Abstract
The objective of this study is to determine the analytic expressions of the Helmholtz free energy, the equilibrium vacancy concentration, the melting temperature, the jumps of volume, enthalpy the mean nearest neighbor distance and entropy at melting point, the Debye temperature for the BCC defective, the limiting temperature of absolute stability for the crystalline state, and for the perfect binary interstitial alloy. The results obtained from the expressions are combined with the statistical moment method, the limiting condition of the absolute stability at the crystalline state, the Clausius–Clapeyron equation, the Debye model and the Gruneisen equation. Our numerical calculations of obtained theoretical results were carried out for alloy WSi under high temperature and pressure. Our calculated melting curve and relation between the melting temperature and the silicon concentration for WSi are in good agreement with other calculations. Our calculations for the jumps of volume, enthalpy and entropy, and the Debye temperature for WSi predict and orient experimental results in the future.
1. Introduction
Metals and interstitial alloys [1,2,3,4,5] have been investigated by several research groups in the last decades due to their applications in various fields [6,7,8,9]. Many theoretical and experimental studies about the mechanical and thermodynamic properties of metals and interstitial alloys gained scientific and technological attention and represent an active area of research that requires integrating modern scientific insights [10,11,12,13,14] from multiple disciplines [15,16,17,18,19].
Different researchers have studied the dependence of the mechanical and thermodynamic properties of materials on the temperature (T), pressure (P), and concentration of components for these materials. It is known that point defects such as vacancies have important contributions to the properties of materials [18,19,20,21,22,23,24]. At the melting point, the equilibrium vacancy concentration of metals changes from 10-4 to 10-2 [20]; therefore, it has a significant influence on the thermodynamic quantities of crystals at high temperatures. W has very high melting temperatures and can provide fundamental information on the equilibrium vacancy concentrations in metals and alloy metals with structured Body-Centered-Cubic (BCC) and their temperature dependences [20,21,22]. At P = 0.1 MPa, W has a BCC structure with lattice constant (a), a = 3.1649 × 10−10 m at T = 300 K and a melting point at T = 3690 K. The melting curve of W was studied by the optical method at P = 5 GPa and T = (4050 ± 200) K with dT/dP = 75 K/GPa [25] and up to P = 90 GPa and T ~ (4000 ± 100) K using the Laser-Heated Diamond Anvil Cell (LHDAC) for optical measurements [26].
In various studies, researchers determined the melting temperature (Tm) of a crystal only from the solid phase and applied the statistical moment method (SMM) [27,28,29,30,31,32,33]. From the SMM method, the stability temperature (TS) can be determined at different pressures, and the corresponding calculations to find Tm from TS, and then TS at crystalline state is defined by . Then, the isothermal compressibility of the crystal is equal to infinite. Several SMM calculations are more consistent with experiments than those obtained from other calculations.
In this research, the melting temperature, the jumps of volume, enthalpy and entropy at the melting point, and the Debye temperature for the BCC defective and perfect binary interstitial alloy by combining the SMM, the limiting condition of the absolute stability of the crystalline state, the Clapeyron-Clausius equation, the Debye model and the Gruneisen equation are studied. The theoretical results are numerically performed for alloy WSi, and some of our calculated results are compared with experiments and other calculations.
2. Content of Research
2.1. Melting Temperature, Jumping of Volume, Enthalpy and Entropy at Melting Point, and Debye Temperature of the BCC Defective and Perfect Binary Interstitial Alloy
In our model, the interstitial alloy AB with the concentration was applied. In it, the A metal has atoms in the center and peaks of cubic, while the atoms of B are in the center of cubic. With the parameters , the cohesive energy with the sphere center at the position of B, radii and are determined as follows [1,2,3,4,29,33]:
where , , and are the displacements of the ith atom in the direction of , the number of atoms on the ith coordination sphere with radius and the nearest neighbor distance between the interstitial atom B and the main metal A in the alloy at temperature T; and are the displacements of atom A1 (atom A in the body center of the cubic unit cell) from the equilibrium position at temperature T, determined from the minimum condition of the cohesive energy , and the nearest neighbor distance between the interstitial atom B and the main metal A in the alloy at temperature 0 K; is the interaction potential between atom A and atom B. The cohesive energy and the alloy parameters for atom A1 (atom A in the body center of the cubic unit cell) in the approximation of three coordination spheres with the sphere center at the position of A1 and the radius of the third coordination sphere are determined as follows [1,2,3,4,29,33]:
where is the nearest neighbor distance between atom A1 and the other atoms in the alloy.
