High-Performance Anion Exchange Chromatography Electrochemical Determination of Uric Acid as a Contamination Marker


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
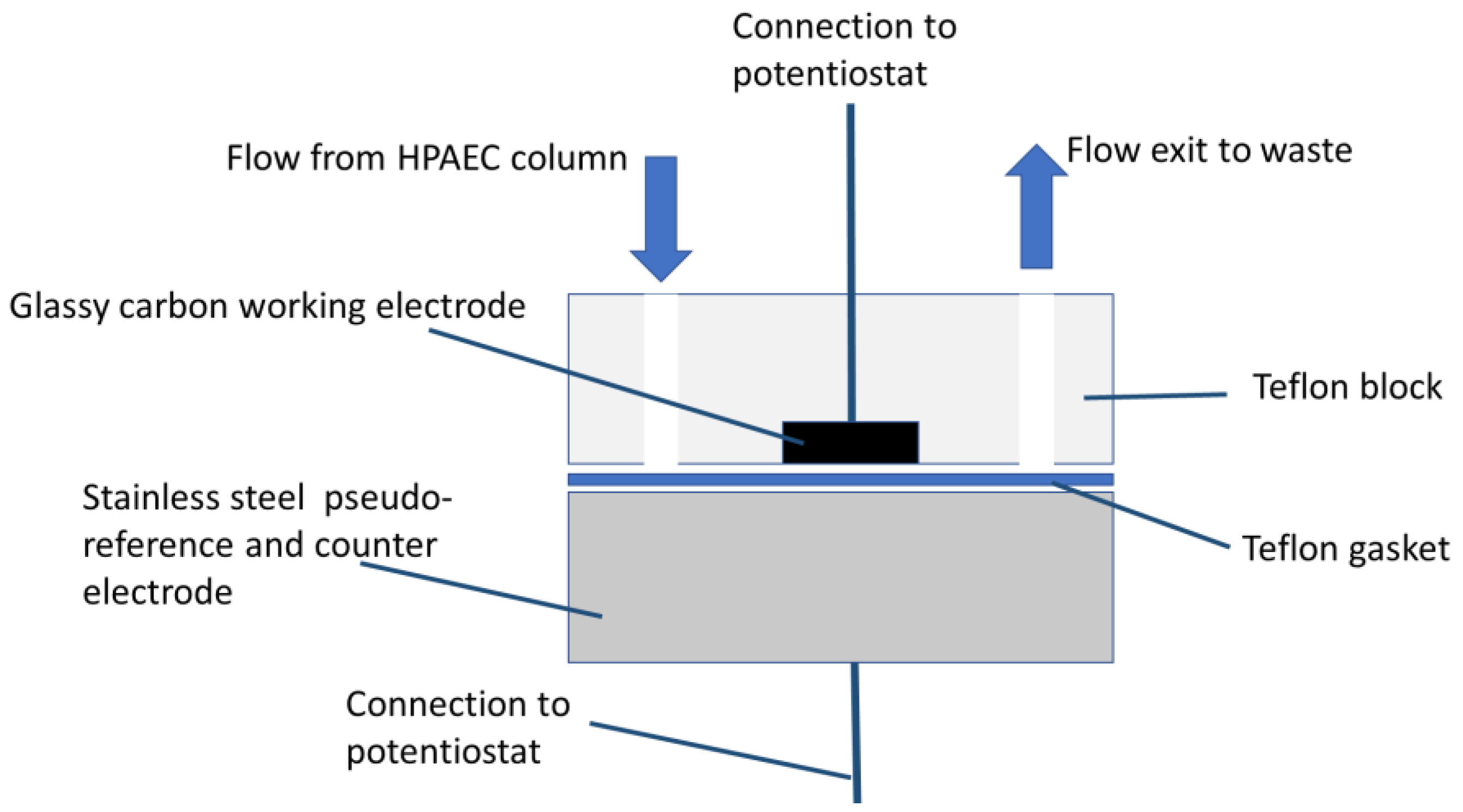
2.2. Apparatus
2.3. Cyclic Voltammetric Procedures
2.4. High-Performance Anion Exchange Liquid Chromatography
2.5. Electrochemical Detection
2.6. Hydrodynamic Voltammetry (HDV)
2.7. Dust Wipe Sampling and Extraction
2.8. Investigation of Precision and Accuracy
3. Results and Discussion
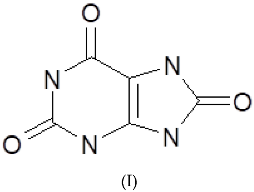
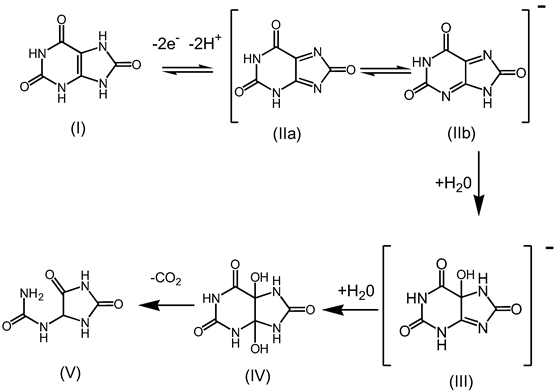
3.1. Cyclic Voltammetric Investigations of Uric Acid
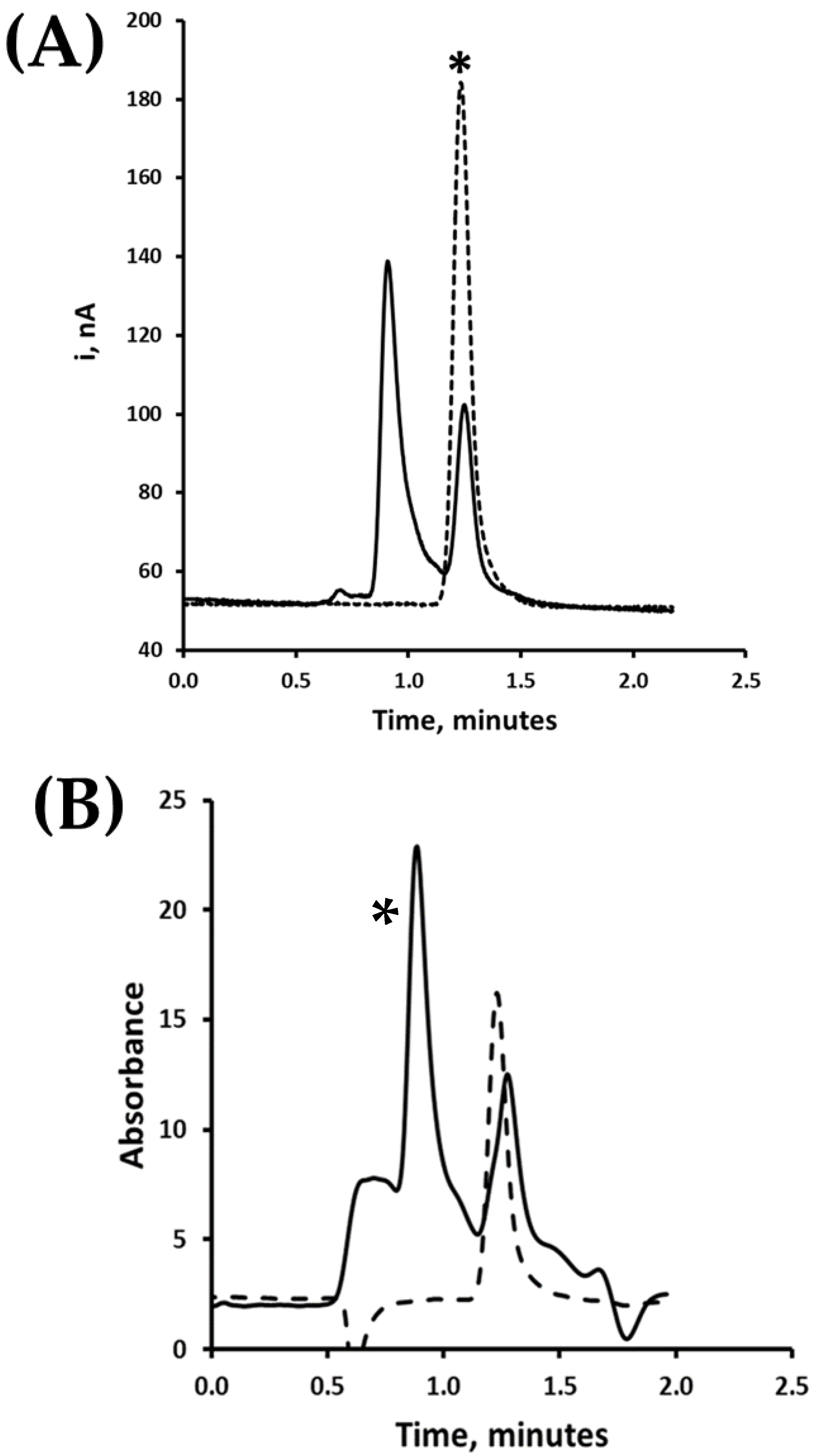
3.2. High-Performance Anion Exchange Liquid Chromatographic Separation of Uric Acid
3.3. Hydrodynamic Voltammetry
3.4. Calibration Curve, Limit of Detection, and Precision
3.5. Studies of Possible Interferences
4. Analytical Application
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CV | Cyclic voltammetry |
| GCE | Glassy carbon electrode |
| HDV | Hydrodynamic voltammetry |
| HPAEC | High-performance anion exchange chromatography |
| ip | Peak current |
| SS | Stianless steel |
| v | Scan rate |
References
- Zhao, F.Y.; Wang, Z.H.; Wang, H.; Zhao, R.; Ding, M.Y. Determination of uric acid in human urine by ion chromatography with conductivity detector. Chin. Chem. Lett. 2011, 22, 342–345. [Google Scholar]
- Kumar, A.N.; Aruna, P.; Naidu, J.N.; Kumar, R.; Srivastava, A.K. Review of concepts and controversies of uric acid as antioxidant and pro-oxidant—An uncertainty. Arch. Med. Rev. J. 2015, 24, 19–40. [Google Scholar]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [PubMed]
- Gérard, B.; Leask, M.; Merriman, T.R.; Bardin, T.; Oehler, E.; Lawrence, A.; Viali, S.; ‘Ofanoa, S.; Te Karu, L.; Stamp, L.K.; et al. Hyperuricaemia and gout in the Pacific. Nat. Rev. Rheumatol. 2025, 21, 197–210. [Google Scholar] [CrossRef]
- Du, L.; Zong, Y.; Li, H.; Wang, Q.; Xie, L.; Yang, B.; Pang, Y.; Zhang, C.; Zhong, Z.; Gao, J. Hyperuricemia and its related diseases: Mechanisms and advances in therapy. Sig. Transduct. Target Ther. 2024, 9, 212. [Google Scholar]
- Urzyk, M.; Pulina, M.; Ajewicz, M.; Zjawiony, I. Uric acid in the surface waters of Spitsbergen and its role in the dissolution processes which affect carbonate rocks. Pol. Polar Res. 2006, 27, 327–340. [Google Scholar]
- Lindeboom, H.J. The Nitrogen Pathway in a Penguin Rookery. Ecology 1984, 65, 269–277. [Google Scholar]
