





Screening and Analysis of Serum Protein Biomarkers Infected by Coronavirus Disease 2019 (COVID-19)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Serum Samples
2.2. Trypsin Digestion
2.3. HPLC Analysis
2.4. LC-MS-MS Analysis
2.5. Database Search
2.6. Gene Ontology Analysis
2.7. Pathway Analysis
2.8. Protein Domain Analysis
2.9. Subcellular Localization
2.10. Enrichment-Based Clustering
2.11. Protein–Protein Interaction Network
3. Results
3.1. Sample and Protein Identification
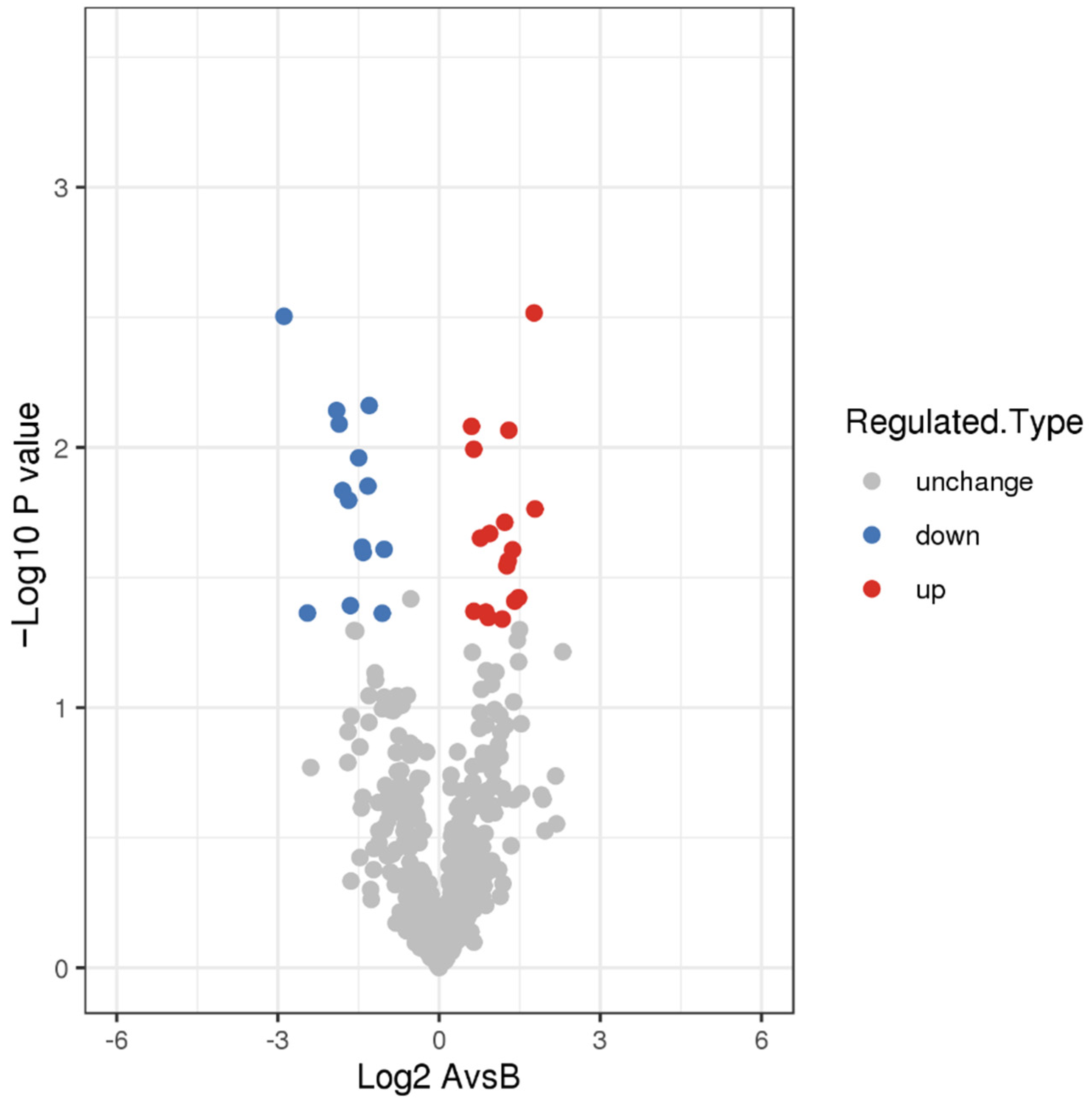
3.2. DEPs and Functional Analysis
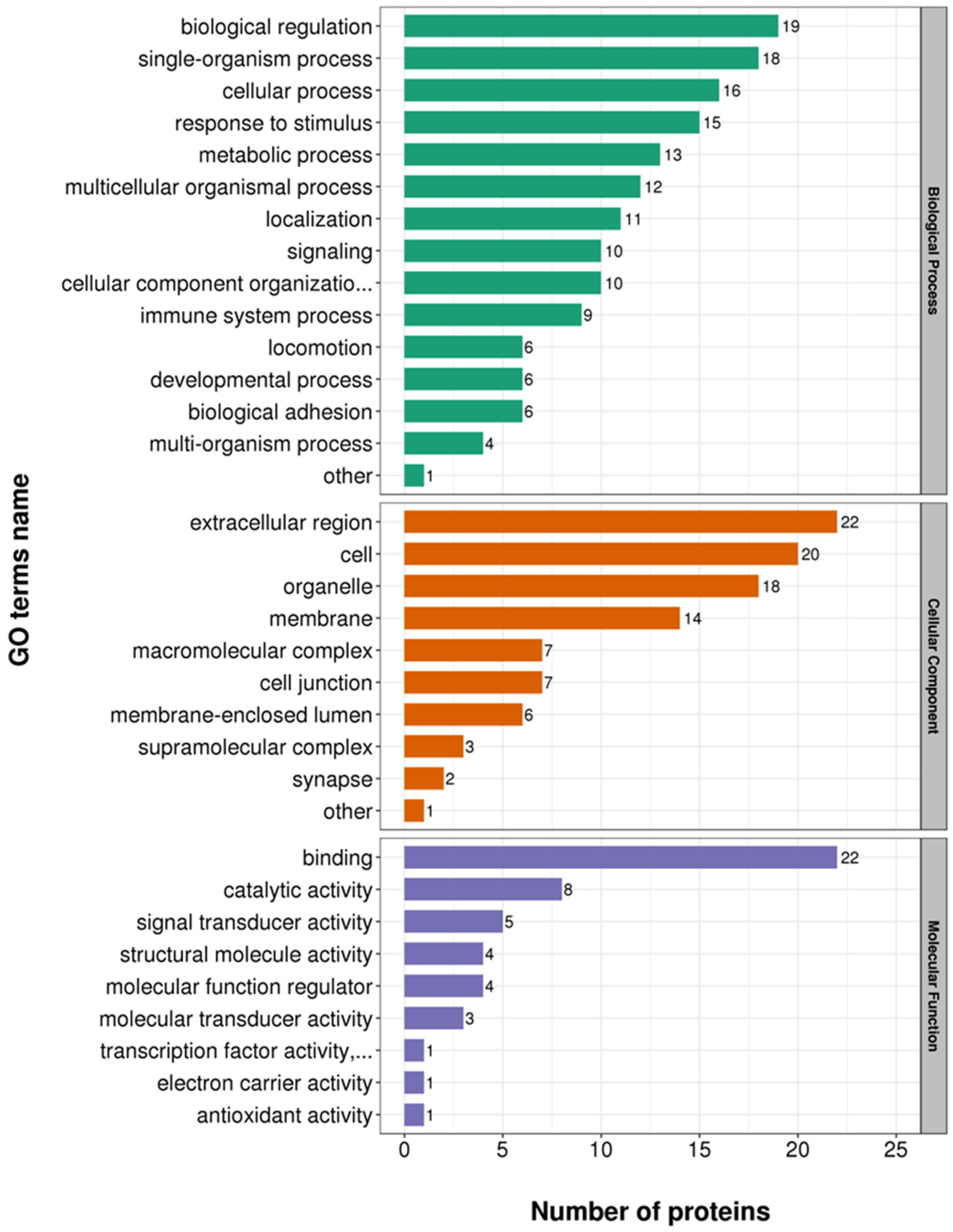
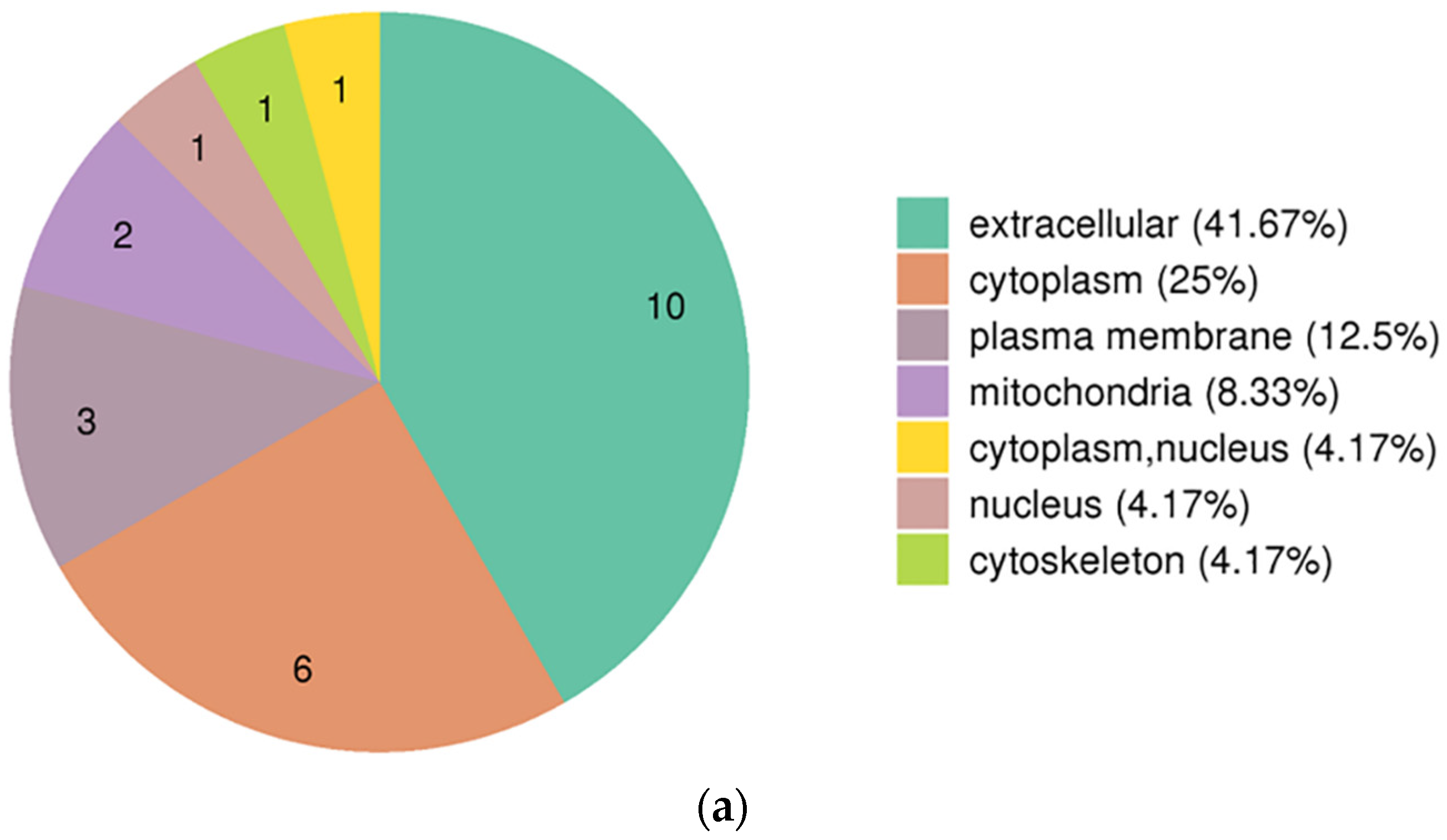
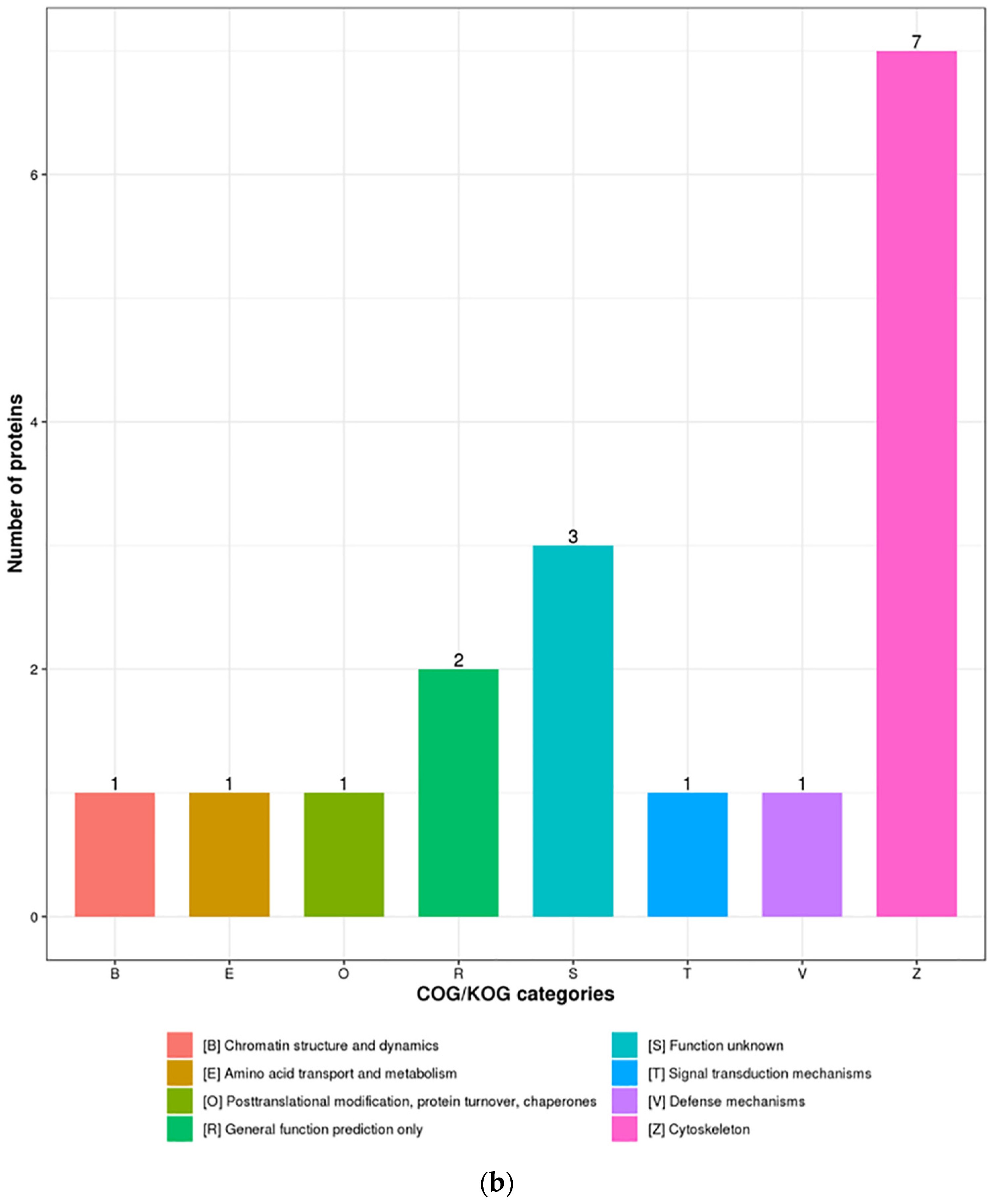
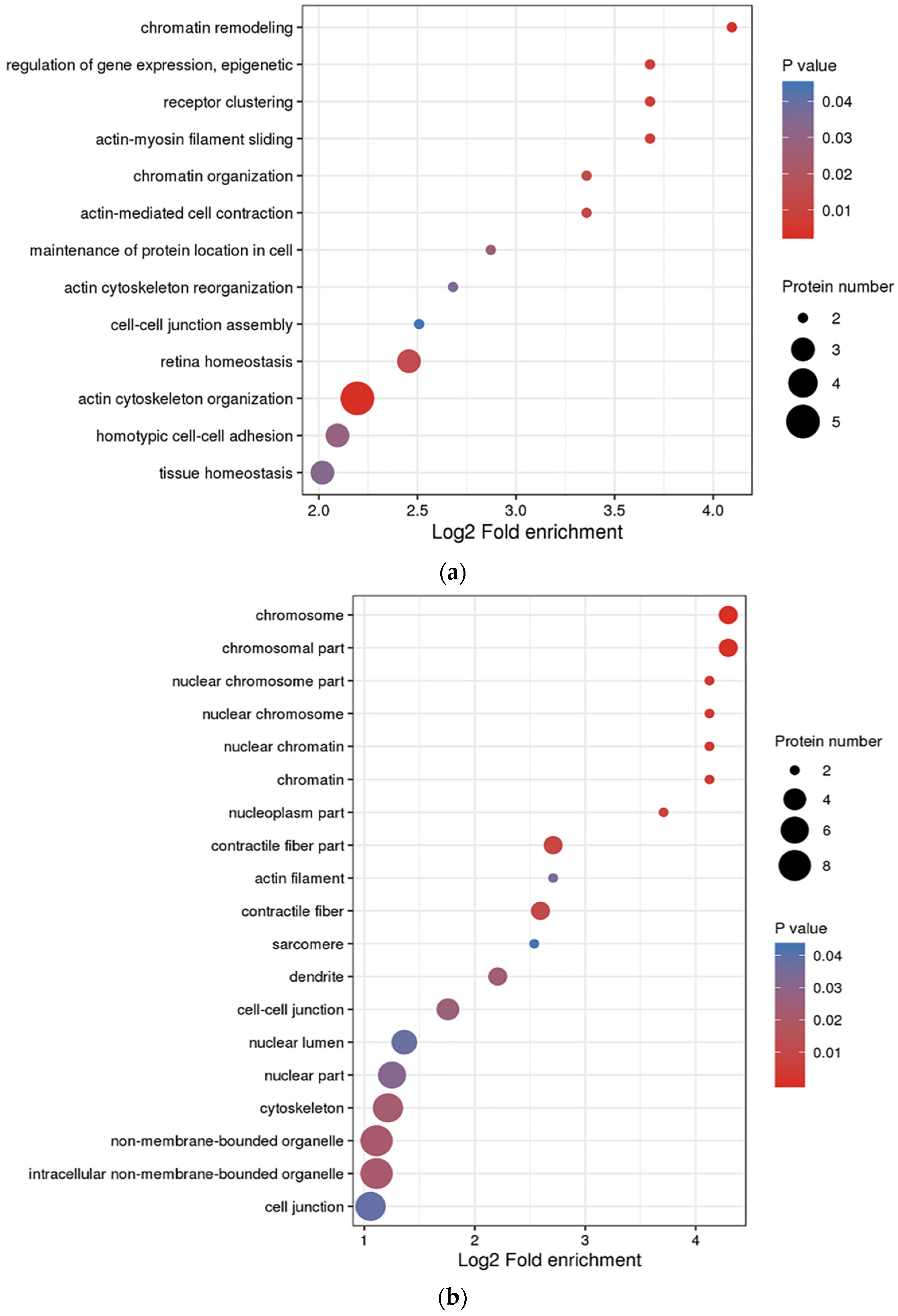
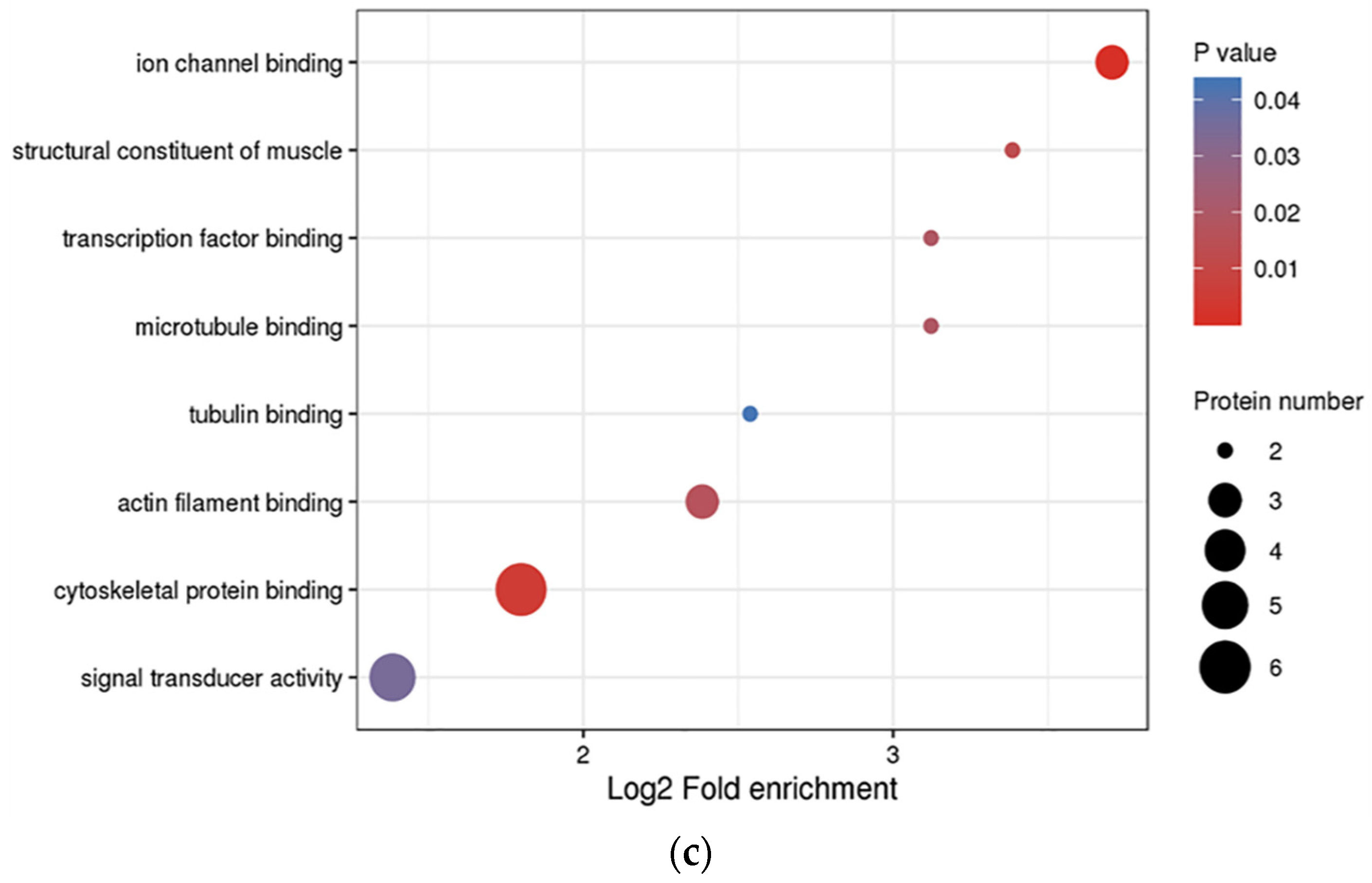
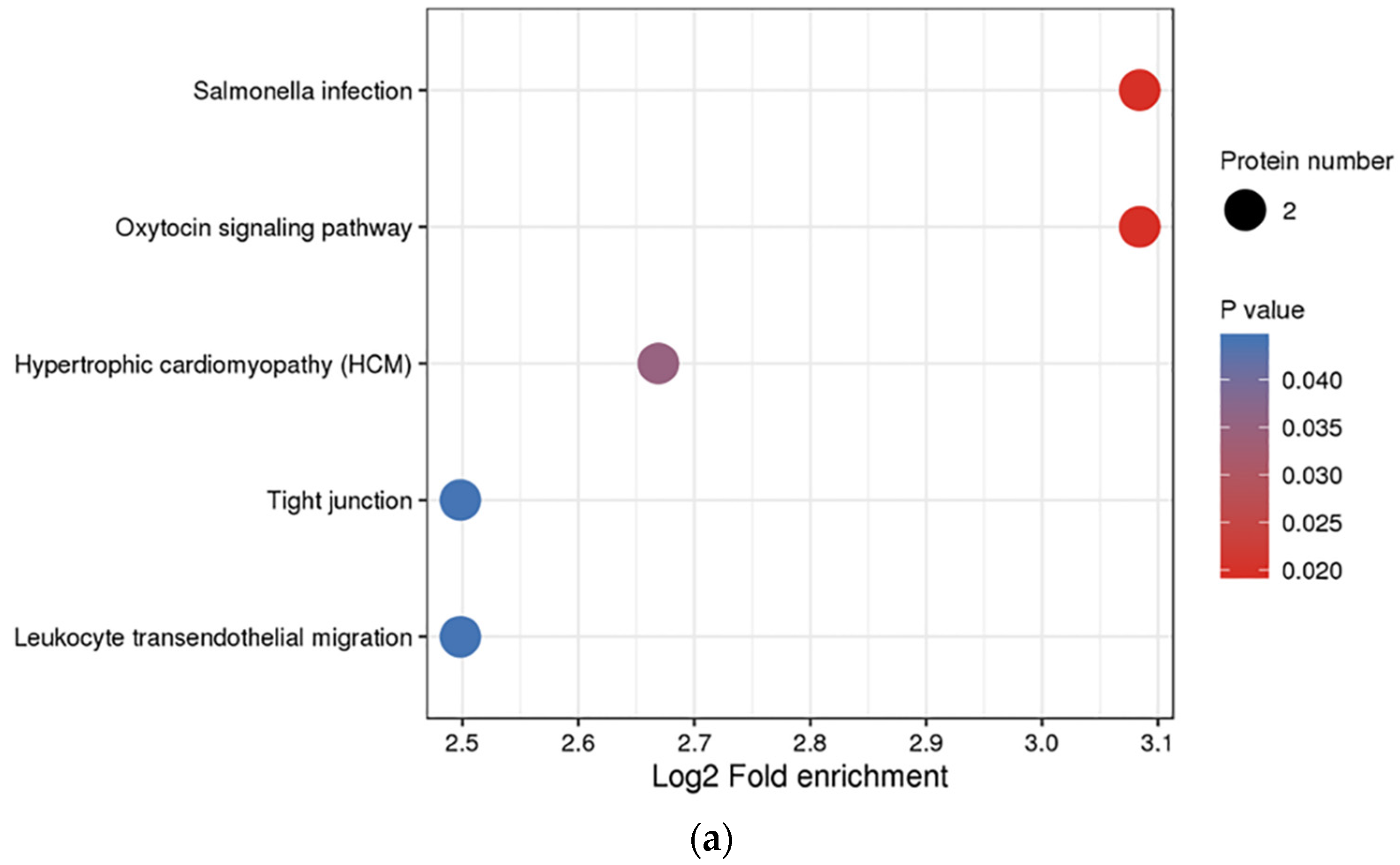
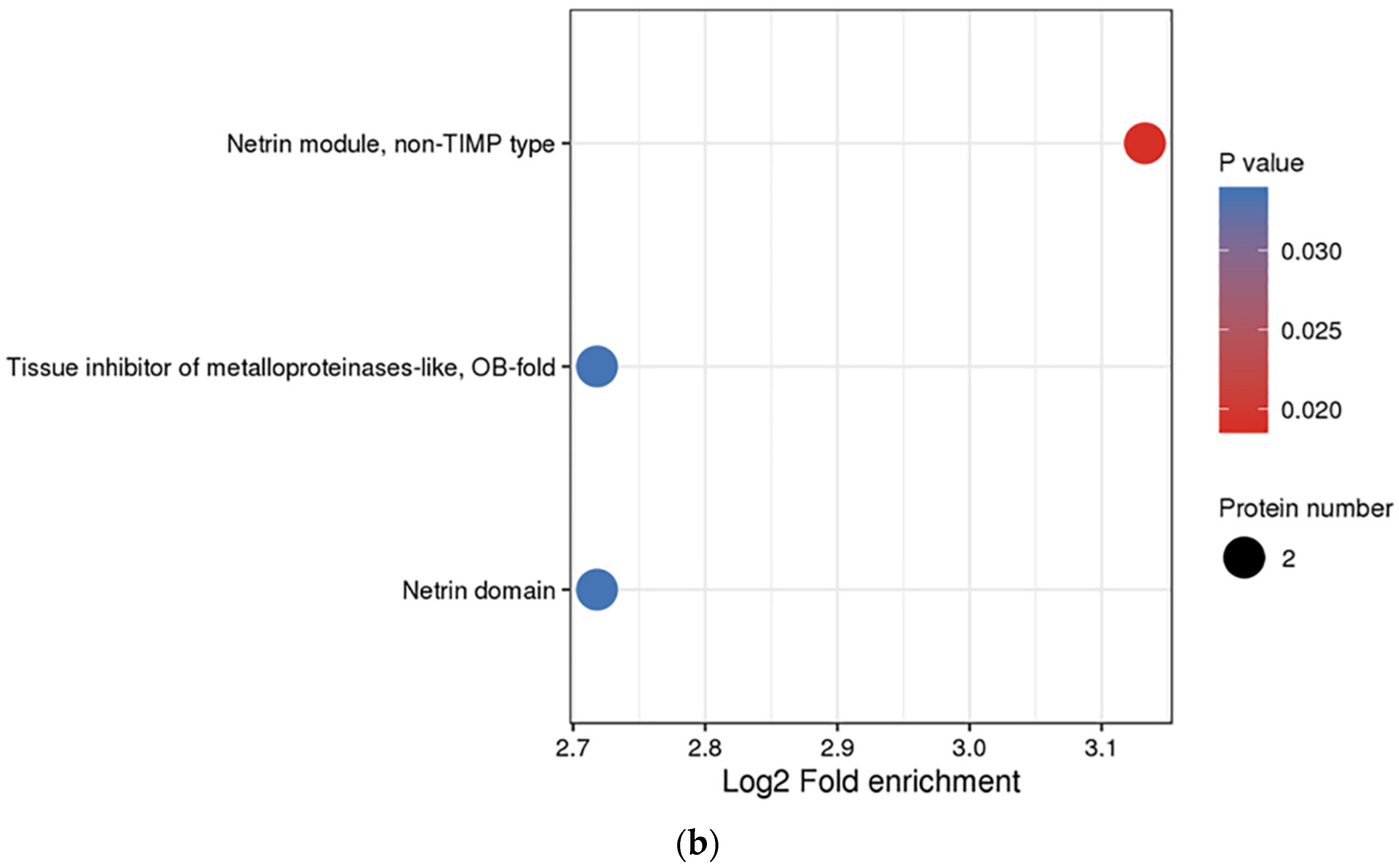
3.3. Functional Enrichment Analysis of DEPs
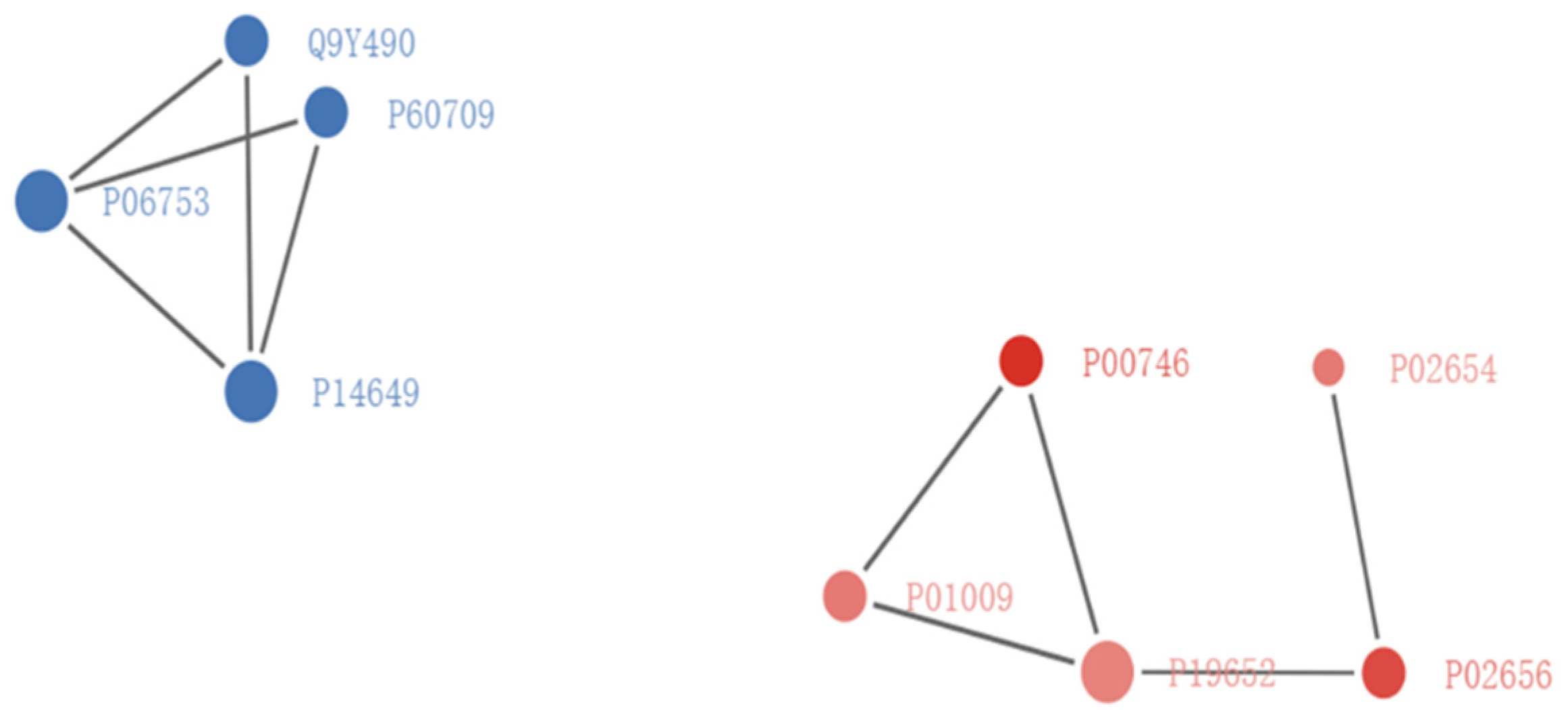
3.4. Protein–Protein Interaction Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Clinical Management of Severe Acute Respiratory Infection When Novel Coronavirus (nCoV) Infection