Abstract
Magnesium diboride gained significant interest in the materials science community after the discovery of its superconductivity, with an unusually high critical temperature of 39 K. Many aspects of the electronic properties and superconductivity of bulk MgB and thin sheets of MgB have been determined; however, a single layer of MgB has not yet been fully theoretically investigated. Here, we present a detailed study of the structural, electronic, vibrational, and elastic properties of monolayer MgB, based on ab initio methods. First-principles calculations reveal the importance of reduction of dimensionality on the properties of MgB and thoroughly describe the properties of this novel 2D material. The presence of a negative Poisson ratio, higher density of states at the Fermi level, and a good dynamic stability under strain make the MgB monolayer a prominent material, both for fundamental research and application studies.
PACS:
71.15.Mb; 74.70.Ad
1. Introduction
Magnesium diboride was first synthesized and had its structure confirmed in 1953 [1]. An interest in its properties has grown ever since 2001, when it was discovered that MgB exhibits the highest superconducting transition temperature T of all metallic superconductors. It is an inter-metallic s-wave compound superconductor with a quasi-two dimensional character [2] and a critical temperature of superconductive transition at T = 39 K. The experimental confirmation of the isotope effect [3] in MgB indicated that it is a phonon-mediated BCS superconductor. A better definition would describe MgB as self-doped semimetal with a crucial -bonding band that is nearly filled [4]. The basic aspects of the electronic structure and pairing is in a rather strong coupling of high frequency boron–boron stretch modes to the bonding electronic boron–boron states at the Fermi surface. The phonon-mediated mechanism with different coupling strengths between a particular phonon mode and selected electronic bands, boron - and -bands [5,6,7,8,9,10,11,12,13], results in the presence of two superconducting gaps at the Fermi level. MgB has already been fabricated in bulk, as single crystals, and as a thin film, and shows potential for practical applications.
The discovery of graphene in 2004 [14] sparked an interest in 2D materials and their properties. A variety of new properties, which distinguished graphene from graphite [14,15,16,17,18,19,20,21,22], inspired a search for other low-dimensional limits of layered materials and possibilities they offered. Interest in a low-dimensional limit of MgB has arisen in past years, showing that it is superconductive even in a monolayer [23,24].
MgB has a distinct layer structure, where boron atoms form a honeycomb layer and magnesium atoms are located above the center of the hexagons, between every boron plane. The boron layers alternate with a triangular lattice of magnesium layers. There is a noticeable structural similarity of MgB to graphite-intercalated compounds (GICs), some of which also exhibit superconductivity [25,26,27,28,29]. Both monolayer and two-layer graphene, decorated/intercalated with atoms of alkali and alkaline earth metals, exhibit superconductivity and have been thoroughly studied using ab initio methods and isotropic and anisotropic Eliashberg theory [30,31,32].
Furthermore, a similarity in the electronic structure between GICs and MgB exists. The peculiar and unique property of MgB is a consequence of the incomplete filling of two bands corresponding to strongly covalent -hybrid bonding within the graphite-like boron layers [33].
Here, we present a comprehensive study of the electronic, vibrational, and mechanical properties of MgB using ab initio methods, in order to provide its detail description.
2. Computational Details