- Brown, L.; Braven, J.; Rhead, M.M.; Evens, R.; Butler, E.I. The use of uric acid as a method of tracing domestic sewage discharges in riverine, estuarine and coastal water environments. Water Res. 1982, 16, 1409–1411. [Google Scholar]
- Braverman, M.M.; Theophil, C.; Masciello, F.; Smith, C. The Contribution to Air Pollution by Pigeons. J. Air Pollut. Control Assoc. 1962, 12, 570–571. [Google Scholar]
- Carlson, M.; Thompson, R.D. Identification of Allantoin, Uric Acid, and Indoxyl Sulfate as Biochemical Indicators of Filth in Food Packaging by LC. J. AOAC Int. 2001, 84, 782–788. [Google Scholar]
- Ghaedian, A.R.; Wehling, R.L. Stability of Uric Acid Used as an Indicator of Insect Contamination During Extrusion of Wheat Flour. Cereal Chem. 1996, 73, 625–627. [Google Scholar]
- Honeychurch, K.C. The determination of uric acid in human saliva by liquid chromatography with electrochemical detection. J. Anal. Bioanal. Sep. Tech. 2017, 2, 1–5. [Google Scholar]
- Dantzler, W.H.; Braun, E.J. Comparative nephron function in reptiles, birds, and mammals. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1980, 239, R197–R213. [Google Scholar]
- Mizutani, H.; Wada, E. High-performance liquid chromatographic determination of uric acid in soil. J. Chromatogr. A 1985, 331, 359–369. [Google Scholar]
- Crouch, N.M.A.; Lynch, V.M.; Clarke, J.A. A re-evaluation of the chemical composition of avian urinary excreta. J. Ornithol. 2020, 161, 17–24. [Google Scholar]
- Otero, X.L.; De La Peña-Lastra, S.; Pérez-Alberti, A.; Ferreira, T.O.; Huerta-Diaz, M.A. Seabird colonies as important global drivers in the nitrogen and phosphorus cycles. Nat. Commun. 2018, 9, 246. [Google Scholar] [PubMed]
- Vasiliu, A.; Buruiana, D. Are Birds a Menace to outdoor Monuments? Int. J. Conserv. Sci. 2010, 1, 83–92. [Google Scholar]
- Tizard, I. Salmonellosis in wild birds. Semin. Avian Exot. Pet Med. 2004, 13, 50–66. [Google Scholar]
- Hogerwerf, L.; Roof, I.; de Jong, M.J.; Dijkstra, F.; van Der Hoek, W. Animal sources for zoonotic transmission of psittacosis: A systematic review. BMC Infect. Dis. 2020, 20, 192. [Google Scholar]
- Tsutsui, T.; Miyazaki, Y.; Kuramochi, J.; Uchida, K.; Eishi, Y.; Inase, N. The Amount of Avian Antigen in Household Dust Predicts the Prognosis of Chronic Bird-related Hypersensitivity Pneumonitis. Ann. Am. Thorac. Soc. 2015, 12, 1013–1021. [Google Scholar]
