First-Principles Investigations of Single Metal Atoms (Sc, Ti, V, Cr, Mn, and Ni) Embedded in Hexagonal Boron Nitride Nanosheets for the Catalysis of CO Oxidation



Abstract
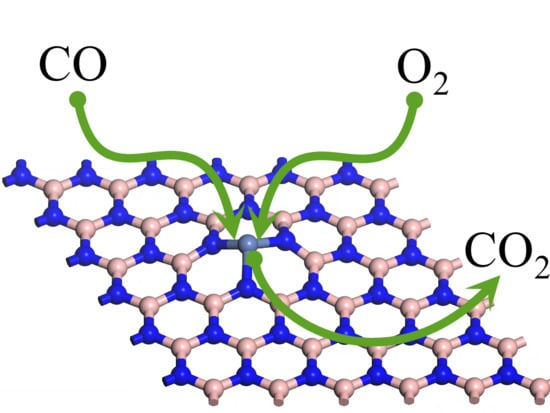
1. Introduction
2. Results and Discussion
2.1. Geometry and Stability
2.2. O2, CO, O, and CO2 Adsorption onto Sc, Cr, and Ni Atoms
2.3. Reaction Mechanism of CO Oxidation
2.4. ER Process
2.5. LH Process
2.6. TER Process
2.7. Effect of Temperature and Entropy
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kummer, J.T. Catalysts for automobile emission control. Prog. Energy Combust. Sci. 1980, 6, 177–199. [Google Scholar] [CrossRef]
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO oxidation as a prototypical reaction for heterogeneous processes. Angew. Chem. Int. Ed. Engl. 2011, 50, 10064–10094. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, K.; Hu, P. A density functional theory study of the interaction between CO and O on a Pt Surface: CO/Pt (111), O/Pt (111), and CO/O/Pt (111). J. Am. Chem. Soc. 1999, 121, 7644–7652. [Google Scholar] [CrossRef]
- Heiz, U.; Sanchez, A.; Abbet, S.; Schneider, W.-D. Catalytic oxidation of carbon monoxide on monodispersed platinum clusters: Each atom counts. J. Am. Chem. Soc. 1999, 121, 3214–3217. [Google Scholar] [CrossRef]
- Allian, A.D.; Takanabe, K.; Fujdala, K.L.; Hao, X.; Truex, T.J.; Cai, J.; Buda, C.; Neurock, M.; Iglesia, E. Chemisorption of CO and mechanism of CO oxidation on supported platinum nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498–4517. [Google Scholar] [CrossRef] [PubMed]
- Bunluesin, T.; Cordatos, H.; Gorte, R. Study of CO oxidation kinetics on Rh/ceria. J. Catal. 1995, 157, 222–226. [Google Scholar] [CrossRef]
- Chen, M.S.; Cai, Y.; Yan, Z.F.; Gath, K.; Axnanda, S.; Goodman, D.W. Highly active surfaces for CO oxidation on Rh, Pd, and Pt. Surf. Sci. 2007, 601, 5326–5331. [Google Scholar] [CrossRef]
- Eichler, A. CO oxidation on transition metal surfaces: Reaction rates from first principles. Surf. Sci. 2002, 498, 314–320. [Google Scholar] [CrossRef]
- Liu, Z.-P.; Hu, P.; Alavi, A. Catalytic role of gold in gold-based catalysts: A density functional theory study on the CO oxidation on gold. J. Am. Chem. Soc. 2002, 124, 14770–14779. [Google Scholar] [CrossRef]
- Lopez, N.; Nørskov, J.K. Catalytic CO oxidation by a gold nanoparticle: A density functional study. J. Am. Chem. Soc. 2002, 124, 11262–11263. [Google Scholar] [CrossRef]
- Lopez, N. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Thomas, J.M.; Raja, R.; Lewis, D.W. Single-site heterogeneous catalysts. Angew. Chem. Int. Ed. Engl. 2005, 44, 6456–6482. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, G.; Gauquelin, N.; Chen, N.; Zhou, J.; Yang, S.; Chen, W.; Meng, X.; Geng, D.; Banis, M.N.; et al. Single-atom Catalysis Using Pt/Graphene Achieved through Atomic Layer Deposition. Sci. Rep. 2013, 3, 1775. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, J.; Guan, P.; Liu, C.; Huang, H.; Xue, F.; Dong, X.; Pennycook, S.J.; Chisholm, M.F. Catalytically active single-atom niobium in graphitic layers. Nat. Commun. 2013, 4, 1924. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Ye, X.; Johnson, R.S.; Guo, H. First-Principles Investigations of Metal (Cu, Ag, Au, Pt, Rh, Pd, Fe, Co, and Ir) Doped Hexagonal Boron Nitride Nanosheets: Stability and Catalysis of CO Oxidation. J. Phys. Chem. C 2013, 117, 17319–17326. [Google Scholar] [CrossRef]
