
Characterization of the Tissue and Strain-Specific Microbiota of Anopheles funestus Giles (Diptera: Culicidae)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mosquitoes
2.2. Tissue Dissections and DNA Extraction
2.3. DNA Amplification and Sequencing
2.4. Bioinformatics
3. Results
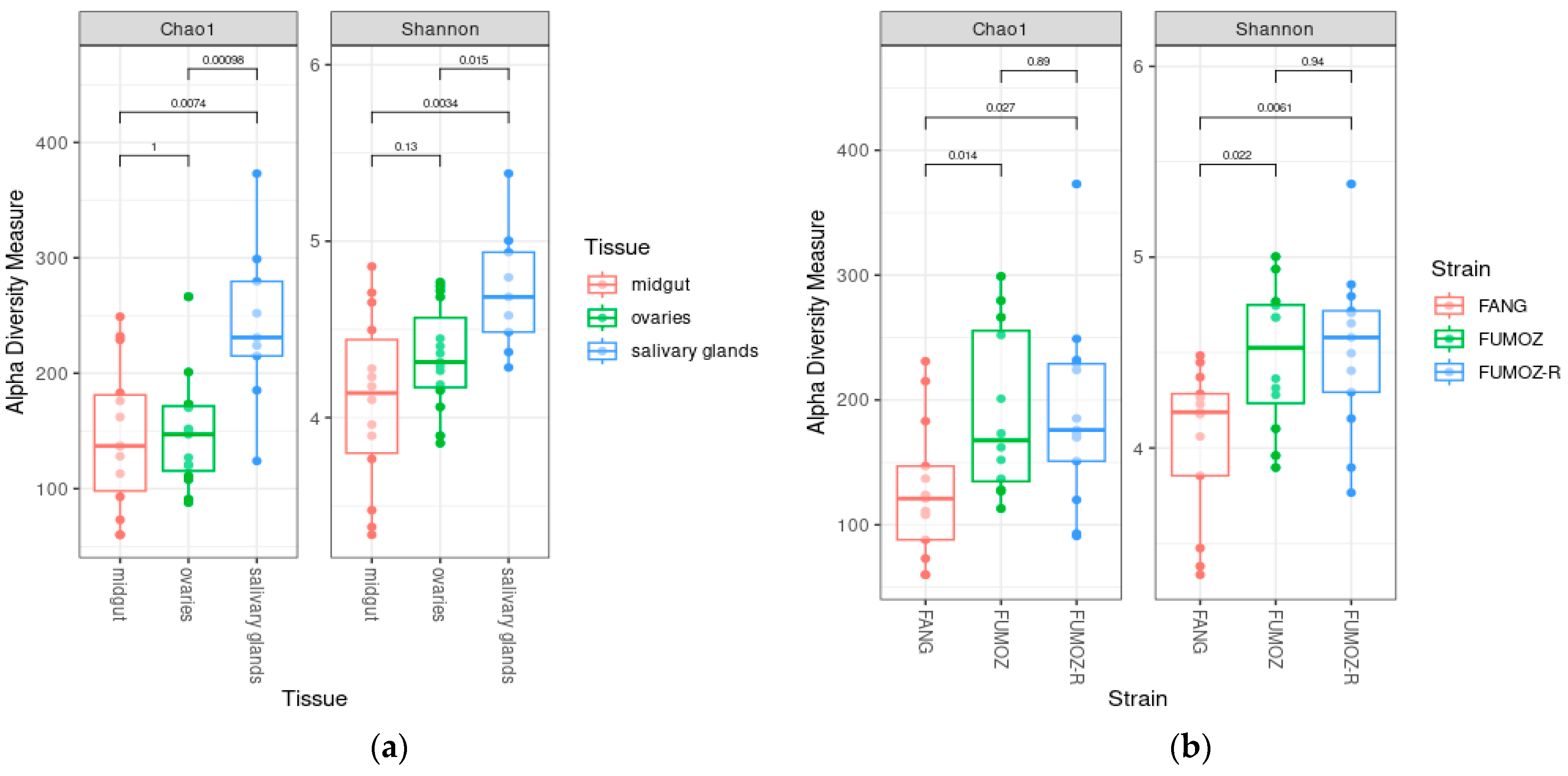
3.1. Alpha Diversity
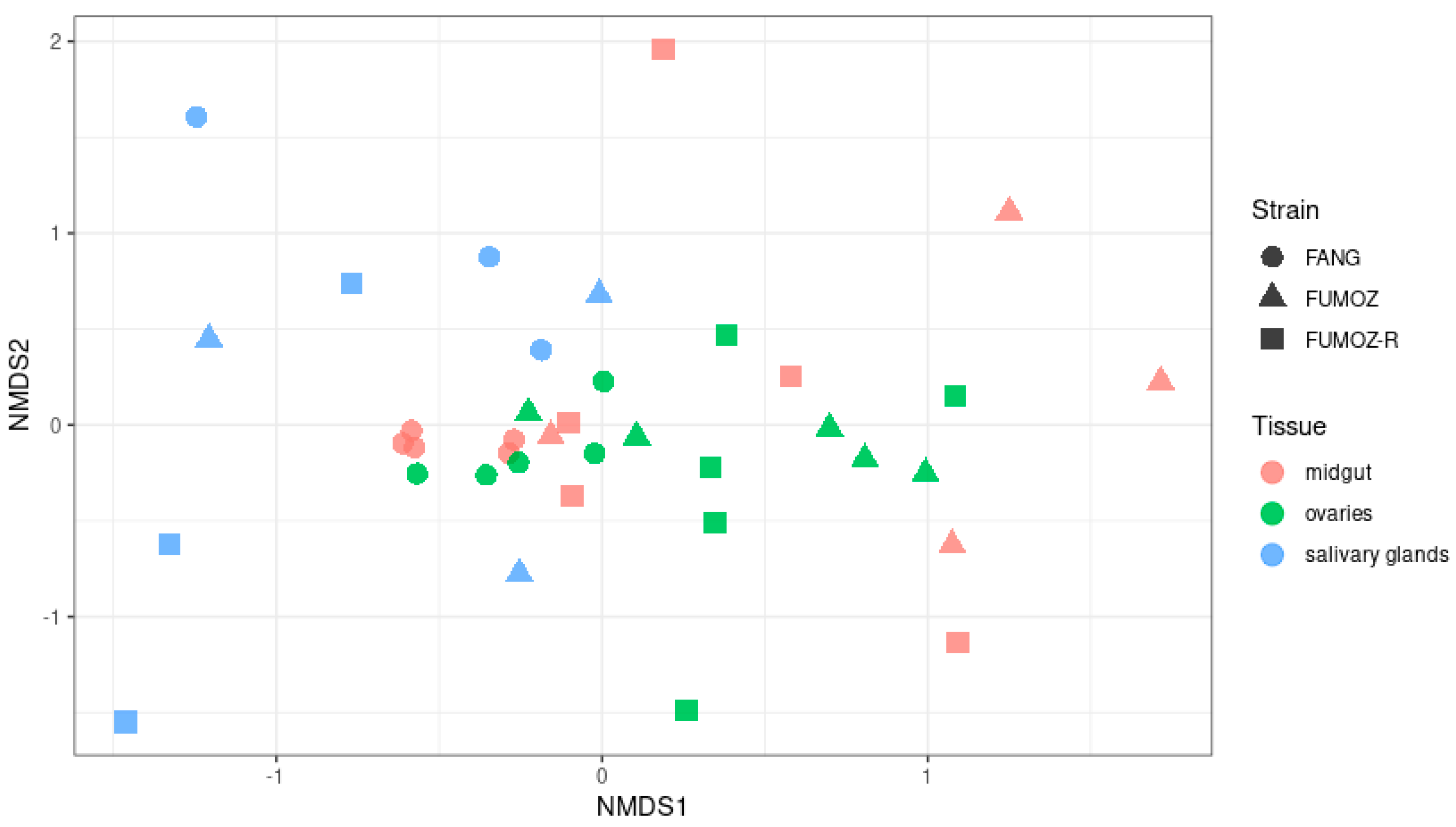
3.2. Beta Diversity
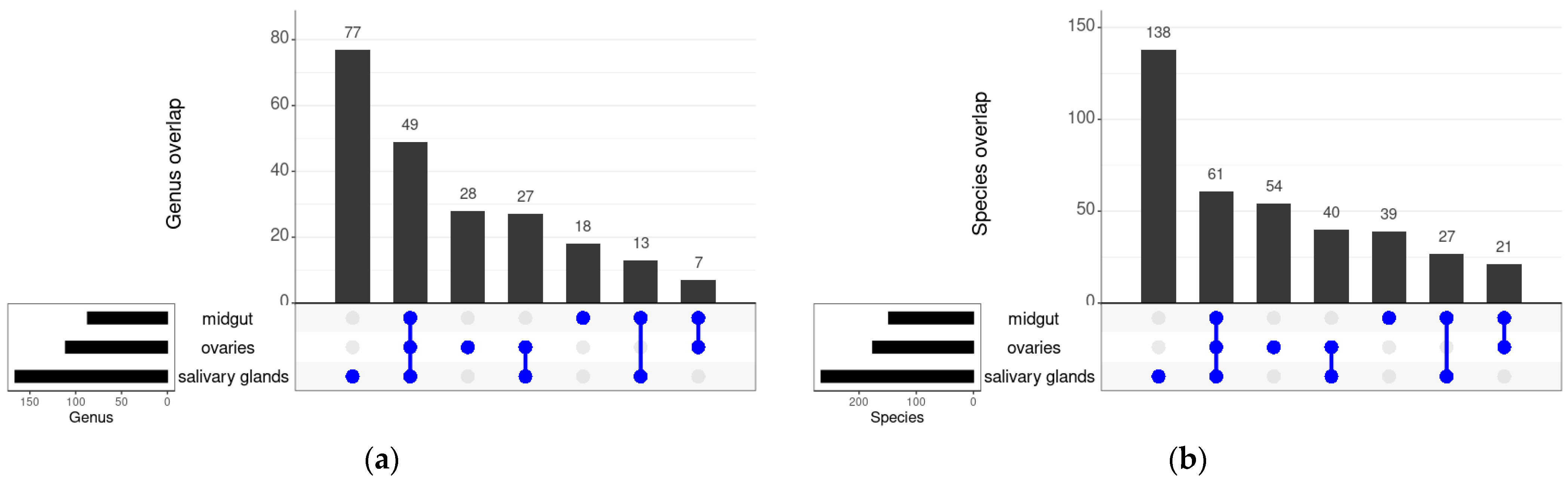
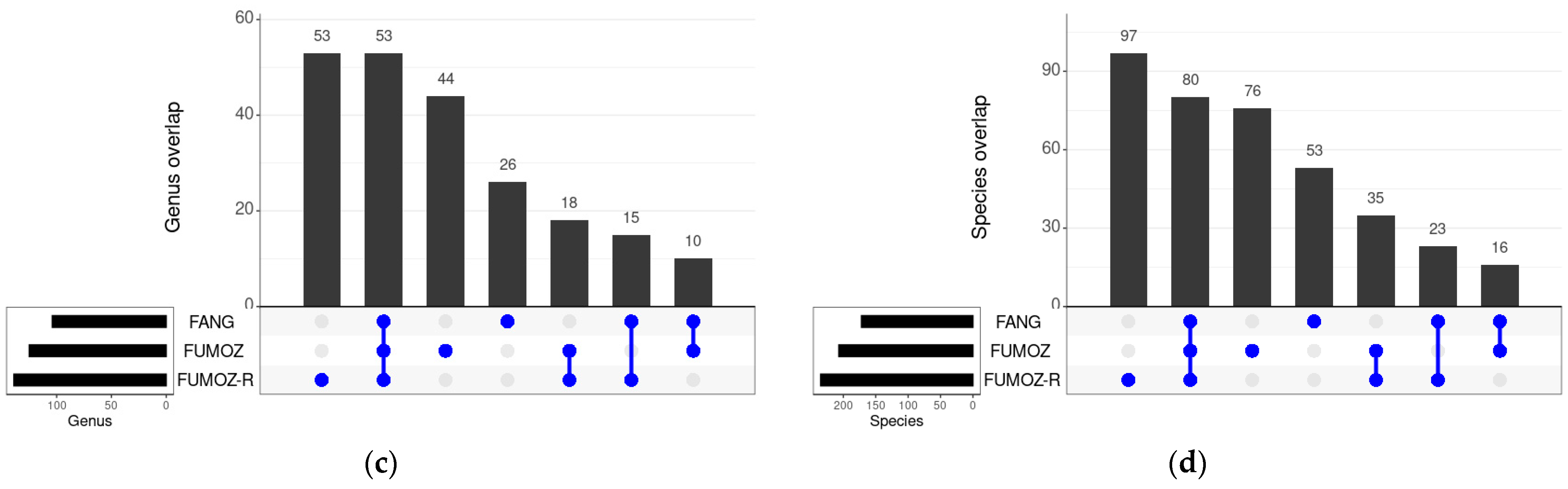
3.3. Shared Bacteria between the Different Tissues and Strains of An. funestus
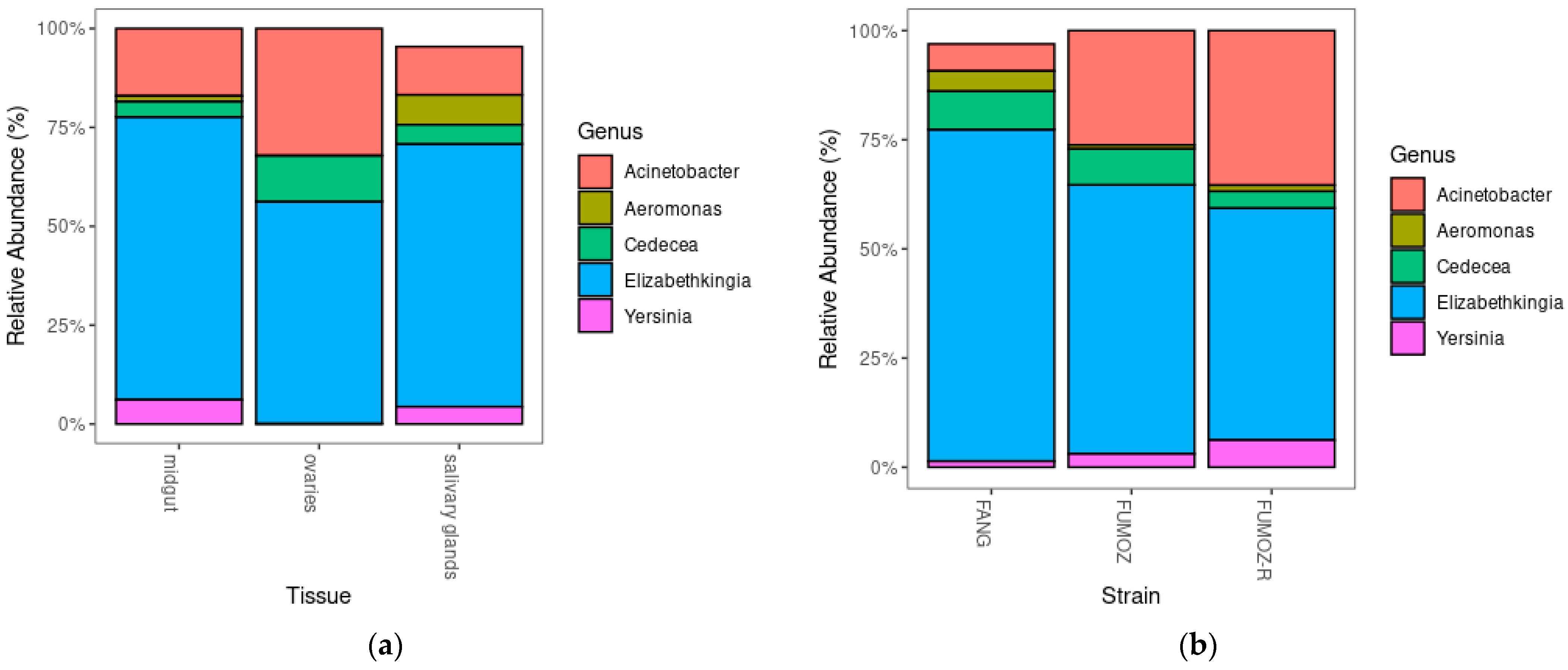
3.4. Relative Abundance
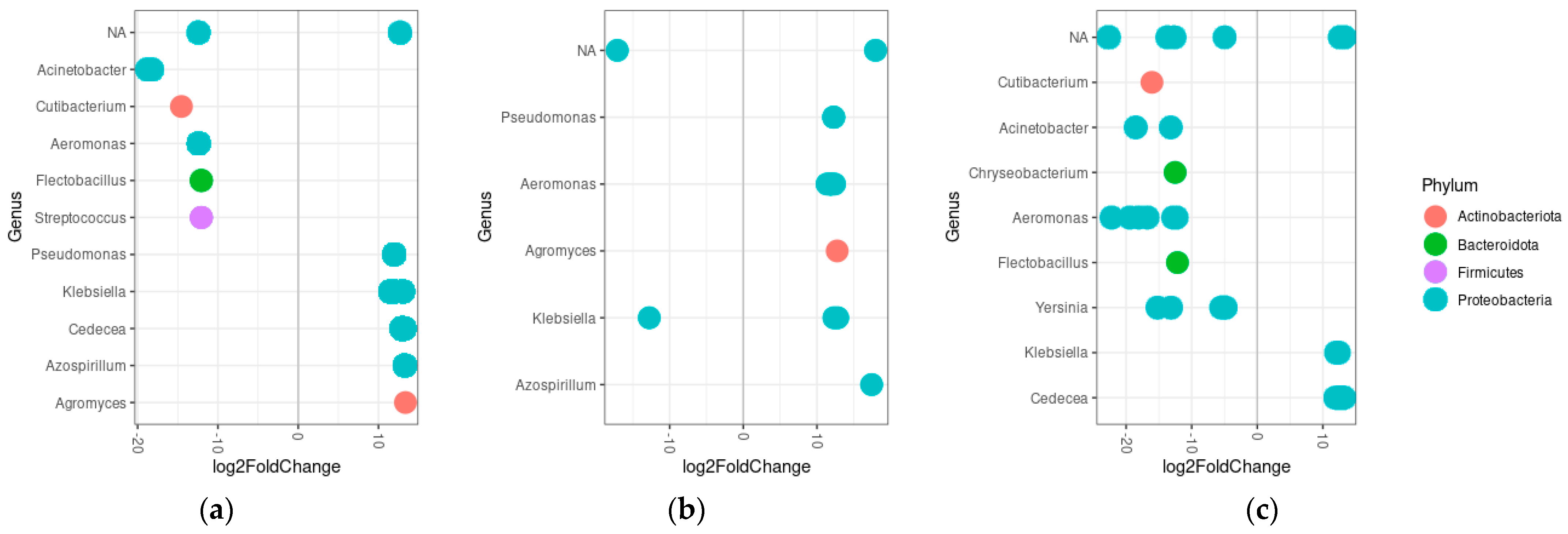
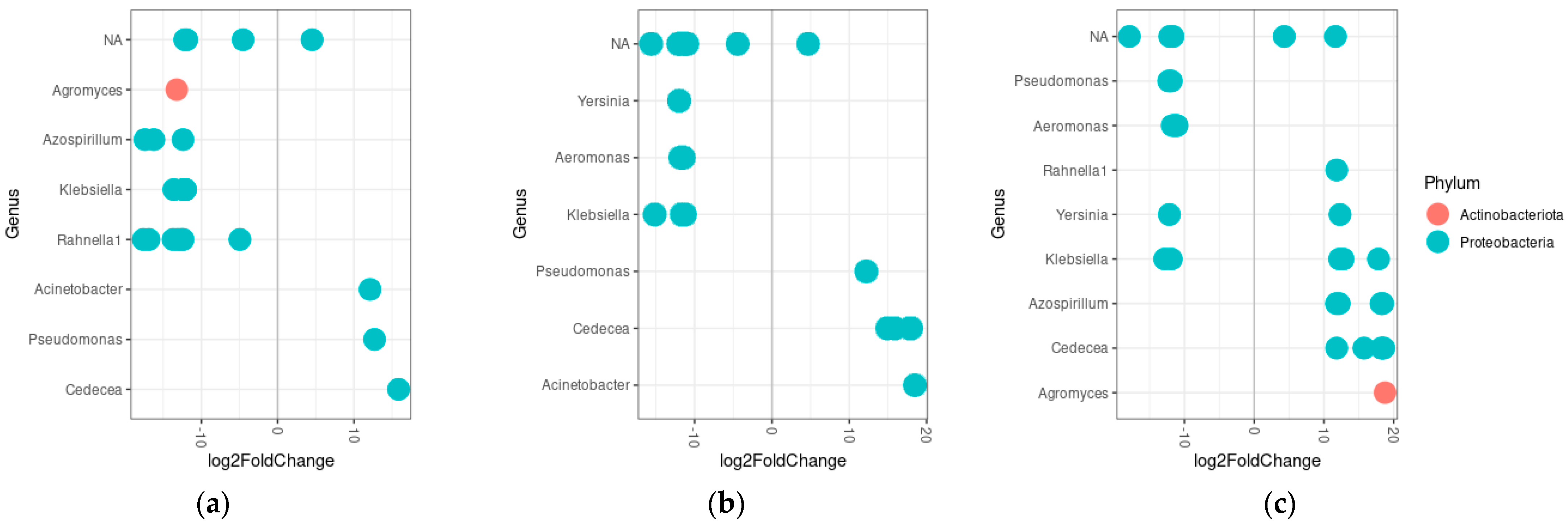
3.5. Differential Abundance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinka, M.E.; Bangs, M.J.; Manguin, S.; Coetzee, M.; Mbogo, C.M.; Hemingway, J.; Patil, A.P.; Temperley, W.H.; Gething, P.W.; Kabaria, C.W.; et al. The dominant Anopheles vectors of human malaria in Africa, Europe and the Middle East: Occurrence data, distribution maps and bionomic précis. Parasites Vectors 2010, 3, 117. [Google Scholar] [CrossRef]
- Sinka, M.E.; Bangs, M.J.; Manguin, S.; Rubio-Palis, Y.; Chareonviriyaphap, T.; Coetzee, M.; Mbogo, C.M.; Hemingway, J.; Patil, A.P.; Temperley, W.H.; et al. A global map of dominant malaria vectors. Parasites Vectors 2012, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Matowo, N.S.; Martin, J.; Kulkarni, M.A.; Mosha, J.F.; Lukole, E.; Isaya, G.; Shirima, B.; Kaaya, R.; Moyes, C.; Hancock, P.A.; et al. An increasing role of pyrethroid-resistant Anopheles funestus in malaria transmission in the Lake Zone, Tanzania. Sci. Rep. 2021, 11, 13457. [Google Scholar] [CrossRef] [PubMed]
- Gillies, M.T.; De Meillon, B. The Anophelinae of Africa South of the Sahara (Ethiopian Zoogeographical Region); CABI: Wallingford, UK, 1968. [Google Scholar]