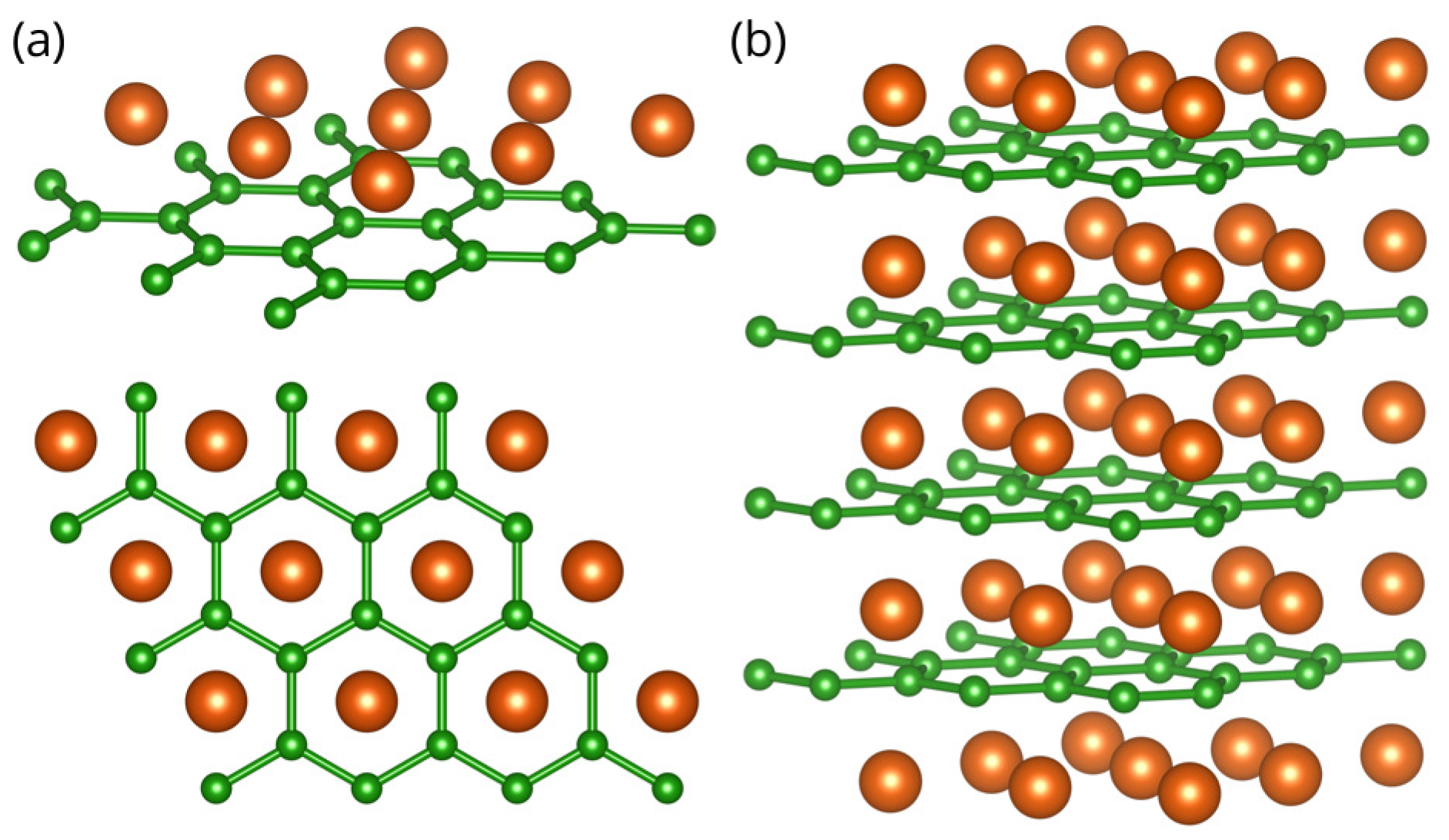
MgB has a hexagonal unit cell and consists of graphite-like B layers stacked with the Mg atoms in between, as shown in Figure 1. The first-principles calculations were performed within the density functional theory (DFT) formalism, using a general gradient approximation (GGA) to calculate the electronic structure. For all electronic and phonon structure, the Quantum Espresso software package [34] was used with ultra-soft pseudopotentials and a plane-wave cutoff energy of 30 Ry. All calculated structures are relaxed to their minimum energy configuration, following the internal force on atoms and stress tensor of the unit cell. We used the Monkhorst-Pack and k-meshes, for the calculations of the electronic structure of the MgB bulk and MgB monolayer, respectively. The phonon frequencies are calculated using Density Functional Perturbation Theory (DPFT) on the and phonon wave vector mesh for the bulk and monolayer structures, respectively. In two-dimensional systems, the van der Waals (vdW) interaction was found to play an important role on the electronic structure [35]; however, as this is study on monolayer MgB, we do not treat vdW interactions, especially since, in this case, the effects are minor and including them would add additional computational costs but would not yield more accurate results.
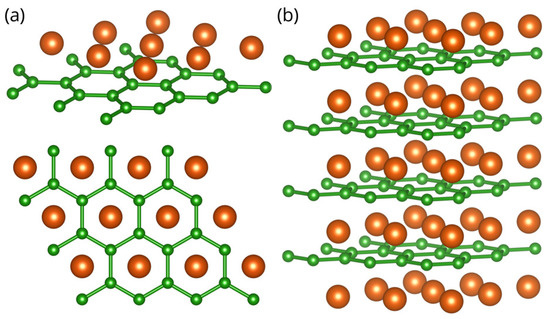
Figure 1.
Crystal structure of the MgB monolayer (a) and bulk MgB (b), with a hexagonal unit cell. Green (orange) spheres represent Boron (Magnesium) atoms. Color online.
The crystal structure of MgB and the MgB monolayer are presented in Figure 1. The lattice parameters for the bulk MgB are in agreement with the experimental results, and [9]. In order to avoid an interlayer interaction due to the periodicity and to simulate a 2D material, an artificial vacuum layer was set to be . When the monolayer is modelled, the structure is geometrically optimized, allowing the atoms to reach a minimum potential energy state. The bond length between neighbouring atoms remained to be 1.78 , but the distance from the boron layer to the Mg atoms changed from to .
For the molecular dynamics (MD) study, the Siesta code was utilized [36]. The super-cell is built by repeating the unit cell three times in both in-plane directions, whereas the lattice vector in the perpendicular direction is 15 , providing a large enough vacuum space between the 2D material and its periodic replica in order to avoid their mutual interaction. The lattice parameters and the geometry of the unit cell are initially optimized using the conjugate gradient method. The Perdew-Burke-Ernzerhof form of the exchange-correlation functional [37], the double-zeta polarized basis set, and the Troulier-Martins pseudopotentials [38] were used in all MD calculations.
The second-order elastic constants were calculated using the ElaStic software package [39]. First, the direction is projected from the strain tensor and total energies for each deformation are calculated. Elastic constants are then calculated using the second derivatives of the energy curves, dependent on the parameter . In our calculations, the maximum positive and negative amplitudes of Lagrangian strain were applied, with a step of .
For the 2D square, rectangular, or hexagonal lattices, the non-zero second-order elastic constants, in Voigt notation, are , , , and . Due to symmetry, in hexagonal structures = and ; so, we have 2 independent elastic constants. The layer modulus, which represents the resistance of a 2D material to stretching, is given as
The 2D Young modulus Y for strains in the (10) and (01) directions, Poisson’s ratio and the shear modulus G are obtained from the following relations,
Units for elastic constants and those parameters are .