- Liu, X.; Duan, T.; Sui, Y.; Meng, C.; Han, Y. Copper atoms embedded in hexagonal boron nitride as potential catalysts for CO oxidation: A first-principles investigation. RSC Adv. 2014, 4, 38750–38760. [Google Scholar] [CrossRef]
- Xu, G.; Wang, R.; Yang, F.; Ma, D. CO oxidation on single Pd atom embedded defect-graphene via a new termolecular Eley-Rideal mechanism. Carbon 2017, 118, 35–42. [Google Scholar] [CrossRef]
- Jiang, Q.G.; Ao, Z.M.; Li, S.; Wen, Z. Density functional theory calculations on the CO catalytic oxidation on Al-embedded graphene. RSC Adv. 2014, 4, 20290–20296. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Z.; Yu, G.; Chen, W.; Chen, Z. CO catalytic oxidation on iron-embedded graphene: Computational quest for low-cost nanocatalysts. J. Phys. Chem. C 2010, 114, 6250–6254. [Google Scholar] [CrossRef]
- Wu, P.; Du, P.; Zhang, H.; Cai, C. Graphyne-supported single Fe atom catalysts for CO oxidation. Phys. Chem. Phys. 2015, 17, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.W.; Li, T.; Wang, Q.; Yang, G.; He, C.; Ma, B.; Lu, Z. Graphyne as a promising substrate for the noble-metal single-atom catalysts. Carbon 2015, 95, 756–765. [Google Scholar] [CrossRef]
- Du, C.; Lin, H.; Lin, B.; Ma, Z.; Hou, T.; Tang, J.; Li, Y. MoS2 supported single platinum atoms and their superior catalytic activity for CO oxidation: A density functional theory study. J. Mater. Chem. A 2015, 3, 23113–23119. [Google Scholar] [CrossRef]
- Deng, D.; Chen, X.; Yu, L.; Wu, X.; Liu, Q.; Liu, Y.; Yang, H.; Tian, H.; Hu, Y.; Du, P.; et al. A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 2015, 1, 1500462. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Dong, J.; Arellano-Jiménez, M.J.; Ye, G.; Kim, N.D.; Samuel, E.L.G.; Peng, Z.; Zhu, Z.; Qin, F.; Bao, J.; et al. Atomic cobalt on nitrogen-doped graphene for hydrogen generation. Nat. Commun. 2015, 6, 8668. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, L.M.; Ganz, E. Mn–graphene single-atom catalyst evaluated for CO oxidation by computational screening. Theor. Chem. Acc. 2018, 137, 98. [Google Scholar] [CrossRef]
- Jin, C.; Lin, F.; Suenaga, K.; Iijima, S. Fabrication of a freestanding boron nitride single layer and its defect assignments. Phys. Rev. Lett. 2009, 102, 195505. [Google Scholar] [CrossRef] [PubMed]
- Alem, N.; Erni, R.; Kisielowski, C.; Rossell, M.D.; Hartel, P.; Jiang, B.; Gannett, W.; Zettl, A. Vacancy growth and migration dynamics in atomically thin hexagonal boron nitride under electron beam irradiation. Phys. Status Solidi-RRL 2011, 5, 295–297. [Google Scholar] [CrossRef]
- Liu, X.; Duan, T.; Mang, C.; Han, Y. Pt atoms stabilized on hexagonal boron nitride as efficient single-atom catalysts for CO oxidation: A first-principles investigation. RSC Adv. 2015, 5, 10452–10459. [Google Scholar] [CrossRef]
- Mao, K.; Li, L.; Zhang, W.; Pei, Y.; Zeng, X.C.; Wu, X.; Yang, J. A theoretical study of single-atom catalysis of CO oxidation using Au embedded two-dimensional h-BN monolayer: A CO-promoted O(2) activation. Sci. Rep. 2014, 4, 5441. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Su, Y.; Zhang, Y.; Li, S.-J. CO catalytic oxidation on iron-embedded hexagonal boron nitride sheet. Chem. Phys. Lett. 2011, 515, 159–162. [Google Scholar] [CrossRef]