- Charlwood, J.D.; Smith, T.; Kihonda, J.; Heiz, B.; Billingsley, P.F.; Takken, W. Density independent feeding success of malaria vectors (Diptera: Culicidae) in Tanzania. Bull. Entomol. Res. 1995, 85, 29–35. [Google Scholar] [CrossRef]
- Coetzee, M.; Kruger, P.; Kruger, P.; Hunt, R.H.; Hunt, R.H.; Durrheim, D.N.; Durrheim, D.N.; Urbach, J.; Urbach, J.; Hansford, C.F.; et al. Malaria in South Africa: 110 years of learning to control the disease. S. Afr. Med. J. 2013, 103, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Riveron, J.M.; Huijben, S.; Tchapga, W.; Tchouakui, M.; Wondji, M.J.; Tchoupo, M.; Irving, H.; Cuamba, N.; Maquina, M.; Paaijmans, K.; et al. Escalation of pyrethroid resistance in the malaria vector Anopheles funestus induces a loss of efficacy of piperonyl butoxide–based insecticide-treated nets in Mozambique. J. Infect. Dis. 2019, 220, 467–475. [Google Scholar] [CrossRef]
- Chanda, J.; Saili, K.; Phiri, F.; Stevenson, J.C.; Mwenda, M.; Chishimba, S.; Mulube, C.; Mambwe, B.; Lungu, C.; Earle, D.; et al. Pyrethroid and carbamate resistance in Anopheles funestus Giles along Lake Kariba in Southern Zambia. Am. J. Trop. Med. Hyg. 2020, 103, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Mugenzi, L.M.J.; Akosah-Brempong, G.; Tchouakui, M.; Menze, B.D.; Tekoh, T.A.; Tchoupo, M.; Nkemngo, F.N.; Wondji, M.J.; Nwaefuna, E.K.; Osae, M.; et al. Escalating pyrethroid resistance in two major malaria vectors Anopheles funestus and Anopheles gambiae (s.l.) in Atatam, Southern Ghana. BMC Infect. Dis. 2022, 22, 799. [Google Scholar] [CrossRef]
- Villegas, L.M.; Pimenta, P.F.P. Metagenomics, paratransgenesis and the Anopheles microbiome: A portrait of the geographical distribution of the anopheline microbiota based on a meta-analysis of reported taxa. Mem. Inst. Oswaldo Cruz 2014, 109, 672–684. [Google Scholar] [CrossRef]
- Mancini, M.V.; Spaccapelo, R.; Damiani, C.; Accoti, A.; Tallarita, M.; Petraglia, E.; Rossi, P.; Cappelli, A.; Capone, A.; Peruzzi, G.; et al. Paratransgenesis to control malaria vectors: A semi-field pilot study. Parasites Vectors 2016, 9, 140. [Google Scholar] [CrossRef]
