NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana †



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material Preparation for Next-Generation Sequencing
2.2. DNA Extraction and Next-Generation Sequencing
2.3. Bioinformatics
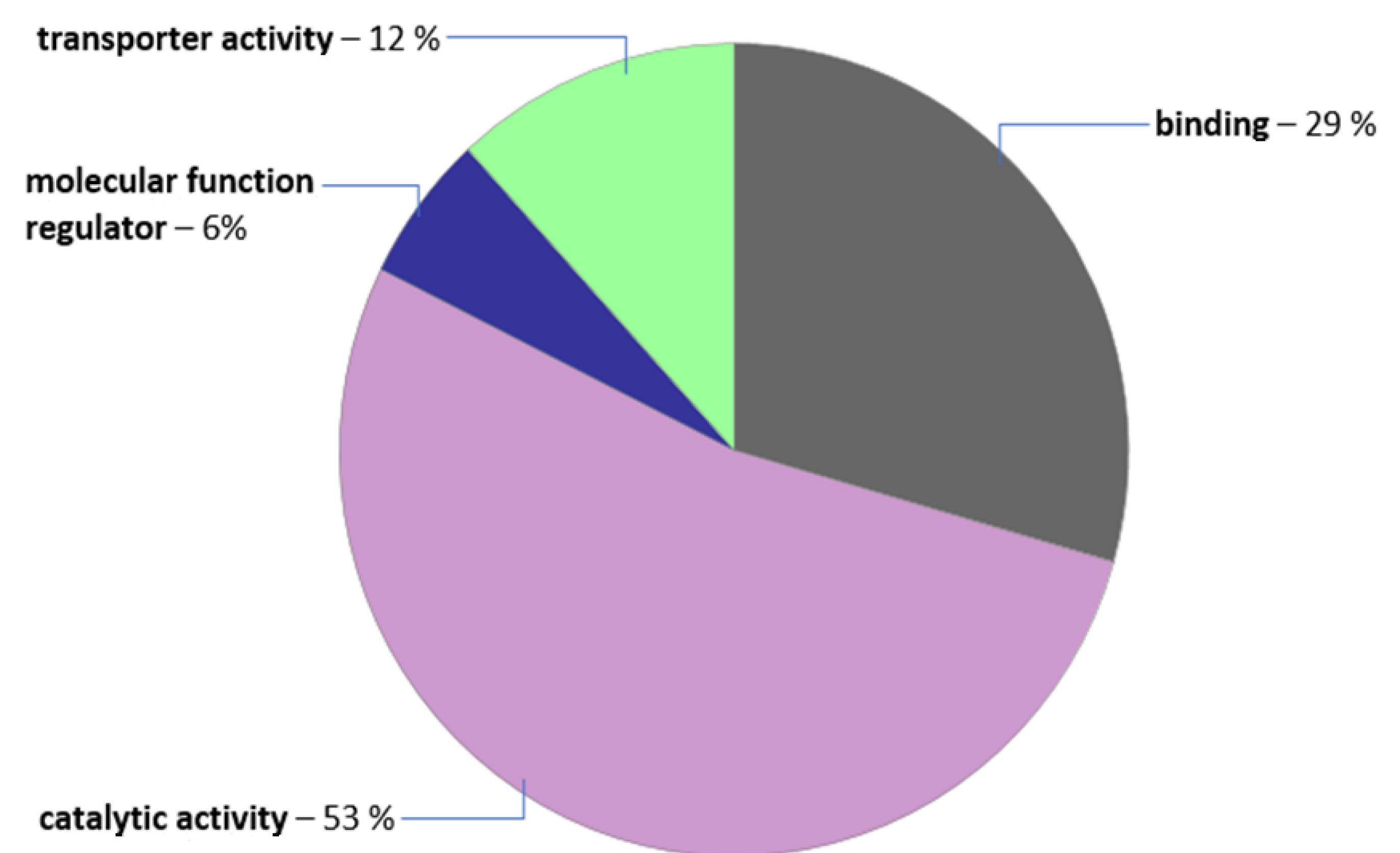
3. Results and Discussion
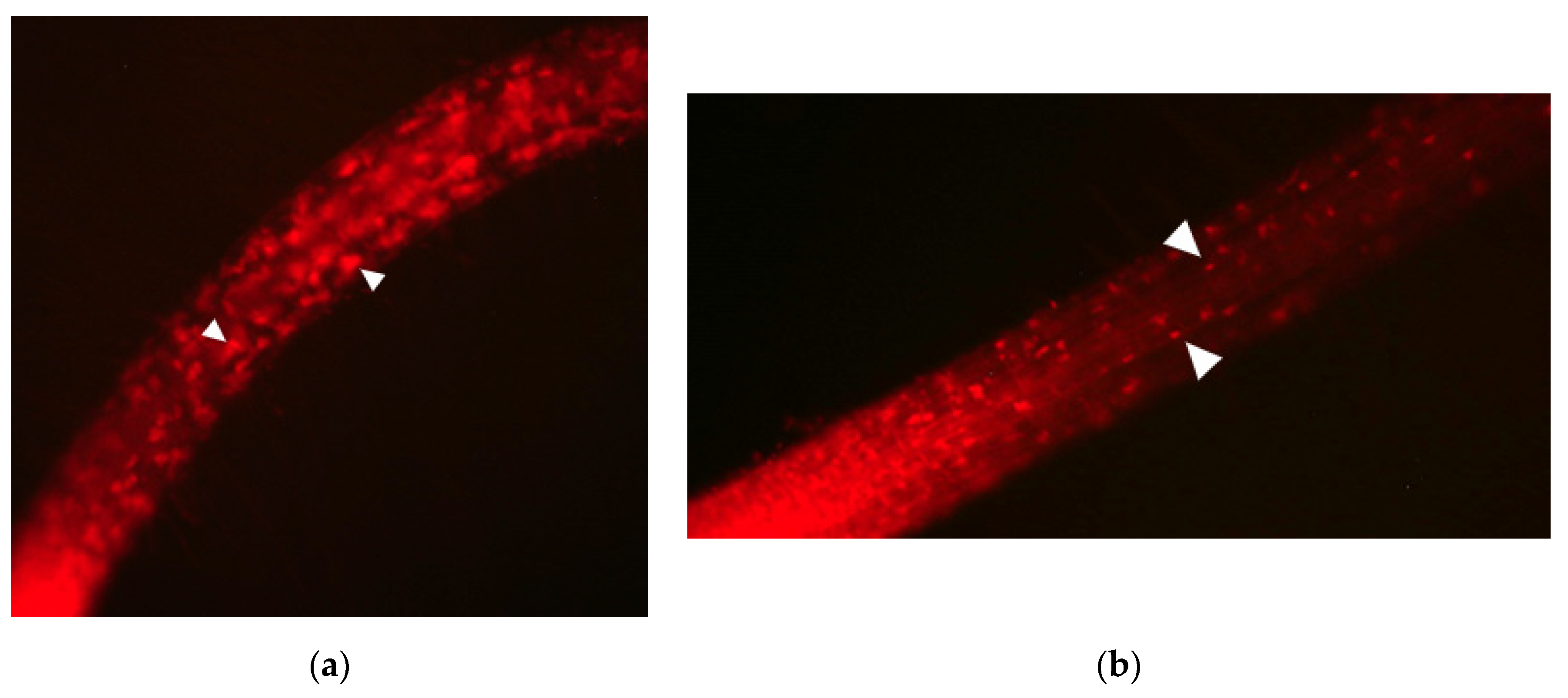
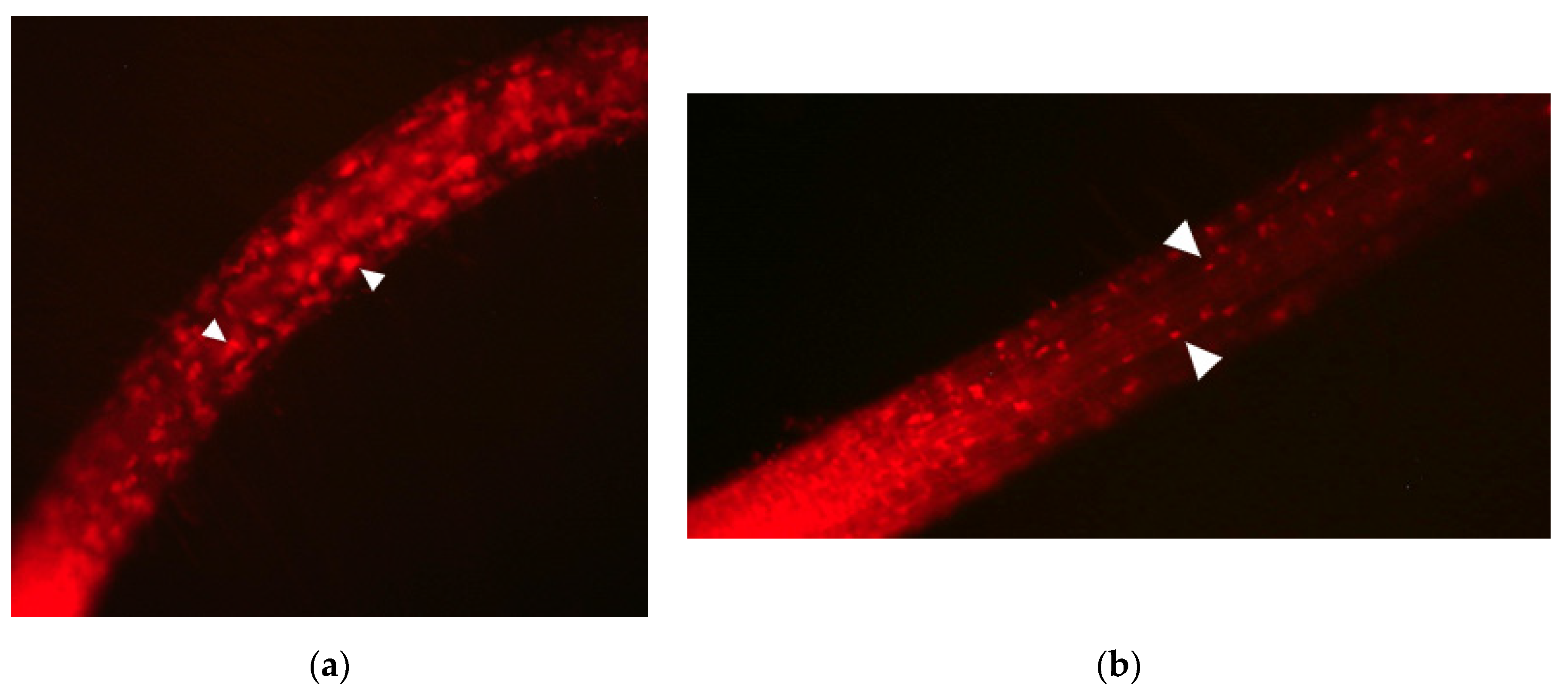
3.1. peup33 Mutants Display Autophagy-Defective Phenotype after FM4-64/E-64d Treatment
3.2. Next-Generation Sequencing for Identifaction of the peup33 Mutation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shibata, M.; Oikawa, K.; Yoshimoto, K.; Kondo, M.; Mano, S.; Yamada, K.; Hayashi, M.; Sakamoto, W.; Ohsumi, Y.; Nishimura, M. Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 2013, 25, 4967–4983. [Google Scholar] [CrossRef]
- Goto-Yamada, S.; Oikawa, K.; Bizan, J.; Shigenobu, S.; Yamaguchi, K.; Mano, S.; Hayashi, M.; Ueda, H.; Hara-Nishimura, I.; Nishimura, M.; et al. Sucrose Starvation Induces Microautophagy in Plant Root Cells. Front. Plant Sci. 2019, 10, 1604. [Google Scholar] [CrossRef]