Is Suspected: Interim Guidance; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Ghelichi-Ghojogh, M.; Kalteh, E.A.; Fararooei, M. Coronavirus disease 2019; epidemiology and recommendations. J. Prev. Epidemiol. 2020, 5, e01. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.F.; Chien, C.S.; Yarmishyn, A.A.; Lin, Y.Y.; Chiou, S.H. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [PubMed]
- Jyl, A.; Zhi, Y.A.; Qw, A.; Zjz, A.; Ye, Q.A.; Rui, L.; Xyg, A. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. 2020, 22, 80–85. [Google Scholar]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-CoV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.; Tang, X.; Shi, Q.; Li, Q.; Deng, H.; Yuan, J.; Hu, J.; Xu, W.; Zhang, Y.; Lv, F.; et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020, 26, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, S.; Yu, X.; Tang, S.; Tang, C. Coronavirus disease 2019 (COVID-19): Current status and future perspectives. Int. J. Antimicrob. Agents 2020, 55, 105951. [Google Scholar] [CrossRef] [PubMed]
- Amirian, E.S. Potential Fecal Transmission of SARS-CoV-2: Current Evidence and Implications for Public Health. Int. J. Infect. Dis. 2020, 95, 363–370. [Google Scholar] [CrossRef]
- World Health Organization. Coronavirus Disease (COVID-19) Situation Report; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Geyer, P.E.; Voytik, E.; Treit, P.V.; Doll, S.; Kleinhempel, A. Plasma Proteome Profiling to detect and avoid sample-related biases in biomarker studies. EMBO Mol. Med. 2019, 11, e10427. [Google Scholar] [CrossRef]
- Amiri-Dashatana, N.; Koushkib, M.; Abbaszadehc, H.A. Proteomics Applications in Health: Biomarker and Drug Discovery and Food Industry. Iran. J. Pharm. Sci. 2018, 17, 1523–1536. [Google Scholar] [CrossRef]
- Gutmann, C.; Takov, K.; Burnap, S.A.; Singh, B.; Ali, H.; Theofilatos, K.; Reed, E.; Hasman, M.; Nabeebaccus, A.; Fish, M.; et al. SARS-CoV-2 RNAemia and proteomic trajectories inform prognostication in COVID-19 patients admitted to intensive care. Nat. Commun. 2021, 12, 3406. [Google Scholar] [CrossRef]