The , are the cohesive energy and the alloy parameters for atom A2 (atom A in the peaks of the cubic unit cell) in the approximation of three coordination spheres, with A2 and radius of the third coordination sphere as follows [1,2,3,4,29,33]:
where and are the nearest neighbor distances between atom A2 and the other atoms in the alloy, and is determined from the minimum condition of the cohesive energy in the displacement of atom B from the equilibrium position at temperature T. In Equations (6)–(15), are the cohesive energy and the metal parameters for atom A in the clean metal A in the approximation of two coordination spheres with the sphere center at the position of A and radii and and have the following forms [29,33]:
The equations of state for the BCC alloy AB at P and T, and at P and T = 0 K, respectively, are determined by the following relations [29,33]:
where r1, , are the nearest neighbor distance between two atoms in the alloy, the volume of the cubic unit cell per atom, and the Boltzmann constant. If the form of the interaction potential between two atoms X (X = A, A1, A2, B) is known, from Equation (22) we can find the nearest neighbor distance between two r01X(P,0) and the alloy parameters for atom X at T = 0 K and P. From that, we can determine the displacement of atom X from the equilibrium position at T and P as follows [29,33]:
The nearest neighbor distance is given by the following relations [1,2,3,4]:
The mean nearest neighbor distance between two atoms A in the alloy is derived from the following expressions [1,2,3,4]:
where , , , and cB are the mean nearest neighbor distances between two atoms A in the clean metal A at pressure P and temperature 0 K; the region containing the interstitial atom C of the alloy at P and T = 0 K, and the concentration of the interstitial atoms B and nearest neighbor distance between two atoms A in the alloy at P and T and at P and T = 0 K. The Helmholtz free energy of the BCC perfect interstitial alloy AB with the condition , respectively, are the concentrations of atoms A and B) can be calculated with the following relations [1,2,3,4]:
where N is the number of atoms in the alloy, is the Helmholtz free energy of atom X in the clean material X, and is the configurational entropy of the interstitial alloy AB.
The concentrations of atoms A, A1, A2 and B are determined by the following relations:
From the condition of absolute stability limit expressed as:
and from the equation of state for the interstitial alloy AB expressed as:
the absolute stability temperature for the crystalline state can be derived in the following expression [3,4,29,32,33]:
where and are the Gruneisen parameters of the alloy. The right side of Equation (30) must be determined at TS. By solving Equation (30) the value of TS can be obtained.
The melting temperature Tm is derived from the absolute stability temperature TS by the following relation [3,4,29,32,33]:
where and Here, approximately is constant in the range of temperature from TS to Tm.
Temperature at zero pressure has the following relation [3,4]:
where are determined at
Temperature at pressure P can be calculated with the following relation [3,4]:
Here, are calculated at Approximately, the Equation (33) can be solved by the approximate iteration method applied at low pressure.
In the case of high pressure, temperature Tm at pressure P is calculated by the following relation [3,4]:
where , , G and are the melting T at P and at zero P, respectively; the bulk modulus and the isothermal elastic modulus are calculated by the following relations [1,2,3,4,29,33]:
where is the strain of the alloy, and are the Poisson ratios of alloy AB and the main metal A, respectively.
The equilibrium vacancy concentration of the alloy is determined from the minimum condition of the real Gibbs thermodynamic potential of the defective alloy AB, and has the following relation form:
where n and are the numbers of vacancies in the alloy and the change in the Gibbs thermodynamic potential when a vacancy is formulated, and is determined from the distribution of atomic concentrations as follows:
where Therefore, and according to ref. [34]:
At constant P and constant interstitial atom concentration, the melting temperature of a defective crystal is the function of the equilibrium vacancy concentration Approximately, the following relations can be applied [2,34]:
where , respectively, are the thermal expansion coefficients of atom X in alloy AB.
The jumping of volume at melting point for the alloy can be found from the following expression [35]:
where is a constant depending on the nature of the alloy and we take the value 0.01 as for metal [36], and is the mean displacement of the main metal atom A from the equilibrium position as in Equation (25). In order to determine the jumping of volume at pressure P and temperature T, it is necessary to determine at pressure P and temperature T. The alloy parameters are determined with respect to at pressure P and temperature T.
After finding the melting temperature Tm and the melting Tm (P), we can calculate the slope of this curve, and the derivative . If Tm, and are known, the jumping of enthalpy at melting point from the Clausius–Clapeyron equation can be derived according to the following relation:
and the jumping of entropy at melting point:
Firstly, we find the isothermal compressibility of the BCC interstitial alloy AB according to Equation (35) and the thermal expansion coefficient of the alloy according to Equation (40). The specific capacity at a constant volume of the alloy is given by the following relations [2,29,33]:
The Gruneisen parameter of the alloy has the following relation [29,33]:
Then, from the mean nearest neighbor distance the quantities and at pressure P and temperature T can be determined. For the BCC lattice, the following relation is known:
Graf et al. [37] proposed the following expression for the Gruneisen parameter:
where and q are material constants and q > 0. Therefore, by using the SMM, can be calculated from , and by using Equation (49) the value of q can be calculated.
According to the Debye model, the Gruneisen parameter is defined as follows:
where is the Debye frequency and is the Debye temperature.