3. Results and Discussion
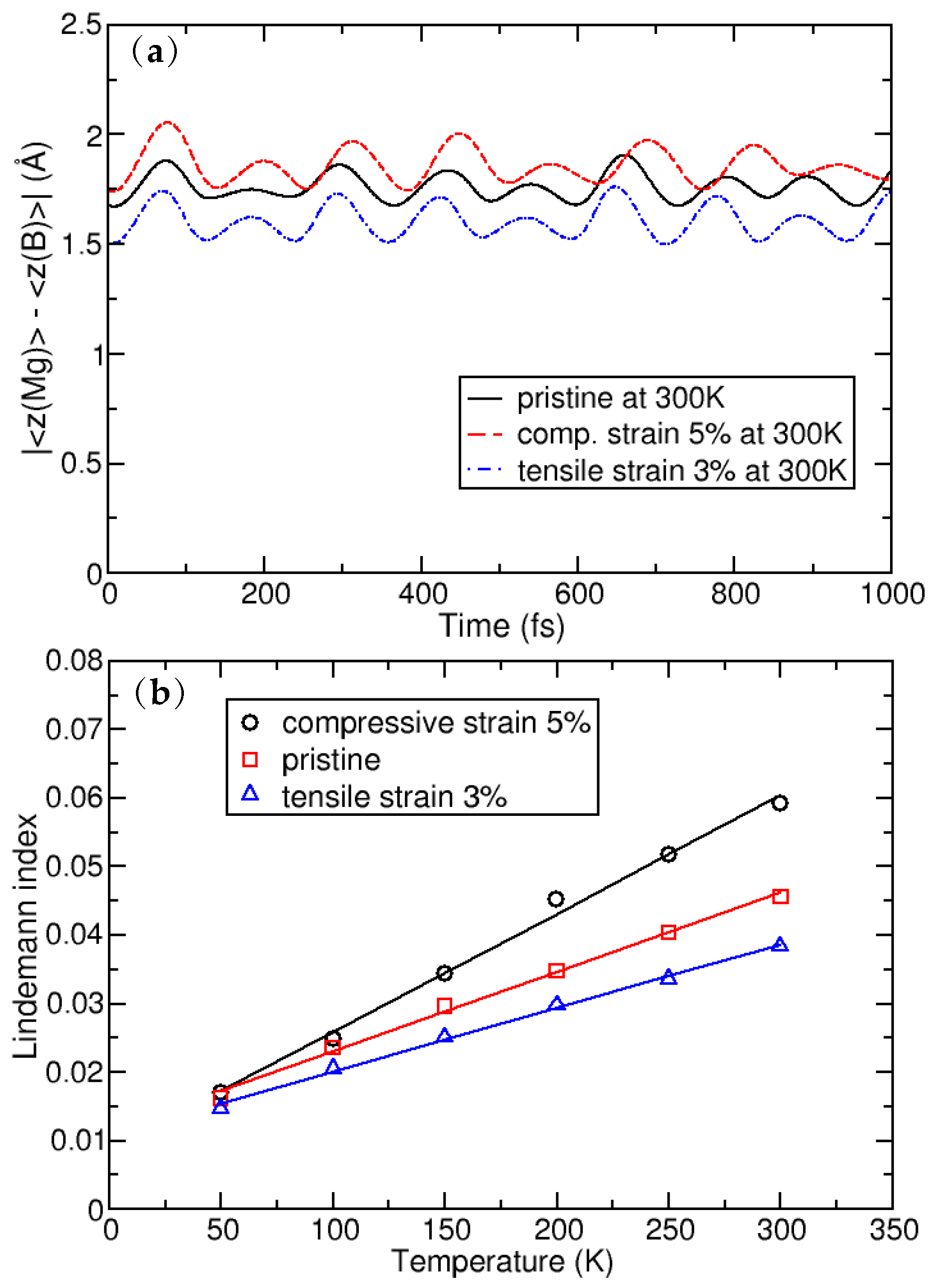
In order to determine the stability of a single layer of MgB, we perform MD simulations based on DFT and the super-cell approach. Besides the system with optimized (pristine) lattice parameters, we also consider a biaxially stretched system (up to 3% of tensile strain) and biaxially compressed system (up to 5% of compressive strain). The MD simulations are conducted in the range of temperatures between 50–300 K, with a step of 50 K, using the Nosé–Hoover thermostat [40].
Figure 2a shows the average distance between Mg and B atomic layers, as evolved over a time of 1 ps. Throughout the simulation time, there is no further evolution of the z-coordinate and the Mg atom shows only oscillatory movement around the equilibrium positions (as is shown in Figure 2) Importantly, the separation indicates that the Mg atoms do not leave the surface of the MgB crystal. The plane in which the Mg atoms reside shifts away from the plane of the B atoms on average by 0.09 in a compressed crystal, while the distance between the planes decreases on average by 0.42 in the stretched system. This (relatively larger) shift in the latter case can be understood by analysing the details of the MgB atomic structure. When the crystal is biaxially stretched, its Mg–B bond lengths increase, which is partially compensated by the nesting of the Mg atoms in the hollow sites closer to the B sublattice. Despite these atomic shifts, the MD simulations show the structural stability of the system. The stability from the MD simulations can be further quantitatively derived from the global Lindemann index, the dependence of which on temperature is shown in Figure 2b. It is calculated for the pristine crystal, with a compressive strain of 5% and a tensile strain of 3%, from the local Lindemann indices, given by the formula
by averaging over all atoms. Here is the local Lindemann index of atom i, N is number of atoms, is a separation between atoms i and j, and the angle brackets denote averaging over time (i.e., MD steps) [41]. The linear behaviour of the Lindemann indices indicate that systems are stable, at least up to room temperature.
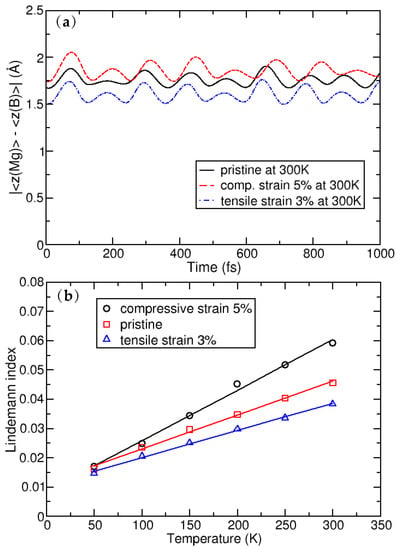
Figure 2.
(a): Average distance between the Mg and B atomic layers; and (b): the dependence of the global Lindemann index as a function of temperature.