- Sedlmaier, N.; Hoppenheidt, K.; Krist, H.; Lehmann, S.; Lang, H.; Büttner, M. Generation of avian influenza virus (AIV) contaminated fecal fine particulate matter (PM): Genome and infectivity detection and calculation of immission. Vet. Microbiol. 2009, 139, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Feddes, J.J.R.; Cook, H.; Zuidhof, M.J. Characterization of airborne dust particles in turkey housing. Can. Agric. Eng. 1992, 34, 273–280. [Google Scholar]
- Centers for Disease Control and Prevention. Reported Human Infections with Avian Influenza A Viruses. Available online: https://www.cdc.gov/bird-flu/php/avian-flu-summary/reported-human-infections.html (accessed on 11 March 2025).
- Cohen, J. Worries about Bird Flu in US Cattle Intensify. Science 2024, 384, 12–13. [Google Scholar] [CrossRef] [PubMed]
- Bindu, P.V.; Manjappa, S.; Kumara Swamy, B.E.; Manjunatha, L.S. Electrochemical Sensing of Uric Acid at Cerium Oxide Poly (Congo Red) Modified Carbon Paste Electrode. Sens. Technol. 2024, 2, 2435833. [Google Scholar]
- Penagos-Llanos, J.; Segura, R.; de la Vega, A.P.; Pichun, B.; Liendo, F.; Riesco, F.; Nagles, E. Electrochemical Determination of Uric Acid Using a Nanocomposite Electrode with Molybdenum Disulfide/Multiwalled Carbon Nanotubes (MoS2@MWCNT). Nanomaterials 2024, 14, 958. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Manh, T.D.; Man, N.Q.; Thoa, P.T.H.; Khieu, D.Q. Electrochemical Detection of Uric Acid and Xanthine in Human Urine Using the Co/UiO-66 Modified Glassy Carbon Electrode. J. Appl. Electrochem. 2024, 54, 2361–2376. [Google Scholar]
- Khan, N.; Ahmad, M.; Sun, W.; Shah, N.S.; Asad, M.; Shah, M.; Ullah, R.; Ibrahim, M.A.; Badshah, A.; Nishan, U. Optical Detection of Uric Acid Based on a Citric Acid Functionalized Copper-Doped Biochar Nanozyme. RSC Adv. 2024, 14, 33007–33018. [Google Scholar] [CrossRef]
- Ke, W.; Kuang, G.; Gu, C.; Wang, J.; Jiang, X.; Zhu, R.; Wang, H.; Zhang, Z. Enzyme cascade nanozyme based colorimetric sensor for detection of uric acid as a biomarker of hyperuricemia. Microchim. Acta 2025, 192, 85. [Google Scholar]
- Moriiwa, Y.; Hatakeyama, K.; Morioka, K.; Inoue, Y.; Murakami, H.; Teshima, N.; Yanagida, A.; Shoji, A. Colorimetric and fluorometric determination of uric acid by a suspension-based assay using enzyme-immobilized micro-sized particles. Anal. Sci. 2024, 40, 951–958. [Google Scholar] [CrossRef]
- Drees, L.R.; Manu, A. Bird urate contamination of atmospheric dust traps. Catena 1996, 27, 287–294. [Google Scholar] [CrossRef]