- Lu, Z.; Lv, P.; Liang, Y.; Ma, D.; Zhang, Y.; Zhang, W.; Yang, X.; Yang, Z. CO oxidation catalyzed by the single Co atom embedded hexagonal boron nitride nanosheet: A DFT-D study. Phys. Chem. Phys. 2016, 18, 21865–21870. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Lv, P.; Yang, Z.; Li, S.; Ma, D.; Wu, R. A promising single atom catalyst for CO oxidation: Ag on boron vacancies of h-BN sheets. Phys. Chem. Phys. 2017, 19, 16795–16805. [Google Scholar] [CrossRef] [PubMed]
- Du, A.; Chen, Y.; Zhu, Z.; Amal, R.; Lu, G.Q.; Smith, S.C. Dots versus antidots: Computational exploration of structure, magnetism, and half-metallicity in boron− nitride nanostructures. J. Am. Chem. Soc. 2009, 131, 17354–17359. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Lv, P.; Xue, J.; Wang, H.; Wang, Y.; Huang, Y.; He, C.; Mac, D.; Yang, Z. Pd1/BN as a promising single atom catalyst of CO oxidation: A dispersion-corrected density functional theory study. RSC Adv. 2015, 5, 84381–84388. [Google Scholar] [CrossRef]
- Huang, C.; Ye, X.; Chen, C.; Lin, S.; Xie, D. A computational investigation of CO oxidation on ruthenium-embedded hexagonal boron nitride nanosheet. Comput. Theor. Chem. 2013, 1011, 5–10. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Species | Adsorption Energy (eV) | Bond Length (Å) | Mulliken Charge of Adsorbed Molecules (|e|) | |
|---|---|---|---|---|---|
| Sc-BN | O2 | 0.97 | O-O | 1.33 | −0.43 |
| CO | 0.63 | C-O | 1.14 | 0.07 | |
| O | 6.62 | Sc-O | 1.92 | −0.55 | |
| CO2 | 0.57 | O-C-O | 1.19, 1.17 | 0.03 | |
| Cr-BN | O2 | 2.47 | O-O | 1.43 | −0.55 |
| CO | 0.99 | C-O | 1.16 | 0.05 | |
| O | 6.18 | Cr-O | 1.60 | −0.53 | |
| CO2 | 0.36 | O-C-O | 1.19, 1.17 | 0.04 | |
| Ni-BN | O2 | 1.34 | O-O | 1.33 | −0.27 |
| CO | 1.23 | C-O | 1.17 | 0.25 | |
| O | 3.79 | Ni-O | 1.65 | −0.44 | |
| CO2 | 0.32 | O-C-O | 1.18, 1.17 | 0 | |
| Reaction Time | Step | |||||||
|---|---|---|---|---|---|---|---|---|
| ER | LH | TER | ||||||
| IS1-MS1 | MS1-FS1 | IS2-FS2 | IS1-MS1 | MS1-FS1 | IS2-FS2 | IS1-MS1 | MS1-FS1 | |
| τ (s) at 298K | 2 × 103 | 6 × 108 | 2 × 10−11 | 3 × 10−11 | 2 × 10−5 | 2 × 10−11 | 9 × 10−5 | 4 × 10−7 |
| τ (s) at 400K | 0.2 | 3 × 103 | 1 × 10−11 | 1 × 10−11 | 4 × 10−7 | 1 × 10−11 | 8 × 10−7 | 1 × 10−8 |
| τ (s) at 500K | 0 | 2.5 | 6 × 10−12 | 8 × 10−12 | 3 × 10−8 | 6 × 10−12 | 5 × 10−8 | 2 × 10−9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Yang, L.-M.; Ganz, E. First-Principles Investigations of Single Metal Atoms (Sc, Ti, V, Cr, Mn, and Ni) Embedded in Hexagonal Boron Nitride Nanosheets for the Catalysis of CO Oxidation. Condens. Matter 2019, 4, 65. https://doi.org/10.3390/condmat4030065
Liu Y, Yang L-M, Ganz E. First-Principles Investigations of Single Metal Atoms (Sc, Ti, V, Cr, Mn, and Ni) Embedded in Hexagonal Boron Nitride Nanosheets for the Catalysis of CO Oxidation. Condensed Matter. 2019; 4(3):65. https://doi.org/10.3390/condmat4030065
Chicago/Turabian StyleLiu, Yi, Li-Ming Yang, and Eric Ganz. 2019. "First-Principles Investigations of Single Metal Atoms (Sc, Ti, V, Cr, Mn, and Ni) Embedded in Hexagonal Boron Nitride Nanosheets for the Catalysis of CO Oxidation" Condensed Matter 4, no. 3: 65. https://doi.org/10.3390/condmat4030065
APA StyleLiu, Y., Yang, L.-M., & Ganz, E. (2019). First-Principles Investigations of Single Metal Atoms (Sc, Ti, V, Cr, Mn, and Ni) Embedded in Hexagonal Boron Nitride Nanosheets for the Catalysis of CO Oxidation. Condensed Matter, 4(3), 65. https://doi.org/10.3390/condmat4030065