper (Boleophthalmus pectinirostris) are under positive selection and respond differently to LPS/Poly(I:C) challenge. Front. Immunol. 2019, 10, 343. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Liao, Z.; Rao, Y.; Li, W.; Yang, C.; Yuan, G.; Feng, H.; Xu, Z.; Shao, J.; Su, J. Thoroughly remold the localization and signaling pathway of TLR22. Front. Immunol. 2020, 10, 3003. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.H.; Gabby Krens, S.F.; Medina Rodriguez, I.A.; He, S.; Bitter, W.; Ewa Snaar- Jagalska, B.; Spaink, H.P. Expression analysis of the Toll-like receptor and TIR domain adaptor families of zebrafish. Mol. Immunol. 2004, 40, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Palti, Y. Toll-like receptors in bony fish: From genomics to function. Dev. Comp. Immunol. 2011, 35, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.W.; Su, J.G. Progresses on three pattern recognition receptor families (TLRs, RLRs and NLRs) in teleost. Dev. Comp. Immunol. 2021, 122, 104131. [Google Scholar] [CrossRef]
- Liao, Z.W.; Yang, C.R.; Jiang, R.; Zhu, W.T.; Zhang, Y.A.; Su, J.G. Cyprinid-specific duplicated membrane TLR5 senses dsRNA as functional homodimeric receptors. Embo Rep. 2022, 23, e54281. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.S.; Bao, C.; Padalko, E.; Lowenstein, C.J. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J. Exp. Med. 2008, 205, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Nestor, B.J.; Bayer, P.E.; Fernandez, C.G.T.; Edwards, D.; Finnegan, P.M. Approaches to increase the validity of gene family identification using manual homology search tools. Genetica 2023, 151, 325–338. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pair-End Libraries | Library Size (bp) | Sequencing Platform | Total Data (Gb) | Sequence Coverage (×) |
|---|---|---|---|---|
| Illumina reads | 350 | Illumina NovaSeq−6000 | 43.88 | 47.40 |
| PacBio reads | 20,000 | PacBio Sequel II | 155.34 | 167.82 |
| Hi-C reads | 350 | Illumina NovaSeq−6000 | 145.69 | 157.39 |
| Transcriptome | 350 | Illumina NovaSeq−6000 | 39.78 | 42.97 |
| Total | 384.69 | 415.58 |
| Statistics | Number of Genes | Percentage (%) |
|---|---|---|
| Complete BUSCOs | 2511 | 97.10% |
| Complete and single-copy BUSCOs | 2441 | 94.40% |
| Complete Duplicated BUSCOs | 70 | 2.70% |
| Fragmented BUSCOs | 44 | 1.70% |
| Missing BUSCOs | 31 | 1.20% |
| Total BUSCO groups searched | 2586 | 100% |
| De Novo & Repbase | TE Proteins | Combined TEs | ||||
|---|---|---|---|---|---|---|
| Length (bp) | % in Genome | Length (bp) | % in Genome | Length (bp) | % in Genome | |
| DNA | 54,528,535 | 6.09 | 5,695,830 | 0.64 | 56,929,089 | 6.36 |
| LINE | 6,820,226 | 0.76 | 12,651,477 | 1.41 | 15,790,457 | 1.76 |
| SINE | 632,665 | 0.07 | 0 | 0 | 632,665 | 0.07 |
| LTR | 339,137,241 | 37.89 | 19,028,647 | 2.13 | 340,522,087 | 38.05 |
| Unknown | 18,886,738 | 2.11 | 0 | 0 | 18,886,738 | 2.11 |
| Total | 411,198,899 | 45.94 | 37,369,252 | 4.18 | 414,869,703 | 46.35 |
| Annotation Database | Number of Annotated Genes | Percentage (%) |
|---|---|---|
| Swissprot | 21,440 | 83.20 |
| Nr | 24,328 | 94.40 |
| KEGG | 21,328 | 82.70 |
| InterPro | 22,481 | 87.20 |
| GO | 16,145 | 62.60 |
| Pfam | 20,160 | 78.20 |
| Annotated | 24,402 | 94.70 |
| Unannotated | 1377 | 5.30 |
| Total | 25,779 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Sun, Y.; Liu, Y.; Song, X.; Wang, S.; Xiao, T.; Nie, P. Chromosome-Level Genome Assembly and Comparative Genomic Analysis of the Barbel Chub (Squaliobarbus curriculus) by Integration of PacBio Sequencing and Hi-C Technology. Fishes 2024, 9, 327. https://doi.org/10.3390/fishes9080327
Zhang B, Sun Y, Liu Y, Song X, Wang S, Xiao T, Nie P. Chromosome-Level Genome Assembly and Comparative Genomic Analysis of the Barbel Chub (Squaliobarbus curriculus) by Integration of PacBio Sequencing and Hi-C Technology. Fishes. 2024; 9(8):327. https://doi.org/10.3390/fishes9080327
Chicago/Turabian StyleZhang, Baidong, Yanling Sun, Yang Liu, Xiaojun Song, Su Wang, Tiaoyi Xiao, and Pin Nie. 2024. "Chromosome-Level Genome Assembly and Comparative Genomic Analysis of the Barbel Chub (Squaliobarbus curriculus) by Integration of PacBio Sequencing and Hi-C Technology" Fishes 9, no. 8: 327. https://doi.org/10.3390/fishes9080327
APA StyleZhang, B., Sun, Y., Liu, Y., Song, X., Wang, S., Xiao, T., & Nie, P. (2024). Chromosome-Level Genome Assembly and Comparative Genomic Analysis of the Barbel Chub (Squaliobarbus curriculus) by Integration of PacBio Sequencing and Hi-C Technology. Fishes, 9(8), 327. https://doi.org/10.3390/fishes9080327