- Yang, Y.D. Simultaneous determination of creatine, uric acid, creatinine and hippuric acid in urine by high performance liquid chromatography. Biomed. Chrom. 1998, 12, 47–49. [Google Scholar]
- Luo, X.; Cai, N.; Cheng, Z. Determination of uric acid in plasma by LC-MS/MS and its application to an efficacy evaluation of recombinant urate oxidase. Anal. Sci. 2013, 29, 709–713. [Google Scholar] [PubMed]
- Kim, K.M.; Henderson, G.N.; Ouyang, X.; Frye, R.F.; Sautin, Y.Y.; Feig, D.I.; Johnson, R.J. A Sensitive and Specific Liquid Chromatography-Tandem Mass Spectrometry Method for the Determination of Intracellular and Extracellular Uric Acid. J. Chromatogr. B 2009, 877, 2032–2038. [Google Scholar]
- Aoki, T.; Yoshiura, M.; Iwamoto, T.; Iriyama, K. Postmortem changes of uric acid in various rat tissues: Determination of uric acid by reversed-phase high-performance liquid chromatography with electrochemical detection. Anal. Biochem. 1984, 143, 113–118. [Google Scholar]
- Howorth, P.J.N.; Zilva, J.F. Determination of uric acid levels in uraemia by enzymatic and colorimetric techniques. J. Clin. Path. 1968, 21, 192–195. [Google Scholar]
- Barham, D.; Trinder, P. Enzymatic determination of Uric acid. Analyst 1972, 97, 142–145. [Google Scholar] [PubMed]
- Cunningham, S.K.; Keaveny, T.V. A two-stage enzymatic method for determination of uric acid and hypoxanthine/xanthine. Clin. Chim. Acta 1978, 86, 217–221. [Google Scholar]
- Galbán, J.; Andreu, Y.; Almenara, M.J.; de Marcos, S.; Castillo, J.R. Direct determination of uric acid in serum by a fluorometric-enzymatic method based on uricase. Talanta 2001, 54, 847–854. [Google Scholar]
- Taei, M.; Jamshidi, M. Highly selective determination of ascorbic acid, epinephrine, and uric acid by differential pulse voltammetry using poly(Adizol Black B)-modified glassy carbon electrode. J. Solid State Electrochem. 2014, 18, 673–683. [Google Scholar]
- Bravo, R.; Brajter-Toth, A. A highly active carbon microdisk electrode surface for the determination of uric acid in physiological buffers. Chem. Anal. 1999, 44, 423–436. [Google Scholar]
- Zhang, Y.; Lei, W.; Xu, Y.; Xia, X.; Hao, Q. Simultaneous Detection of Dopamine and Uric Acid Using a Poly(l-lysine)/Graphene Oxide Modified Electrode. Nanomaterials 2016, 6, 178. [Google Scholar] [CrossRef]
- Gilmartin, M.A.T.; Hart, J.P.; Birch, B. Voltammetric and amperometric behaviour of uric acid at bare and surface-modified screen-printed electrodes: Studies towards a disposable uric acid sensor. Analyst 1992, 117, 1299–1303. [Google Scholar]