- Caragata, E.P.; Tikhe, C.V.; Dimopoulos, G. Curious entanglements: Interactions between mosquitoes, their microbiota, and arboviruses. Curr. Opin. Virol. 2019, 37, 26–36. [Google Scholar] [CrossRef]
- Steven, B.; Hyde, J.; LaReau, J.C.; Brackney, D.E. The axenic and gnotobiotic mosquito: Emerging models for microbiome host interactions. Front. Microbiol. 2021, 12, 714222. [Google Scholar] [CrossRef] [PubMed]
- Minard, G.; Tran, F.H.; Raharimalala, F.N.; Hellard, E.; Ravelonandro, P.; Mavingui, P.; Valiente Moro, C. Prevalence, genomic and metabolic profiles of Acinetobacter and Asaia associated with field-caught Aedes albopictus from Madagascar. FEMS Microbiol. Ecol. 2013, 83, 63–73. [Google Scholar] [CrossRef]
- Gao, H.; Cui, C.; Wang, L.; Jacobs-Lorena, M.; Wang, S. Mosquito microbiota and implications for disease control. Trends Parasitol. 2020, 36, 98–111. [Google Scholar] [CrossRef]
- Favia, G.; Ricci, I.; Damiani, C.; Raddadi, N.; Crotti, E.; Marzorati, M.; Rizzi, A.; Urso, R.; Brusetti, L.; Borin, S.; et al. Bacteria of the genus Asaia stably associate with Anopheles stephensi, an Asian malarial mosquito vector. Proc. Natl. Acad. Sci. USA 2007, 104, 9047–9051. [Google Scholar] [CrossRef]
- Damiani, C.; Ricci, I.; Crotti, E.; Rossi, P.; Rizzi, A.; Scuppa, P.; Esposito, F.; Bandi, C.; Daffonchio, D.; Favia, G. Paternal transmission of symbiotic bacteria in malaria vectors. Curr. Biol. 2008, 18, R1087–R1088. [Google Scholar] [CrossRef]
- Damiani, C.; Ricci, I.; Crotti, E.; Rossi, P.; Rizzi, A.; Scuppa, P.; Capone, A.; Ulissi, U.; Epis, S.; Genchi, M.; et al. Mosquito-bacteria symbiosis: The case of Anopheles gambiae and Asaia. Microb. Ecol. 2010, 60, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Crotti, E.; Damiani, C.; Pajoro, M.; Gonella, E.; Rizzi, A.; Ricci, I.; Negri, I.; Scuppa, P.; Rossi, P.; Ballarini, P.; et al. Asaia, a versatile acetic acid bacterial symbiont, capable of cross-colonizing insects of phylogenetically distant Genera and Orders. Environ. Microbiol. 2009, 11, 3252–3264. [Google Scholar] [CrossRef] [PubMed]
