Synthesis of New bis 1-Substituted 1H-Tetrazoles via Efficient Heterocyclizations from Symmetric Dianilines, Methyl Orthoester, and Sodium Azide †


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
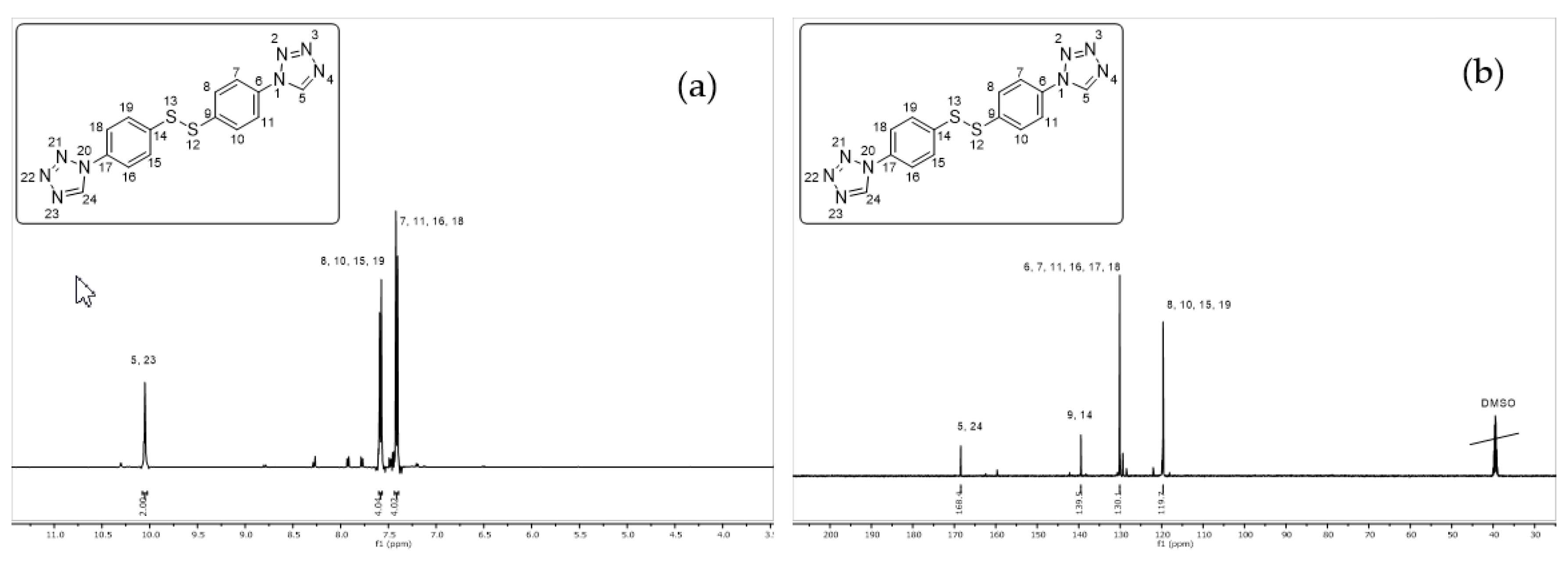
2.1. Synthesis of 1,2-bis(4-(1H-tetrazol-1-yl)phenyl)disulfane (2)
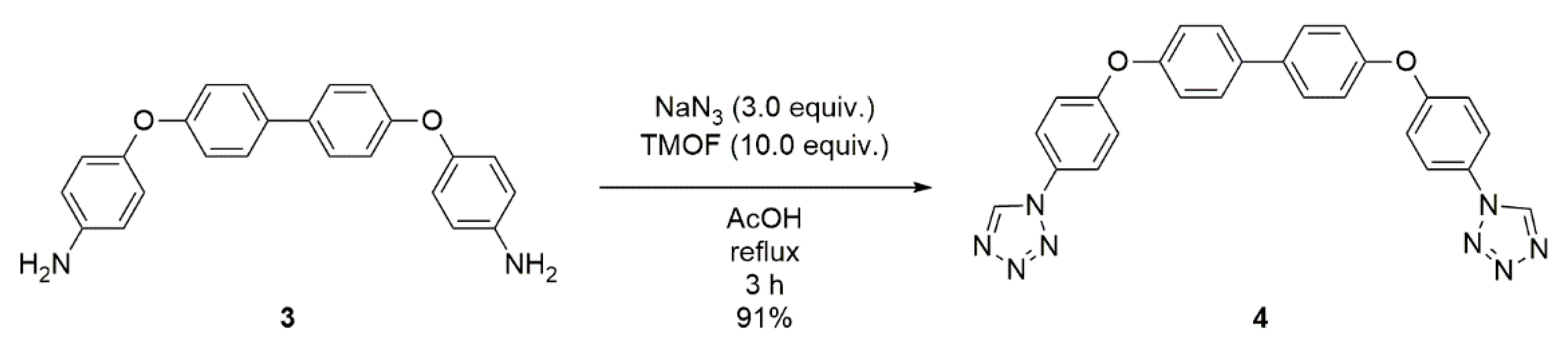
2.2. Synthesis of 4,4’-bis(4-(1H-tetrazol-1-yl)phenoxy)-1,1’-biphenyl (4)
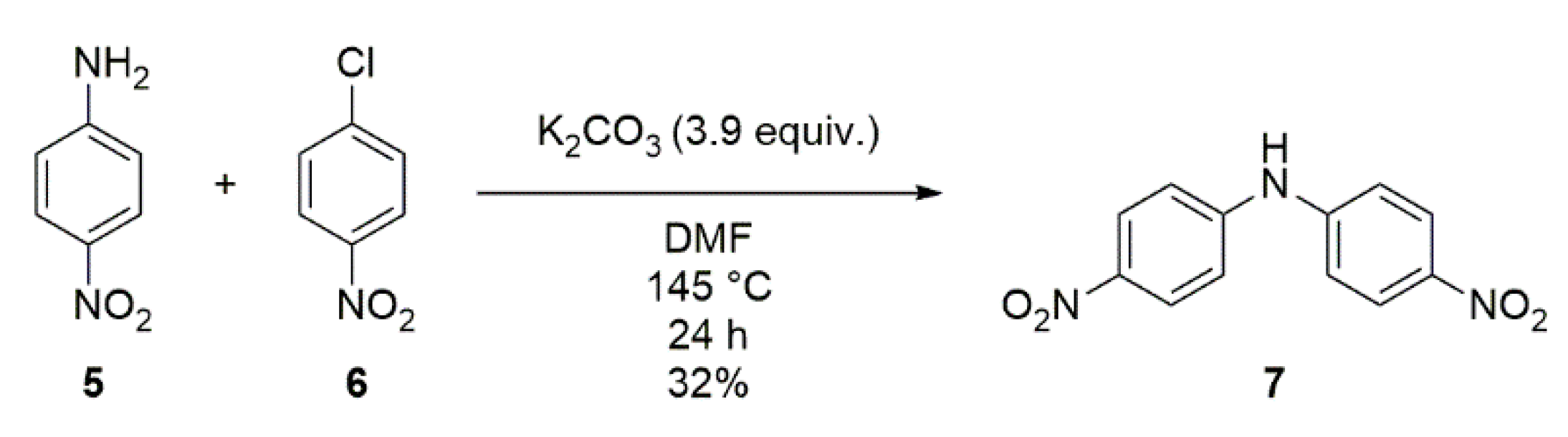
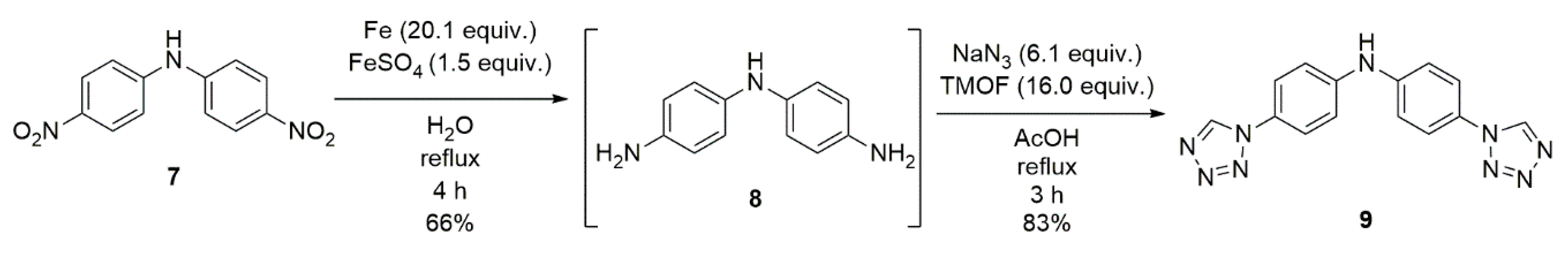
2.3. Synthesis of bis(4-(1H-tetrazol-1-yl)phenyl)amine (9)
3. Conclusions
4. Experimental Section
4.1. General Information, Instrumentation, and Chemicals
4.2. Synthesis of 1,2-bis(4-(1H-tetrazol-1-yl)phenyl)disulfane (2)
4.3. Synthesis of 4,4’-bis(4-(1H-tetrazol-1-yl)phenoxy)-1,1’-biphenyl (4)
4.4. Synthesis of bis(4-nitrophenyl)amine (7)
4.5. Synthesis of N1-(4-aminophenyl)benzene-1,4-diamine (8)
4.6. Synthesis of bis(4-(1H-tetrazol-1-yl)phenyl)amine (9)
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Izatt, R.M. Macrocyclic and Supramolecular Chemistry; Wiley: Chichester, UK, 2016. [Google Scholar] [CrossRef]
- Lee, J.; Farha, O.K.; Roberts, J.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal-organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houk, R.J. Luminescent metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Tavra, C.; Baxter, E.F.; Tian, T.; Bennett, T.D.; Slater, N.K.; Cheetham, A.K.; Fairen-Jimenez, D. Amorphous metal–organic frameworks for drug delivery. Chem. Commun. 2015, 51, 13878–13881. [Google Scholar] [CrossRef] [PubMed]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.; Van Duyne, R.P.; Hupp, J.T. Metal–organic framework materials as chemical sensors. Chem. Rev. 2012, 112, 1105–1125. [Google Scholar] [CrossRef] [PubMed]