- Peng, X.; Giltrow, D.; Bowdler, P.; Honeychurch, K.C. Liquid chromatography electrochemical determination of nicotine in third-hand smoke. Electroanalysis 2017, 29, 374–379. [Google Scholar]
- Struck, W.A.; Elving, P.J. Electrolytic Oxidation of Uric Acid: Products and Mechanism. Biochemistry 1965, 4, 1343–1353. [Google Scholar]
- Wang, Y. The electrochemistry of uric acid at a gold electrode modified with L-cysteine, and its application to sensing uric urine. Microchim. Acta 2011, 172, 419–424. [Google Scholar]
- Goyal, R.N.; Mittal, A.; Agarwal, D. Electrochemical Oxidation and Kinetics of the Decay of UV-Absorbing Intermediate of Uric-Acid Oxidation at Pyrolytic-Graphite Electrodes. Can. J. Chem. 1994, 72, 1668–1674. [Google Scholar]
- Dryhurst, G.; Nguyen, N.T.; Wrona, M.Z.; Goyal, R.N.; Brajter-Toth, A.; Owens, J.L.; Marsh, H.A. Elucidation of the Biological Redox Chemistry of Purines Using Electrochemical Techniques. J. Chem. Ed. 1983, 60, 315–319. [Google Scholar]
- Mautjana, N.A.; Estes, J.; Eyler, J.R.; Brajter-Toth, A. One-Electron Oxidation and Sensitivity of Uric Acid in On-Line Electrochemistry and in Electrospray Ionization Mass Spectrometry. Electroanalysis 2008, 20, 2501–2508. [Google Scholar]
- Lakshmi, D.; Whitcombe, M.J.; Davis, F.; Sharma, P.S.; Prasad, B.B. Electrochemical Detection of Uric Acid in Mixed and Clinical Samples: A Review. Electroanalysis 2011, 23, 305–320. [Google Scholar]
- Goyal, R.N.; Brajter-Toth, A.; Dryhurst, G. Further Insights into the Electrochemical Oxidation of Uric Acid. J. Electroanal. Chem. Interfacial Electrochem. 1982, 131, 181–202. [Google Scholar]
- Van der Horst, C.; de Souza Gil, E.; Somerset, V. Electrochemical Sensing of Uric Acid Using Bismuth-Silver Bimetallic Nanoparticles Modified Sensor. Eclét. Quím. 2024, 49, e-1497. [Google Scholar] [CrossRef]
- Li, Y.; Zhai, X.; Liu, X.; Ji, X.; Wang, L. Voltammetric determination of uric acid in the presence of ascorbic acid and dopamine using chitosan/ionic liquid composite electrode. Ionics 2014, 20, 1247–1254. [Google Scholar]
- Rastogi, P.K.; Gupta, R.; Prakash, S.; Ganesan, V. Non-enzymatic Determination of Uric Acid by Ion Exchange Voltammetry at a Permselective Electrochemical Sensing Platform. Anal. Chem. Lett. 2015, 5, 127–134. [Google Scholar]
- Wopschall, R.H.; Sharin, I. Adsorption characteristics of the methylene blue system using stationary electrode polarography. Anal. Chem. 1967, 39, 1527–1534. [Google Scholar]