- De Freece, C.; Damiani, C.; Valzano, M.; D’amelio, S.; Cappelli, A.; Ricci, I.; Favia, G. Detection and isolation of the α-Proteobacterium Asaia in Culex mosquitoes. Med. Vet. Entomol. 2014, 28, 438–442. [Google Scholar] [CrossRef]
- Ramos-Nino, M.E.; Fitzpatrick, D.M.; Eckstrom, K.M.; Tighe, S.; Hattaway, L.M.; Hsueh, A.N.; Stone, D.M.; Dragon, J.A.; Cheetham, S. Metagenomic analysis of Aedes aegypti and Culex quinquefasciatus mosquitoes from Grenada, West Indies. PLoS ONE 2020, 15, e0231047. [Google Scholar] [CrossRef]
- Capone, A.; Ricci, I.; Damiani, C.; Mosca, M.; Rossi, P.; Scuppa, P.; Crotti, E.; Epis, S.; Angeletti, M.; Valzano, M.; et al. Interactions between Asaia, Plasmodium and Anopheles: New insights into mosquito symbiosis and implications in malaria symbiotic control. Parasites Vectors 2013, 6, 182. [Google Scholar] [CrossRef]
- Cappelli, A.; Damiani, C.; Mancini, M.V.; Valzano, M.; Rossi, P.; Serrao, A.; Ricci, I.; Favia, G. Asaia activates immune genes in mosquito eliciting an anti-Plasmodium response: Implications in malaria control. Front. Genet. 2019, 10, 836. [Google Scholar] [CrossRef] [PubMed]
- Cirimotich, C.M.; Dong, Y.; Clayton, A.M.; Sandiford, S.L.; Souza-Neto, J.A.; Mulenga, M.; Dimopoulos, G. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 2011, 332, 855–858. [Google Scholar] [CrossRef]
- Wang, S.; Ghosh, A.K.; Bongio, N.; Stebbings, K.A.; Lampe, D.J.; Jacobs-Lorena, M. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc. Natl. Acad. Sci. USA 2012, 109, 12734–12739. [Google Scholar] [CrossRef]
- Wang, S.; Dos-Santos, A.L.A.; Huang, W.; Liu, K.C.; Oshaghi, M.; Wei, G.; Agre, P.; Jacobs-Lorena, M. Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science 2017, 357, 1399–1402. [Google Scholar] [CrossRef]