- Allen, R.; Nakasugi, K.; Doran, R.L.; Millar, A.A.; Waterhouse, P.M. Facile mutant identification via a single parental backcross method and application of whole genome sequencing based mapping pipelines. Front. Plant Sci. 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- James, G.V.; Patel, V.; Nordström, K.J.V.; Klasen, J.R.; Salomé, P.A.; Weigel, D.; Schneeberger, K. User guide for mapping-by-sequencing in Arabidopsis. Genome Biol. 2013, 14, R61. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC. A quality control tool for high throughput sequence data. Babraham Bioinforma. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 November 2020).
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Moriyasu, Y.; Ohsumi, Y. Autophagy in Tobacco Suspension-Cultured Cells in Response to Sucrose Starvation. Plant Physiol. 1996, 111, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Dashti, M.J.S.; Gamieldien, J. A practical guide to filtering and prioritizing genetic variants. Biotechniques 2017, 62, 18–30. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sieńko, K.; Żukowski, K.; Yamada, K.; Goto-Yamada, S. NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana. Proceedings 2021, 76, 7. https://doi.org/10.3390/IECGE-07155
Sieńko K, Żukowski K, Yamada K, Goto-Yamada S. NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana. Proceedings. 2021; 76(1):7. https://doi.org/10.3390/IECGE-07155
Chicago/Turabian StyleSieńko, Katarzyna, Kacper Żukowski, Kenji Yamada, and Shino Goto-Yamada. 2021. "NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana" Proceedings 76, no. 1: 7. https://doi.org/10.3390/IECGE-07155
APA StyleSieńko, K., Żukowski, K., Yamada, K., & Goto-Yamada, S. (2021). NGS Screening for Identification of Novel Pexophagy-Related Mutation in Arabidopsis thaliana. Proceedings, 76(1), 7. https://doi.org/10.3390/IECGE-07155