- Dong, H.; Li, J.; Lv, Y.; Zhou, Y.; Wang, G.; Hu, S.; He, X.; Yang, P.; Zhou, Z.; Xiang, X.; et al. Comparative analysis of the alveolar macrophage proteome in ALI/ARDS patients between the exudative phase and recovery phase. BMC Immunol. 2013, 14, 25. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Meng, Q.; Wang, L.; Cui, Y. TRIM27 protects against cardiac ischemia-reperfusion injury by suppression of apoptosis and inflammation via negatively regulating p53. Biochem. Biophys. Res. Commun. 2021, 557, 127–134. [Google Scholar] [CrossRef]
- Ercan, H.; Schrottmaier, W.C.; Pirabe, A.; Schmuckenschlager, A.; Pereyra, D.; Santol, J.; Pawelka, E.; Traugott, M.T.; Schörgenhofer, C.; Seitz, T.; et al. Platelet Phenotype Analysis of COVID-19 Patients Reveals Progressive Changes in the Activation of Integrin αIIbβ3, F13A1, the SARS-CoV-2 Target EIF4A1 and Annexin A5. Front. Cardiovasc. Med. 2021, 8, 779073. [Google Scholar] [CrossRef]
- Ghosh, S.; Parikh, S.; Nissa, M.U.; Acharjee, A.; Singh, A.; Patwa, D.; Makwana, P.; Athalye, A.; Barpanda, A.; Laloraya, M.; et al. Semen Proteomics of COVID-19 Convalescent Men Reveals Disruption of Key Biological Pathways Relevant to Male Reproductive Function. ACS Omega 2022, 65, 8601–8612. [Google Scholar] [CrossRef]