- Dada, N.; Sheth, M.; Liebman, K.; Pinto, J.; Lenhart, A. Whole metagenome sequencing reveals links between mosquito microbiota and insecticide resistance in malaria vectors. Sci. Rep. 2018, 8, 2084. [Google Scholar] [CrossRef] [PubMed]
- Dada, N.; Lol, J.C.; Benedict, A.C.; López, F.; Sheth, M.; Dzuris, N.; Padilla, N.; Lenhart, A. Pyrethroid exposure alters internal and cuticle surface bacterial communities in Anopheles albimanus. ISME J. 2019, 13, 2447–2464. [Google Scholar] [CrossRef] [PubMed]
- Omoke, D.; Kipsum, M.; Otieno, S.; Esalimba, E.; Sheth, M.; Lenhart, A.; Njeru, E.M.; Ochomo, E.; Dada, N. Western Kenyan Anopheles gambiae showing intense permethrin resistance harbour distinct microbiota. Malar. J. 2021, 20, 77. [Google Scholar] [CrossRef]
- Pelloquin, B.; Kristan, M.; Edi, C.; Meiwald, A.; Clark, E.; Jeffries, C.L.; Walker, T.; Dada, N.; Messenger, L.A. Overabundance of Asaia and Serratia bacteria is associated with deltamethrin insecticide susceptibility in Anopheles coluzzii from Agboville, Côte d’Ivoire. Microbiol. Spectr. 2021, 9, e00157-21. [Google Scholar] [CrossRef]
- Sharma, P.; Sharma, S.; Maurya, R.K.; De, T.D.; Thomas, T.; Lata, S.; Singh, N.; Pandey, K.C.; Valecha, N.; Dixit, R. Salivary glands harbor more diverse microbial communities than gut in Anopheles culicifacies. Parasites Vectors 2014, 7, 235. [Google Scholar] [CrossRef]
- Brooke, B.D.; Kloke, G.; Hunt, R.H.; Koekemoer, L.L.; Tem, E.A.; Taylor, M.E.; Small, G.; Hemingway, J.; Coetzee, M. Bioassay and biochemical analyses of insecticide resistance in Southern African Anopheles funestus (Diptera: Culicidae). Bull. Entomol. Res. 2001, 91, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.V.; Lyons, C.L.; Brooke, B.D. The effect of blood feeding on insecticide resistance intensity and adult longevity in the major malaria vector Anopheles funestus (Diptera: Culicidae). Sci. Rep. 2022, 12, 3877. [Google Scholar] [CrossRef]