- Li, J.R.; Kuppler, R.J.; Zhou, H.C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination Polymers: Design, Analysis and Application; Royal Society of Chemistry: Cambridge, UK, 2009. [Google Scholar] [CrossRef]
- Liu, X.; Akerboom, S.; Jong, M.D.; Mutikainen, I.; Tanase, S.; Meijerink, A.; Bouwman, E. Mixed-lanthanoid metal–organic framework for ratiometric cryogenic temperature sensing. Inorg. Chem. 2015, 54, 11323–11329. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Deria, P.; Bury, W.; Hod, I.; Kung, C.W.; Karagiaridi, O.; Hupp, J.T.; Farha, O.K. MOF functionalization via solvent-assisted ligand incorporation: Phosphonates vs. carboxylates. Inorg. Chem. 2015, 54, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.P.; Zhang, Y.B.; Lin, J.B.; Chen, X.M. Metal azolate frameworks: From crystal engineering to functional materials. Chem. Rev. 2012, 112, 1001–1033. [Google Scholar] [CrossRef] [PubMed]
- Herr, R.J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: Medicinal chemistry and synthetic methods. Bioorg. Med. Chem. 2002, 10, 3379–3393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Qu, Z.R.; Ye, H.Y.; Xiong, R.G. In situ hydrothermal synthesis of tetrazole coordination polymers with interesting physical properties. Chem. Soc. Rev. 2008, 37, 84–100. [Google Scholar] [CrossRef]
- Kuznetsov, Y.I.; Kazansky, L.P. Physicochemical aspects of metal protection by azoles as corrosion inhibitors. Russ. Chem. Rev. 2008, 77, 219. [Google Scholar] [CrossRef]
- Gao, H.; Shreeve, J.N.M. Azole-based energetic salts. Chem. Rev. 2011, 111, 7377–7436. [Google Scholar] [CrossRef] [PubMed]
- Franke, P.L.; Haasnoot, J.G.; Zuur, A.P. Tetrazoles as ligands. Part IV. Iron (II) complexes of monofunctional tetrazole ligands, showing high-spin (p5T2g) ⇋ low-spin transitions. Inorg. Chim. Acta 1982, 59, 5–9. [Google Scholar] [CrossRef]