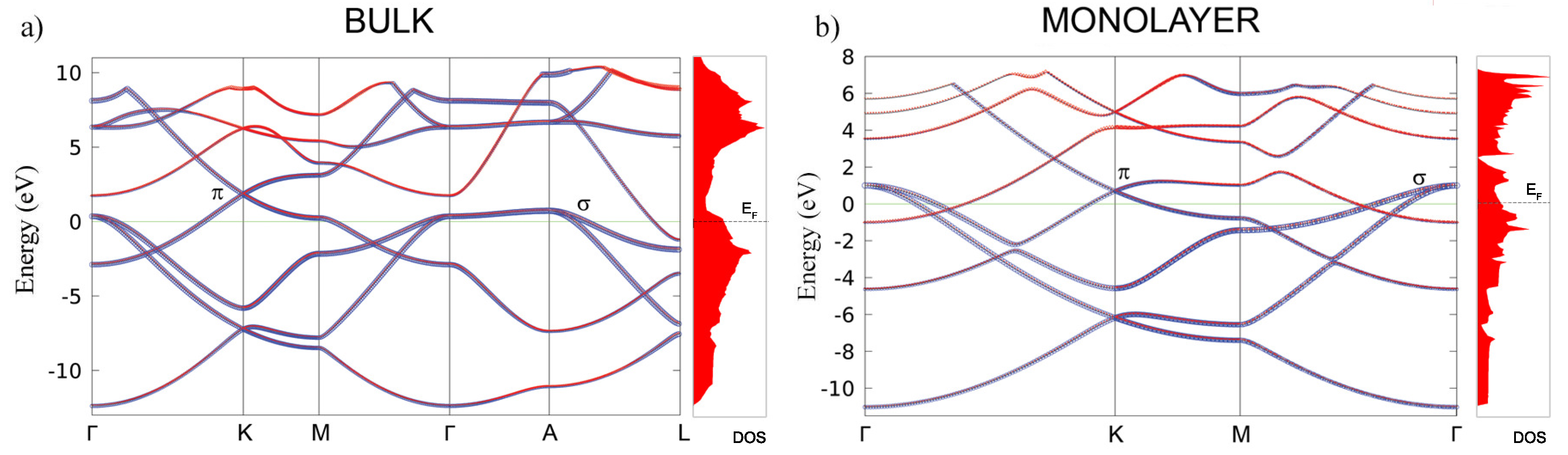
The calculated second-order elastic constants and other structural parameters for monolayer are given in Table 1. All elastic constants related to the bulk material (those that have 3, 4, or 5 in their subscripts), are calculated close to zero, as is expected for the monolayer. Compared to similar 2D materials, the layer modulus of of 30.18 is relatively small (in the range of Silicene and Germanene), roughly five times smaller than that of graphene or h–BN, for example [42,43]. Similar results are obtained for the Young modulus. Compared to borophene (two-dimensional boron sheets with rectangular structures) [44], which is a hard and brittle 2D material that exhibits an extremely large Young’s modulus of 398 N/m along the a direction [45], the MgB monolayer has a significantly smaller value of 63.29 N/m. The most interesting observation in the elastic properties of the monolayer is that the constant is negative, which gives a negative Poisson ratio in the a and b directions, too—although, with a very small negative value of −0.05. However, compared to 2D borophene, which has an out-of-plane negative Poisson’s ratio (that effectively holds the strong boron bonds lying along the a direction and makes the boron sheet show superior mechanical flexibility along the b direction [46]), we obtain similar values [45]. For comparison, graphene has a Young modulus of 352.2 and a Poisson ratio of 0.185 [42]. After confirming its stability and determining the elastic properties of the MgB monolayer, we study its electronic properties. In Figure 3, the electronic structures of bulk MgB and the MgB monolayer are presented. The band structures for the bulk along the high-symmetry points -K-M--A-L, and for the monolayer along -K-M- were calculated. The Fermi level is set to zero. The band structure of the bulk is in full agreement with previous studies [10,47,48,49]. The two bands crossing the Fermi level play a crucial role in the electronic properties of MgB. The density of the states around E are predominantly related to the B atoms and their p-orbitals, whereas the Mg atom contribution is negligible in this region. Previous studies described Mg as fully ionized and showed that the electrons donated to the system are not localized on the anion but, rather, are distributed over the whole crystal [6]. A similarity to graphite can be observed, with three bands, corresponding to the in-plane () hybridization in the boron layer and two -bands of boron orbitals [33]. Boron and orbitals contribute as and states. Analysing projected DOS, one concludes that the states are considerably involved in the total density of states at the Fermi level, while the states have only a partial contribution. It is worth emphasizing that the bulk bands of this material at the K-point above the Fermi level present a formation similar to the Dirac cones in graphene.

Table 1.
The calculated elastic stiffness constants, layer modulus , Young’s modulus Y, Poisson’s ratio , and shear modulus G for the MgB monolayer. All parameters are in units of N/m.
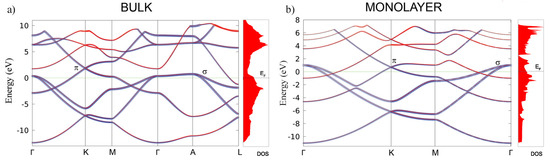
Figure 3.
The electronic band structure and total density of states in bulk MgB (a) and the MgB monolayer (b). The blue and red colors represent the B and Mg atoms contributions to the electronic dispersion, respectively.
In the monolayer, there is an increase in the total density of states at the Fermi level from N(E) = 0.72 states/eV to N(E) = 0.97 states/eV. In the same manner as in the bulk, the monolayer Mg atoms negligibly contribute to the density of states at the Fermi level, and the main contribution comes from the B p-orbitals. The characteristic Dirac cone-like structure is still present and closer to the Fermi level. Dg77, as the symmetry group of the MgB monolayer, hosts a Dirac-like dispersion in the vicinity of the K-point in the hexagonal Brillouin zone, if the orbital wave functions belong to the 2D representation E of the C point group of the wave vector [50,51]. In the tight-binding case, the and orbitals of two boron ions give rise to one E-representation (and to two one-dimensional representations), while the s-orbitals form a basis for one E-representation and -orbitals form a basis for one E-representation as well. This explains the presence of the Dirac cones at the K-point in the band structure of the MgB monolayer (as shown in Figure 3b).