- Hunt, R.H.; Brooke, B.D.; Pillay, C.; Koekemoer, L.L.; Coetzee, M. Laboratory Selection for and characteristics of pyrethroid resistance in the malaria vector Anopheles funestus. Med. Vet. Entomol. 2005, 19, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and Next-Generation Sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Tchioffo, M.T.; Boissière, A.; Abate, L.; Nsango, S.E.; Bayibéki, A.N.; Awono-Ambéné, P.H.; Christen, R.; Gimonneau, G.; Morlais, I. Dynamics of bacterial community composition in the malaria mosquito’s epithelia. Front. Microbiol. 2016, 6, 1500. [Google Scholar] [CrossRef]
- Rani, A.; Sharma, A.; Rajagopal, R.; Adak, T.; Bhatnagar, R.K. Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi—An asian malarial vector. BMC Microbiol. 2009, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Boissière, A.; Tchioffo, M.T.; Bachar, D.; Abate, L.; Marie, A.; Nsango, S.E.; Shahbazkia, H.R.; Awono-Ambene, P.H.; Levashina, E.A.; Christen, R.; et al. Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 2012, 8, e1002742. [Google Scholar] [CrossRef] [PubMed]
- Ngo, C.T.; Aujoulat, F.; Veas, F.; Jumas-Bilak, E.; Manguin, S. Bacterial diversity associated with wild caught Anopheles mosquitoes from Dak Nong Province, Vietnam using culture and DNA fingerprint. PLoS ONE 2015, 10, e0118634. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Allam, M.; Kwenda, S.; Khumalo, Z.T.H.; Ismail, A.; Oliver, S.V. The dynamic gut microbiota of zoophilic members of the Anopheles gambiae complex (Diptera: Culicidae). Sci. Rep. 2022, 12, 1495. [Google Scholar] [CrossRef] [PubMed]
- Gimonneau, G.; Tchioffo, M.T.; Abate, L.; Boissière, A.; Awono-Ambéné, P.H.; Nsango, S.E.; Christen, R.; Morlais, I. Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Infect. Genet. Evol. 2014, 28, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Shaw, W.R.; Marcenac, P.; Catteruccia, F. Plasmodium development in Anopheles: A tale of shared resources. Trends Parasitol. 2022, 38, 124–135. [Google Scholar] [CrossRef]