- Aromí, G.; Barrios, L.A.; Roubeau, O.; Gamez, P. Triazoles and tetrazoles: Prime ligands to generate remarkable coordination materials. Coord. Chem. Rev. 2011, 255, 485–546. [Google Scholar] [CrossRef]
- Huang, Z.; Song, H.B.; Du, M.; Chen, S.T.; Bu, X.H.; Ribas, J. Coordination polymers assembled from angular dipyridyl ligands and CuII, CdII, CoII salts: Crystal structures and properties. Inorg. Chem. 2004, 43, 931–944. [Google Scholar] [CrossRef]
- Jin, T.; Kamijo, S.; Yamamoto, Y. Synthesis of 1-substituted tetrazoles via the acid-catalyzed [3+2] cycloaddition between isocyanides and trimethylsilyl azide. Tetrahedron Lett. 2004, 45, 9435–9437. [Google Scholar] [CrossRef]
- Gaponik, P.N.; Karavai, V.P.; Grigor’ev, Y.V. Synthesis of 1-substituted tetrazoles by heterocyclization of primary amines, orthoformic ester, and sodium azide. Chem. Heterocycl. Compd. 1985, 21, 1255–1258. [Google Scholar] [CrossRef]
- Grigoriev, Y.V.; Voitekhovich, S.V.; Karavai, V.P.; Ivashkevich, O.A. Synthesis of tetrazole and its derivatives by heterocyclization reaction involving primary amines, orthoesters, and azides. Chem. Heterocycl. Compd. 2017, 53, 670–681. [Google Scholar] [CrossRef]
- Muttenthaler, M.; Bartel, M.; Weinberger, P.; Hilscher, G.; Linert, W. Synthesis and characterisation of new ditetrazole-ligands as more rigid building blocks of envisaged iron (II) spin-crossover coordination polymers. J. Mol. Struct. 2005, 741, 159–169. [Google Scholar] [CrossRef]
- Kamiya, T.; Saito, Y. Process for Preparing 1H-tetrazole Compounds. U.S. Patent 3,767,667; U.S. Patent and Trademark Office, Washington, DC, USA, 23 October 1973. [Google Scholar]