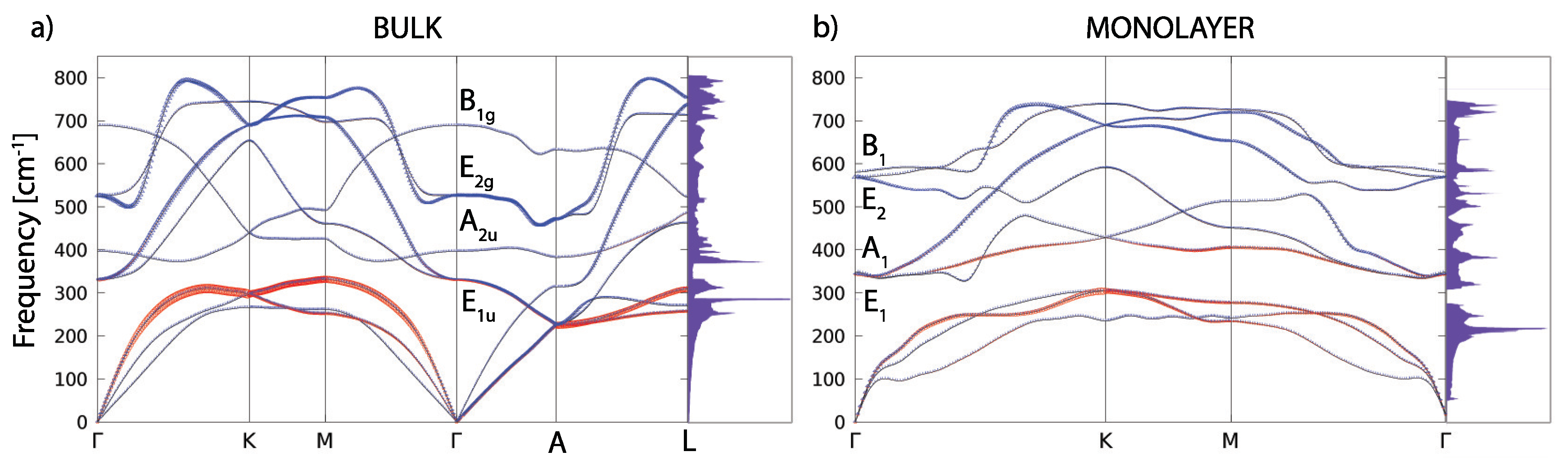
Figure 4 shows the phonon dispersions for both the bulk and monolayer. For the bulk (in Figure 4a), there are four optical modes at the point. Due to the light atomic mass of the B atoms and the strong B–B coupling, the two high-frequency modes almost have a pure boron character. The in-plane stretching mode E and the out-of-plane mode (where the atoms move in opposite directions B) are the boron atom modes. E is a doubly-degenerate Raman active mode and experimental studies [6,9] showed that this mode is very sensitive to structural changes and it has a strong electron-phonon coupling. The low-frequency modes (A) and double degenerate (E) are infrared active and they do not involve changes on in-plane bonds. In Figure 4b, the phonon dispersion of the MgB monolayer is presented. In the phonon spectrum there are no imaginary frequencies, which confirms, once again, the dynamical stability of the system (also demonstrated earlier by the MD calculations).
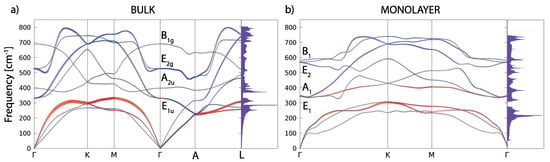
Figure 4.
The phonon dispersion and the phonon density of states for the MgB bulk (a) and monolayer (b). The blue and red colours represent the B and Mg atom contributions in the phonon dispersion, respectively.
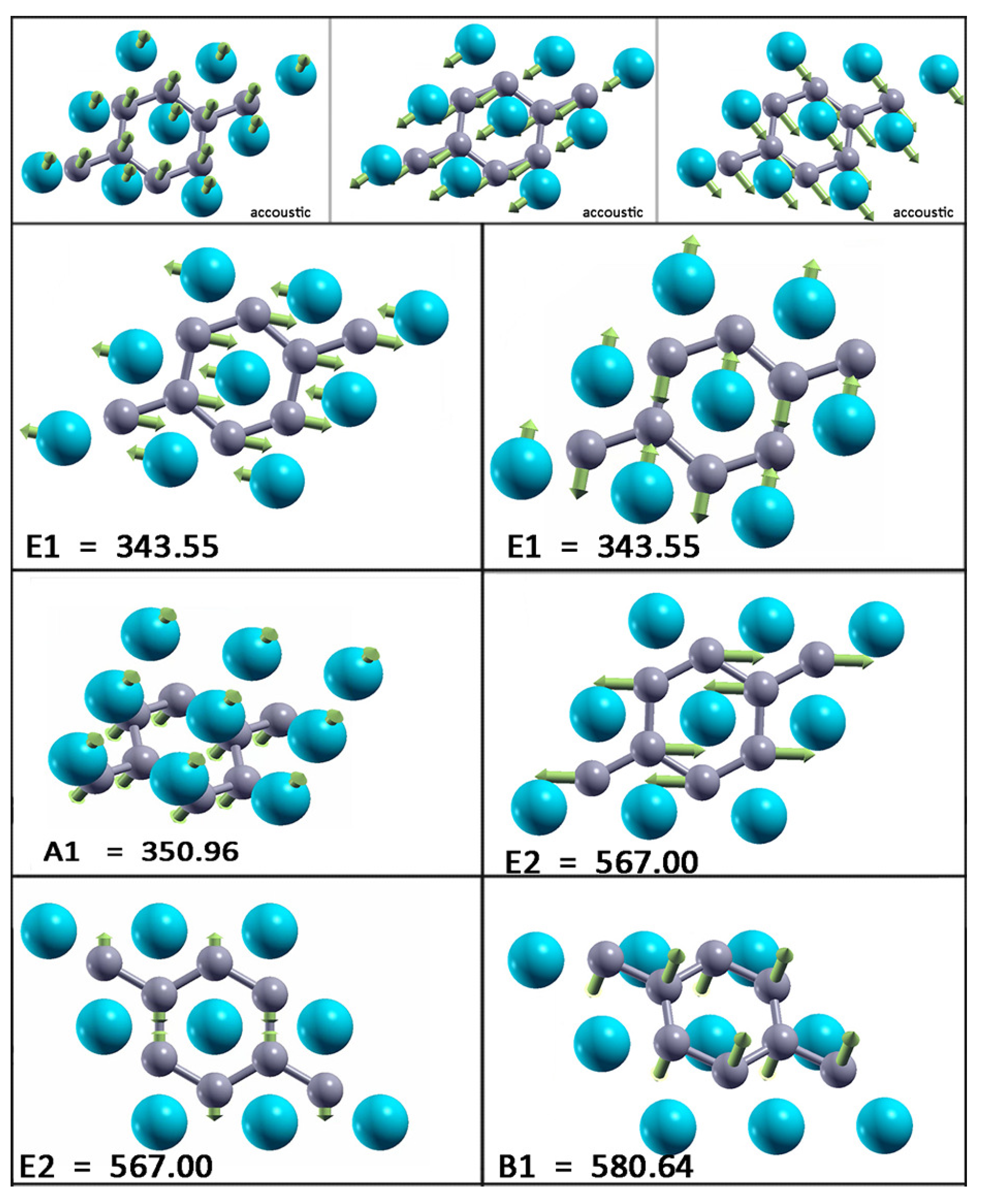
At the point, there are three acoustic and six optical modes (from which two pairs are doubly degenerate). The optical modes A, B, E, and E are related to the optical modes of the parent material. Two significant differences between the bulk and monolayer spectrum can be observed: The E and A mode become energy degenerate in the monolayer, resulting in either a slight softening (hardening) of the modes which leads to nearly equal frequencies, which opens a gap in the phonon density of states (DOS) between the acoustic and optical modes. A more significant effect concerns the softening of the B mode and hardening of the E mode. As in the bulk E mode, the monolayer E mode is strongly coupled to electrons, causing the superconductivity in the monolayer in a similar fashion as in the bulk. In Figure 5, the vibrational frequencies and normal coordinates for the MgB monolayer are presented. The symmetry group is , and the