- Shapiro, L.L.M.; Whitehead, S.A.; Thomas, M.B. Quantifying the effects of temperature on mosquito and parasite traits that determine the transmission potential of human malaria. PLoS Biol. 2017, 15, e2003489. [Google Scholar] [CrossRef]
- Lindh, J.M.; Borg-Karlson, A.-K.; Faye, I. Transstadial and horizontal transfer of bacteria within a colony of Anopheles gambiae (Diptera: Culicidae) and oviposition response to bacteria-containing water. Acta Trop. 2008, 107, 242–250. [Google Scholar] [CrossRef]
- Wang, Y.; Iii, T.M.G.; Kukutla, P.; Yan, G.; Xu, J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE 2011, 6, e24767. [Google Scholar] [CrossRef]
- Akhouayri, I.G.; Habtewold, T.; Christophides, G.K. Melanotic pathology and vertical transmission of the gut commensal Elizabethkingia meningoseptica in the major malaria vector Anopheles gambiae. PLoS ONE 2013, 8, e77619. [Google Scholar] [CrossRef]
- Kukutla, P.; Lindberg, B.G.; Pei, D.; Rayl, M.; Yu, W.; Steritz, M.; Faye, I.; Xu, J. Insights from the genome annotation of Elizabethkingia anophelis from the malaria vector Anopheles gambiae. PLoS ONE 2014, 9, e97715. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, C.J.; Glöckner, V.; Abdelmohsen, U.R.; Scheuermayer, M.; Fischer, R.; Hentschel, U.; Pradel, G. 16S rRNA gene-based identification of Elizabethkingia meningoseptica (Flavobacteriales: Flavobacteriaceae) as a dominant midgut bacterium of the Asian malaria vector Anopheles stephensi (Dipteria: Culicidae) with antimicrobial activities. J. Med. Entomol. 2013, 50, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Terenius, O.; Lindh, J.M.; Eriksson-Gonzales, K.; Bussière, L.; Laugen, A.T.; Bergquist, H.; Titanji, K.; Faye, I. Midgut bacterial dynamics in Aedes Aegypti. FEMS Microbiol. Ecol. 2012, 80, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Luckhart, S.; Vodovotz, Y.; Cui, L.; Rosenberg, R. The Mosquito Anopheles stephensi limits malaria parasite development with inducible synthesis of nitric oxide. Proc. Natl. Acad. Sci. USA 1998, 95, 5700–5705. [Google Scholar] [CrossRef]