- Nicholson, R.S.; Shain, I. Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem. 1964, 36, 706–723. [Google Scholar]
- McCrudden, F.H. Uric Acid: The Chemistry, Physiology and Pathology of Uric Acid and the Physiologically Important Purin Bodies, with a Discussion of the Metabolism in Gout; BiblioBazaar: Charleston, SC, USA, 2008; ISBN 978-0-554-61991-0. [Google Scholar]
- Buckberry, J.; Telford, R.; Castells Navarro, L.; Snaith, J.; Swinson, D.; Healey, A.; Brickley, M.B. High-performance liquid chromatography (HPLC) as a means of assessing the presence of uric acid in archeological human remains: Challenges and future directions. Am. J. Biol. Anthropol. 2024, 184, e24938. [Google Scholar]
- Lin, X.; Wang, Y.; Sun, J.; Xiao, D.; Luo, W.; Li, H. Determination of uric acid in human plasma and urine by microemulsion electrokinetic chromatography. Anal. Methods 2013, 5, 5201–5207. [Google Scholar]
- Aafria, S.; Kumari, P.; Sharma, S.; Yadav, S.; Batra, B.; Rana, J.S.; Sharma, M. Electrochemical biosensing of uric acid: A review. Microchem. J. 2022, 182, 107945. [Google Scholar]
- Ruiz-Guerrero, C.D.; Estrada-Osorio, D.V.; Gutiérrez, A.; Espinosa-Lagunes, F.I.; Escalona-Villalpando, R.A.; Luna-Bárcenas, G.; Molina, A.; Arenillas, A.; Arriaga, L.G.; Ledesma-García, J. Novel Cobalt-Based Aerogels for Uric Acid Detection in Fluids at Physiological pH. Biosens. Bioelectron. 2025, 267, 116850. [Google Scholar]
- Ardakani, M.M.; Sheikhmohseni, M.A.; Beitollahi, H.; Benvidi, A.; Naeimi, H. Simultaneous determination of dopamine, uric acid, and folic acid by a modified TiO2 nanoparticles carbon paste electrode. Turk. J. Chem. 2011, 35, 6. [Google Scholar]
- Tawade, A.K.; Kamble, B.B.; Sharma, K.K.K.; Tayade, S.N. Simultaneous electrochemical investigations of dopamine and uric acid by in situ amino functionalized reduced graphene oxide. SN Appl. Sci. 2020, 2, 1082. [Google Scholar] [CrossRef]
- Hsu, D.-S.; Chen, S.S. Determination of uric acid by an anion-exchange column chromatography. Anal. Biochem. 1987, 163, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Young, I.S.; Woodside, J.V. Antioxidants in health and disease. J. Clin. Pathol. 2001, 54, 176–186. [Google Scholar] [CrossRef]