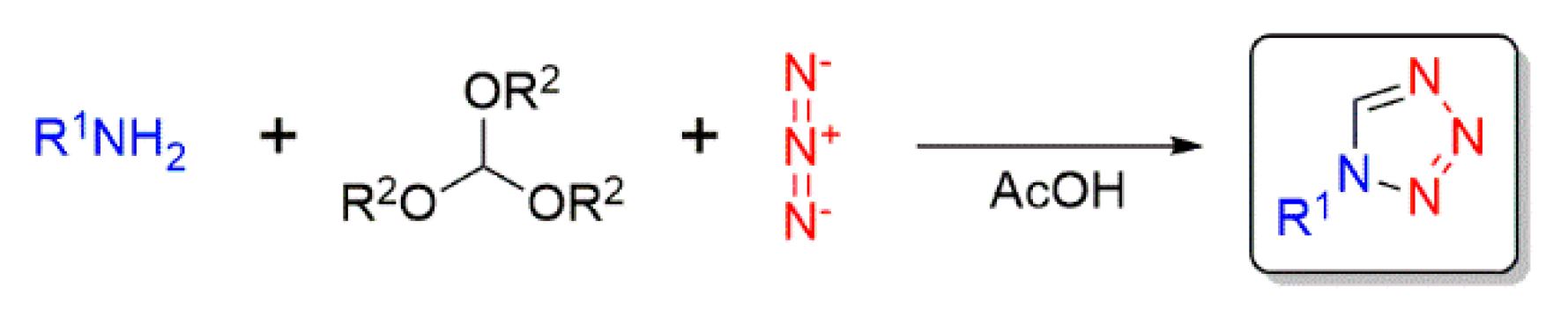




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Reyes, J.C.; Blanco-Carapia, R.E.; López-Olvera, A.; Islas-Jácome, P.; Medina-Martínez, Y.; Rincón-Guevara, M.A.; Ibarra, I.A.; Lomas-Romero, L.; González-Zamora, E.; Islas-Jácome, A. Synthesis of New bis 1-Substituted 1H-Tetrazoles via Efficient Heterocyclizations from Symmetric Dianilines, Methyl Orthoester, and Sodium Azide. Proceedings 2019, 41, 26. https://doi.org/10.3390/ecsoc-23-06521
Flores-Reyes JC, Blanco-Carapia RE, López-Olvera A, Islas-Jácome P, Medina-Martínez Y, Rincón-Guevara MA, Ibarra IA, Lomas-Romero L, González-Zamora E, Islas-Jácome A. Synthesis of New bis 1-Substituted 1H-Tetrazoles via Efficient Heterocyclizations from Symmetric Dianilines, Methyl Orthoester, and Sodium Azide. Proceedings. 2019; 41(1):26. https://doi.org/10.3390/ecsoc-23-06521
Chicago/Turabian StyleFlores-Reyes, Julio C., Roberto E. Blanco-Carapia, Alfredo López-Olvera, Perla Islas-Jácome, Yizrell Medina-Martínez, Mónica A. Rincón-Guevara, Ilich A. Ibarra, Leticia Lomas-Romero, Eduardo González-Zamora, and Alejandro Islas-Jácome. 2019. "Synthesis of New bis 1-Substituted 1H-Tetrazoles via Efficient Heterocyclizations from Symmetric Dianilines, Methyl Orthoester, and Sodium Azide" Proceedings 41, no. 1: 26. https://doi.org/10.3390/ecsoc-23-06521
APA StyleFlores-Reyes, J. C., Blanco-Carapia, R. E., López-Olvera, A., Islas-Jácome, P., Medina-Martínez, Y., Rincón-Guevara, M. A., Ibarra, I. A., Lomas-Romero, L., González-Zamora, E., & Islas-Jácome, A. (2019). Synthesis of New bis 1-Substituted 1H-Tetrazoles via Efficient Heterocyclizations from Symmetric Dianilines, Methyl Orthoester, and Sodium Azide. Proceedings, 41(1), 26. https://doi.org/10.3390/ecsoc-23-06521