- Hillyer, J.F.; Estévez-Lao, T.Y. Nitric oxide is an essential component of the hemocyte-mediated mosquito immune response against bacteria. Dev. Comp. Immunol. 2010, 34, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ngo, C.T.; Romano-Bertrand, S.; Manguin, S.; Jumas-Bilak, E. Diversity of the bacterial microbiota of Anopheles mosquitoes from Binh Phuoc Province, Vietnam. Front. Microbiol. 2016, 7, 205911. [Google Scholar] [CrossRef][Green Version]
- Chen, S.; Bagdasarian, M.; Walker, E.D. Elizabethkingia anophelis: Molecular manipulation and interactions with mosquito hosts. Appl. Environ. Microbiol. 2015, 81, 2233–2243. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Chan, W.-Y.; Ismail, A.; Oliver, S.V. Characterization of the Tissue and Strain-Specific Microbiota of Anopheles funestus Giles (Diptera: Culicidae). Trop. Med. Infect. Dis. 2024, 9, 84. https://doi.org/10.3390/tropicalmed9040084
Chen C-Y, Chan W-Y, Ismail A, Oliver SV. Characterization of the Tissue and Strain-Specific Microbiota of Anopheles funestus Giles (Diptera: Culicidae). Tropical Medicine and Infectious Disease. 2024; 9(4):84. https://doi.org/10.3390/tropicalmed9040084
Chicago/Turabian StyleChen, Chia-Yu, Wai-Yin Chan, Arshad Ismail, and Shüné V. Oliver. 2024. "Characterization of the Tissue and Strain-Specific Microbiota of Anopheles funestus Giles (Diptera: Culicidae)" Tropical Medicine and Infectious Disease 9, no. 4: 84. https://doi.org/10.3390/tropicalmed9040084
APA StyleChen, C.-Y., Chan, W.-Y., Ismail, A., & Oliver, S. V. (2024). Characterization of the Tissue and Strain-Specific Microbiota of Anopheles funestus Giles (Diptera: Culicidae). Tropical Medicine and Infectious Disease, 9(4), 84. https://doi.org/10.3390/tropicalmed9040084