- Dong, X.; Penrice-Randal, R.; Goldswain, H.; Prince, T.; Randle, N.; Donovan-Banfield, I.; Salguero, F.J.; Tree, J.; Vamos, E.; Nelson, C.; et al. Analysis of SARS-CoV-2 known and novel subgenomic mRNAs in cell culture, animal model, and clinical samples using LeTRS, a bioinformatic tool to identify unique sequence identifiers. Gigascience 2022, 11, giac045. [Google Scholar] [CrossRef]
- Tong, T.R. Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). In Perspectives in Medical Virology; Tabor, E., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 16, pp. 43–95. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Zhang, X.; Lin, F.; Ge, F. Quantitative Proteomics Analysis Reveals BAG3 as a Potential Target to Suppress Severe Acute Respiratory Syndrome Coronavirus Replication. J. Virol. 2010, 84, 6050–6059. [Google Scholar] [CrossRef]
- Poon, T.C.; Pang, R.T.; Chan, K.C.; Lee, N.L.; Chiu, R.W.; Tong, Y.K.; Chim, S.S.; Ngai, S.M.; Sung, J.J.; Lo, Y.M. Proteomic analysis reveals platelet factor 4 and beta-thromboglobulin as prognostic markers in severe acute respiratory syndrome. Electrophoresis 2012, 33, 1894–1900. [Google Scholar] [CrossRef]