acoustic modes are A and E. The optical modes at the point are A, B, E, and E, where the infrared-active ones are A and E. The Raman-active modes are A, E, and E, and B is silent. In Table 2, the Raman tensor for the MgB monolayer is presented [52]. Similar to graphene, the phonon eigenvectors and the normal coordinates at the -point are determined by symmetry rules and, therefore, are a model independent.
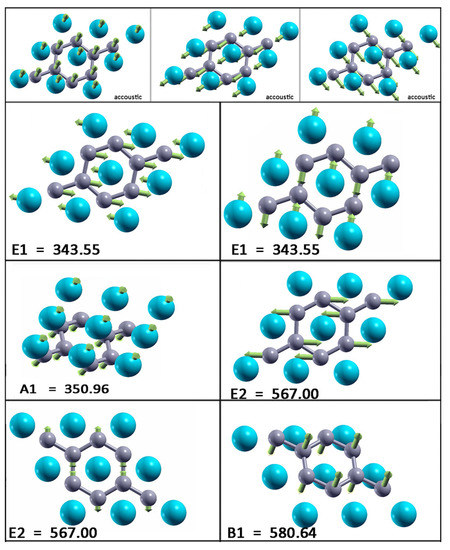
Figure 5.
Vibrational frequencies (in wavenumbers) and the vibration normal coordinates at for the MgB monolayer.
Table 2.
Raman tensor of the MgB monolayer.
4. Conclusions
The electronic band structure, density of states, phonon dispersion, and elastic constants have been calculated for the MgB monolayer and compared to the bulk material, using first-principles calculations within the DFT framework. We demonstrated an increase of electronic density of states at the Fermi level in the monolayer (compared to the bulk) and determined its stability under various strains. These two features are crucial for the enhancement of electron–phonon coupling and they enable significant mechanical modification that increases the critical superconducting temperature. Establishing stability and offering insight into this novel 2D material, we focus on the effects of ultimate lowering of the dimensionality. The question of reduction of dimensionality to its limit, a truly atomic-scale 2D system, and the consequences of this [53,54,55,56,57,58,59,60,61] are highly relevant, not only to fundamental science but also to applications in nanotechnology.
Author Contributions
Conceptualization, J.P. and R.G.; Validation, K.H., M.B., R.G.; Investigation, J.P., I.P., A.Š. and V.D.; Writing—Original Draft Preparation, J.P., I.P., A.Š. and V.D.; Writing—Review & Editing, J.P.; Supervision, R.G.; Funding Acquisition, K.H., M.B. and R.G.
Funding
This research is supported by Serbian Ministry of Education, Science and Technological Development under projects OI 171005, III 45018, and III 45016 and by the Qatar National Research Fund, cycle 11, under grant number NPRP 11S-1126-170033. K.H. acknowledges the support of the European Commission under the H2020 grant TWINFUSYON.GA692034.