- Ford, E.; Hughes, M.N.; Wardman, P. Kinetics of the Reactions of Nitrogen Dioxide with Glutathione, Cysteine, and Uric Acid at Physiological pH. Free Radic. Biol. Med. 2002, 32, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Simic, M.G.; Jovanovic, S.V. Antioxidation Mechanisms of Uric Acid. J. Am. Chem. Soc. 1989, 111, 5778–5782. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Linear Range, μg/mL | Limit of Detection, μg/mL | Selectivity | Ref. |
|---|---|---|---|---|
| HPAEC | 0.1–100 | 0.866 | Nitrite, nitrate, acetate, chloride, and sulphate were found not to interfere | Present study |
| HILC with UV and MS detection | 0.0011–21 | 5.0 (UV detection) and 0.05 (MS) | Longer retention time (>15 min) needed for uric acid response to be free from interference from sample components. | [58] |
| Microemulsion electrokinetic chromatography | 5–200 | 0.5 | Requires sample extraction. | [59] |
| Cobalt-based aerogel modified electrodes | Up to 42.0 | 9.8 | Glucose, urea, lactic acid, and ascorbic acid cause changes in amperometric signal. | [61] |
| TiO2 nanoparticle-modified carbon paste electrode | 2.0–151 | 2 | Able to determine uric acid, dopamine, and folic acid simultaneously. | [62] |
| Glassy carbon amino functionalized reduced graphene oxide-modified electrode | 34.3–342 | 28.1 | Notably higher detection limit. | [63] |
| Sample | Comments | µg/m2 |
|---|---|---|
| 1 | Inside of window of office | nd |
| 2 | Inside of window of door to garden | nd |
| 3 | Outside of door windowpane | nd |
| 4 | Outside of window of entrance security lodge to carpark | 42.8 |
| 5 | Metro Bus touchscreen (outside sample) | nd |
| 6 | Outside door windowpane | 87.8 |
| 7 | Outside windowpane (opposite feral pigeon roost) | 3980 |
| 8 | Outside door windowpane | 32.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honeychurch, K.C. High-Performance Anion Exchange Chromatography Electrochemical Determination of Uric Acid as a Contamination Marker. Sci 2025, 7, 40. https://doi.org/10.3390/sci7020040
Honeychurch KC. High-Performance Anion Exchange Chromatography Electrochemical Determination of Uric Acid as a Contamination Marker. Sci. 2025; 7(2):40. https://doi.org/10.3390/sci7020040
Chicago/Turabian StyleHoneychurch, Kevin C. 2025. "High-Performance Anion Exchange Chromatography Electrochemical Determination of Uric Acid as a Contamination Marker" Sci 7, no. 2: 40. https://doi.org/10.3390/sci7020040
APA StyleHoneychurch, K. C. (2025). High-Performance Anion Exchange Chromatography Electrochemical Determination of Uric Acid as a Contamination Marker. Sci, 7(2), 40. https://doi.org/10.3390/sci7020040