- Luan, J.; Zhu, X.; Yu, L.; Li, Y.; He, X.; Chen, L.; Zhang, Y. Construction of magnetic covalent organic frameworks functionalized by benzoboroxole for efficient enrichment of glycoproteins in the physiological environment. Talanta 2022, 251, 123772. [Google Scholar] [CrossRef]
- Tu, C.; Rudnick, P.A.; Martinez, M.Y.; Cheek, K.L.; Stein, S.E.; Slebos, R.J.C.; Liebler, D.C. Depletion of Abundant Plasma Proteins and Limitations of Plasma Proteomics. J. Proteome Res. 2010, 9, 4982–4991. [Google Scholar] [CrossRef]
- Markowitz, J.; Carson, W.E. Review of S100A9 biology and its role in cancer. Biochim. Biophys. Acta BBA—Rev. Cancer 2013, 1835, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Ehrchen, J.M.; Sunderkötter, C.; Foell, D.; Vogl, T.; Roth, J. The endogenous Toll–like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukocyte Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, R.; Wang, Z.; Jing, Z.; Wang, S.; Ma, J. S100A8/A9 in Inflammation. Front. Immunol. 2018, 9, 1298. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Secretory IgA: Designed for Anti-Microbial Defense. Front. Immunol. 2013, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, J.; Eggeling, C. Super-Resolution STED Microscopy-Based Mobility Studies of the Viral Env Protein at HIV-1 Assembly Sites of Fully Infected T-Cells. Viruses 2021, 13, 608. [Google Scholar] [CrossRef]
- Newton, A.H.; Cardani, A.; Braciale, T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016, 38, 471–482. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef]
- Soto, K.; Wu, Y.L.; Ortiz, A.; Aparício, S.R.; Yu, C.Y. Familial C4B deficiency and immune complex glomerulonephritis. Clin. Immunol. 2010, 137, 166–175. [Google Scholar] [CrossRef]
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in disease: A defence system turning offensive. Nat. Rev. Nephrol. 2016, 12, 383–401. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a16295. [Google Scholar] [CrossRef]
- Narazaki, M.; Kishimoto, T. The Two-Faced Cytokine IL-6 in Host Defense and Diseases. Int. J. Mol. Sci. 2018, 19, 3528. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, Z.; Pan, Y.; Liu, Y.; Zhao, J.; Peng, X.; Lu, G.; Shi, W.; Zhang, D.; Cui, S. Screening and Analysis of Serum Protein Biomarkers Infected by Coronavirus Disease 2019 (COVID-19). Trop. Med. Infect. Dis. 2022, 7, 397. https://doi.org/10.3390/tropicalmed7120397
Feng Z, Pan Y, Liu Y, Zhao J, Peng X, Lu G, Shi W, Zhang D, Cui S. Screening and Analysis of Serum Protein Biomarkers Infected by Coronavirus Disease 2019 (COVID-19). Tropical Medicine and Infectious Disease. 2022; 7(12):397. https://doi.org/10.3390/tropicalmed7120397
Chicago/Turabian StyleFeng, Zhaomin, Yang Pan, Yimeng Liu, Jiachen Zhao, Xiaomin Peng, Guilan Lu, Weixian Shi, Daitao Zhang, and Shujuan Cui. 2022. "Screening and Analysis of Serum Protein Biomarkers Infected by Coronavirus Disease 2019 (COVID-19)" Tropical Medicine and Infectious Disease 7, no. 12: 397. https://doi.org/10.3390/tropicalmed7120397
APA StyleFeng, Z., Pan, Y., Liu, Y., Zhao, J., Peng, X., Lu, G., Shi, W., Zhang, D., & Cui, S. (2022). Screening and Analysis of Serum Protein Biomarkers Infected by Coronavirus Disease 2019 (COVID-19). Tropical Medicine and Infectious Disease, 7(12), 397. https://doi.org/10.3390/tropicalmed7120397