Acknowledgments
The DFT calculations were performed using the computational resources at Johannes Kepler University, Linz, Austria. This work was supported by the Serbian Ministry of Education, Science and Technological Development under projects OI 171005, III 45018, and III 45016.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jones, M.E.; Marsh, R.E. The preparation and structure of magnesium boride, MgB2. J. Am. Chem. Soc. 1953, 76, 5. [Google Scholar] [CrossRef]
- Nagamatsu, J.; Nakagawa, N.; Muranaka, T.; Zenitani, Y.; Akimitsu, J. Superconductivity at 39 K in magnesium diboride. Nature 2001, 410, 63. [Google Scholar] [CrossRef] [PubMed]
- Bud’ko, S.L.; Lapertot, G.; Petrovic, C.; Cunningham, C.E.; Anderson, N.; Canfield, P.C. Boron Isotope Effect in Superconducting MgB2. Phys. Rev. Lett. 2001, 86, 1877. [Google Scholar] [CrossRef] [PubMed]
- Pickett, W. Superconductivity: 2D Physics, Unknown Mechanisms, Current Puzzles. Emerg. Phenom. Correl. Matter Lect. Notes Autumn School Corr. Electron. 2013, 2013, 45. [Google Scholar]
- Choi, H.J.; Roundy, D.; Sun, H.; Cohen, M.L.; Steven Louie, G. The origin of the anomalous superconducting properties of MgB2. Nature 2002, 418, 758. [Google Scholar] [CrossRef]
- Kortus, J.; Mazin, I.I.; Belaachenko, K.D.; Antropov, V.P.; Boyer, L.L. Superconductivity of Metallic Boron in MgB2. Phys. Rev. Lett. 2001, 86, 4656. [Google Scholar] [CrossRef]
- An, J.M.; Pickett, W.E. Superconductivity of MgB2: Covalent Bonds Driven Metallic. Phys. Rev. Lett. 2001, 86, 4366. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Mazin, I.I.; Kortus, J. Beyond Eliashberg Superconductivity in MgB2: Anharmonicity, Two-Phonon Scattering, and Multiple Gaps. Phys. Rev. Lett. 2001, 87, 087005. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Dolgov, O.V.; Jepsen, O.; Andersen, O.K. Electron-phonon interaction in the normal and superconducting states of MgB2. Phys. Rev. B 2001, 64, 020501. [Google Scholar] [CrossRef]
- Bohnen, K.-P.; Heid, R.; Renker, B. Phonon Dispersion and Electron-Phonon Coupling in MgB2 and AlB2. Phys. Rev. Lett. 2001, 86, 5771. [Google Scholar] [CrossRef] [PubMed]
- Kunc, K.; Loa, I.; Syassen, K.; Kremer, R.K.; Ahn, K. MgB2 under pressure: phonon calculations, Raman spectroscopy, and optical reflectance. J. Phys. Condens. Matter 2001, 13, 9945. [Google Scholar] [CrossRef]
- Choi, H.J.; Roundy, D.; Sun, H.; Cohen, M.L.; Louie, S.G. First-principles calculation of the superconducting transition in MgB2 within the anisotropic Eliashberg formalism. Phys. Rev. B 2002, 66, 020513. [Google Scholar] [CrossRef]
- Canfield, P.C.; Crabtree, G.W. Magnesium Diboride: Better Late than Never. Phys. Today 2003, 56, 34. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Katsnelson, M.I.; Novoselov, K.S.; Geim, A.K. Chiral tunnelling and the Klein paradox in graphene. Nat. Phys. 2006, 2, 620–625. [Google Scholar] [CrossRef]
- Katsnelson, M.I. Zitterbewegung, chirality, and minimal conductivity in graphene. Eur. Phys. J. B 2006, 51, 157–160. [Google Scholar] [CrossRef]
- Rusin, T.M.; Zawadzki, W. Zitterbewegung of electrons in graphene in a magnetic field. Phys. Rev. B 2008, 78, 125419. [Google Scholar] [CrossRef]
- Pisana, S.; Lazzeri, M.; Casiraghi, C.; Novoselov, K.S.; Geim, A.K.; Ferrari, A.C.; Mauri, F. Breakdown of the adiabatic Born-Oppenheimer approximation in graphene. Nat. Mater. 2007, 6, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Piscanec, S.; Lazzeri, M.; Mauri, F.; Ferrari, A.C.; Robertson, J. Kohn Anomalies and Electron-Phonon Interactions in Graphite. Phys. Rev. Lett. 2004, 93, 85503. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Jiang, Z.; Zhang, Y.; Morozov, S.V.; Stormer, H.L.; Zeitler, U.; Maan, J.C.; Boebinger, G.S.; Kim, P.; Geim, A.K. Room-temperature quantum Hall effect in graphene. Science 2007, 315, 1379. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Gweon, G.-H.; Fedorov, A.V.; First, P.N.; de Heer, W.A.; Lee, D.-H.; Guinea, F.; Castro Neto, A.H.; Lanzara, A.; et al. Substrate-induced bandgap opening in epitaxial graphene. Nat. Mater. 2007, 6, 770–775. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, Y.; Stormer, H.L.; Kim, P. Experimental observation of the quantum Hall effect and Berry’s phase in graphene. Nature 2005, 438, 201–204. [Google Scholar] [CrossRef]
- Bekaert, J.; Aperis, A.; Partoens, B.; Oppeneer, P.M.; Milošević, M.V. Evolution of multigap superconductivity in the atomically thin limit: Strain-enhanced three-gap superconductivity in monolayer MgB2. Phys. Rev. B 2017, 96, 094510. [Google Scholar]
- Morshedloo, T.; Roknabadi, M.R.; Behdani, M. First-principles study of the superconductivity in MgB2 bulk and in its bilayer thin film based on electron–phonon coupling. Physica C 2015, 509. [Google Scholar] [CrossRef]
- Calandra, M.; Profeta, G.; Mauri, F. Superconductivity in metal-coated graphene. Phys. Status Solidi (b) 2012, 249, 2544. [Google Scholar] [CrossRef]
- Ludbrook, B.M.; Levy, G.; Nigge, P.; Zonno, M.; Schneider, M.; Dvorak, D.J.; Veenstra, C.N.; Zhdanovich, S.; Wong, D.; Dosanjh, P.; et al. Evidence for superconductivity in Li-decorated monolayer graphene. Proc. Natl. Acad. Sci. USA 2015, 112, 11795. [Google Scholar] [CrossRef] [PubMed]
- Profeta, G.; Calandra, M.; Mauri, F. Phonon-mediated superconductivity in graphene by lithium deposition. Nat. Phys. 2012, 8, 131–134. [Google Scholar] [CrossRef]
- Pešić, J.; Gajić, R.; Hingerl, K.; Belić, M. Strain-enhanced superconductivity in Li-doped graphene. Europhys. Lett. 2014, 108, 67005. [Google Scholar] [CrossRef]
- Szczesniak, D.; Durajski, A.P.; Szczesniak, R. Influence of lithium doping on the thermodynamic properties of graphene based superconductors. J. Phys-Condens. Mat. 2014, 26, 255701. [Google Scholar] [CrossRef]
- Durajski, A.; Skoczylas, K.; Szczesniak, R. Superconductivity in bilayer graphene intercalated with alkali and alkaline earth metals. Phys. Chem. Chem. Phys. 2019, 21, 5925. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-J.; Margine, E.R. First-principles calculations of the superconducting properties in Li-decorated monolayer graphene within the anisotropic Migdal-Eliashberg formalism. Phys. Rev. B 2016, 94, 064509. [Google Scholar] [CrossRef]
- Margine, E.R.; Lambert, H.; Giustino, F. Electron-phonon interaction and pairing mechanism in superconducting Ca-intercalated bilayer graphene. Sci. Rep. 2016, 6, 21414. [Google Scholar] [CrossRef]
- Mazin, I.I.; Antropov, V.P. Electronic structure, electron–phonon coupling, and multiband effects in MgB2. Phys. C Supercond. 2003, 385, 49–65. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno, N.M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Quantum espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condensed Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Guo, H.; Zhuo, Z.; Wang, L.; Wu, X.; Zeng, X.C. Twisted MX2/MoS2 heterobilayers: effect of van der Waals interaction on the electronic structure. Nanoscale 2017, 9, 19131–19138. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter. 2002, 14, 2745. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Golesorkhtabar, R.; Pavone, P.; Spitaler, J.; Puschnig, P.; Draxl, C. ElaStic: A tool for calculating second-order elastic constants from first principles. Comput. Phys. Commun. 2013, 184, 1861–1873. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511. [Google Scholar] [CrossRef]
- Lindemann, F.A. The calculation of molecular vibration frequencies. Phys. Z. 1910, 11, 609. [Google Scholar]
- Andrew, R.C.; Mapasha, R.E.; Ukpong, A.M.; Chetty, N. Mechanical properties of graphene and boronitrene. Phys. Rev. B 2012, 85, 125428. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Penev, E.S.; Yakobson, B.I. Elasticity, Flexibility, and Ideal Strength of Borophenes. Adv. Func. Mater. 2017, 27, 1605059. [Google Scholar] [CrossRef]
- Zhong, H.; Huang, K.; Yu, G.; Yuan, S. Electronic and mechanical properties of few-layer borophene. Phys. Rev. B 2018, 98, 054104. [Google Scholar] [CrossRef]
- Mannix, A.J.; Zhou, X.F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Gao, Y.; Miao, F.; Zhou, X.-F.; Wan, X.G. Strain effects on borophene: Ideal strength, negative Possion’s ratio and phonon instability. New J. Phys. 2016, 18, 073016. [Google Scholar] [CrossRef]
- De la Pena-Seaman, O.; de Cross, R.; Heid, R.; Bohnen, K.-P. Effects of Al and C doping on the electronic structure and phonon renormalization in MgB2. Phys. Rev. B 2009, 79, 134523. [Google Scholar] [CrossRef]
- Ponce, S.; Margine, E.R.; Verdi, C.; Giustino, F. EPW: Electron-phonon coupling, transport and superconducting properties using maximally localized Wannier functions. Comp. Phys. Commun. 2016, 209, 116–133. [Google Scholar] [CrossRef]
- Margine, E.R.; Giustino, F. Anisotropic Migdal-Eliashberg theory using Wannier functions. Phys. Rev. B 2013, 87, 024505. [Google Scholar] [CrossRef]
- Damljanovic, V.; Gajic, R. Existence of Dirac cones in the Brillouin zone of diperiodic atomic crystals according to group theory. J. Phys. Condens. Matter 2016, 28, 085502. [Google Scholar] [CrossRef]
- Damljanovic, V.; Gajic, R. Addendum to ‘Existence of Dirac cones in the Brillouin zone of diperiodic atomic crystals according to group theory’. J. Phys. Condens. Matter 2016, 28, 439401. [Google Scholar] [CrossRef]
- Poulet, H.; Mathieu, J.P. Vibration Spectra and Symmetry of Crystals; Gordon and Breach: New York, NY, USA, 1976. [Google Scholar]
- Szalowski, K. Critical temperature of MgB2 ultrathin superconducting films: BCS model calculations in the tight-binding approximation. Phys. Rev. B 2006, 74, 094501. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, Y.; Liu, Z.-H.; Feng, Q.-R.; Gan, Z.-Z. Suppression of superconductivity in epitaxial MgB2 ultrathin films. J. Appl. Phys. 2013, 114, 023903. [Google Scholar] [CrossRef]
- Ao, B.; Zhang, Z.; Tang, T.; Zhao, Y. Potential enhancement of superconductivity in MgB2 nanosheets: First-principles calculations. Chem. Phys. Lett. 2014, 591, 185–188. [Google Scholar] [CrossRef]
- Romero-Bermudez, A.; Garcıa-Garcıa, A.M. Shape resonances and shell effects in thin-film multiband superconductors. Phys. Rev. B 2014, 89, 024510. [Google Scholar] [CrossRef]
- Romero-Bermudez, A.; Garcıa-Garcıa, A.M. Size effects in superconducting thin films coupled to a substrate. Phys. Rev. B 2014, 89, 064508. [Google Scholar] [CrossRef]
- Acharya, N.; Wolak, M.A.; Cunnane, D.P.; Karasik, B.S.; Xi, X.X. MgB2 ultrathin films fabricated by hybrid physical chemical vapor deposition and ion milling. APL Mater. 2016, 4, 086114. [Google Scholar] [CrossRef]
- Valentinis, D.; van der Marel, D.; Berthod, C. Rise and fall of shape resonances in thin films of BCS superconductors. Phys. Rev. B 2016, 94, 054516. [Google Scholar] [CrossRef]
- Narlikar, A.V. Small Superconductors: Introduction. In The Oxford Handbook of Small Superconductors, 1st ed.; Narlikar, A.V., Ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Gariglio, S.; Scheurer, M.; Schmalian, J.; Monteiro, A.M.R.V.L.; Goswami, S.; Caviglia, A. Surface and Interface Superconductivity. In The Oxford Handbook of Small Superconductors, 1st ed.; Narlikar, A.V., Ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).