By substituting the Gruneisen parameter from Equation (47) into Equation (48) and taking the integration, we derived the dependence of the Debye temperature at P on the Debye temperature , and the Gruneisen parameter at zero P, as well as the volume ratio [7]:
The Debye temperature TD0 of the alloy at zero pressure is given by the following relation:
The Debye frequency at zero pressure and temperature T is related to the Einstein frequency at zero pressure and temperature T by the following relation [38]:
where k(0,T) is the harmonic parameter of the alloy at zero pressure and temperature T. Therefore, it can be obtained through the following expression:
Equations (1)–(40) are used in our previous papers [1,2,3,4,5,6,7,8,9] on elastic, thermodynamic and melting properties of metals and interstitial alloys. Equations (41)–(52) only are used to study the jumps of volume, enthalpy and entropy, and the Debye temperature of metals. In this study, for the first time, we apply Equations (41)–(52) to study the jumps of volume, enthalpy and entropy, and the Debye temperature of the BCC interstitial alloy AB.
2.2. Numerical Results and Discussions for Alloy WSi
In order to study alloy WSi, we applied the Mie–Lennard-Jones (MLJ) pair interaction potential as follows [39]:
where D, r0, m and n are the depths of potential well corresponding to the equilibrium distance—they are determined empirically. Then, the potential parameters for the interaction W–Si are determined by the following relations [28]:
We find nW-Si, mW-Si by fitting the experimental data and the theoretical result for the Young modulus of the interstitial alloy WSi at room temperature. The Mie–Lennard-Jones potential’s parameters for the interactions of W–W and Si–Si are given in Table 1.

Table 1.
Mie–Lennard-Jones potential’s parameters for the interactions W–W and Si–Si.
Our computed results are summarised from Table 2, Table 3, Table 4, Table 5, Table 6, Table 7, Table 8 and Table 9 and are illustrated in Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8 and Figure 9. We calculated the silicon concentration and pressure of the volume, the isothermal compressibility, the Gruneisen parameter and the Debye temperature, the thermal expansion coefficient, the specific heat at constant volume in Table 2, Table 3, Table 4, Table 5, Table 6 and Table 7 and in Figure 1, Figure 2, Figure 3, Figure 4, Figure 5 and Figure 6. According to our obtained results, for WSi at the same temperature and silicon concentration when pressure increases, the volume, the isothermal compressibility, the thermal expansion coefficient, the Gruneisen parameter decreases, the Debye temperature increases, and the specific heat at constant volume. For WSi at the same temperature and pressure when the silicon concentration increases, the volume, the specific heat at constant volume, the Gruneisen parameter increases, the thermal expansion coefficient, the Debye temperature decreases and the isothermal compressibility.
Table 2.
Si concentration and pressure dependence of volume V for WSi at T = 300 K.
Table 3.
Si concentration and pressure dependence of isothermal compressibility for WSi at T = 300 K.
Table 4.
Si concentration and pressure dependence of thermal expansion coefficient for WSi at T = 300 K.
Table 5.
Si concentration and pressure dependence of specific capacity at constant volume for WSi at T = 300 K.
Table 6.
Si concentration and pressure dependence of the Gruneisen parameter γG. for WSi at T = 300 K.
Table 7.
Si concentration and pressure dependence of Debye temperature for WSi at T = 300 K.
Table 8.
Si concentration and pressure dependence of volume jumping for WSi at T = 300 K.
Table 9.
Jumping of volume, and enthalpy and entropy at melting point (using the melting curve calculated by the SMM for the defective model).
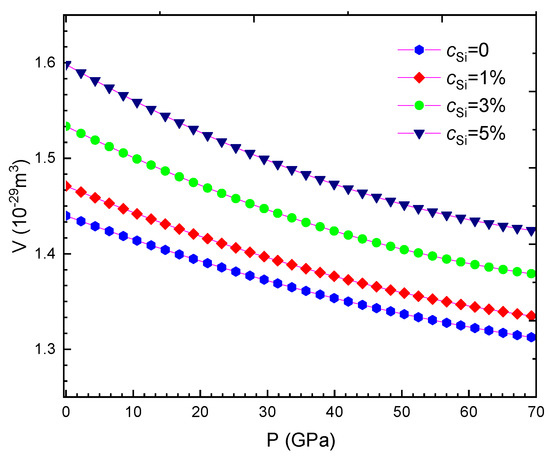
Figure 1.
For WSi at T = 300 K.
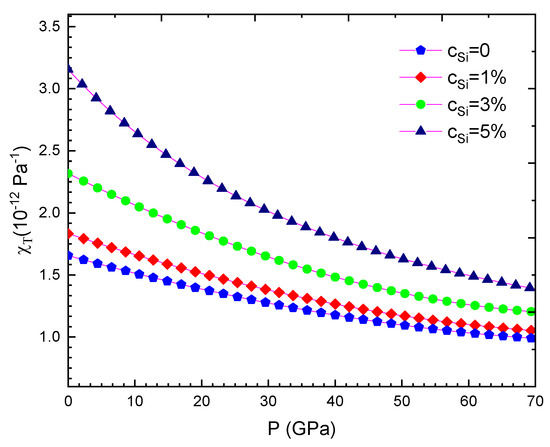
Figure 2.
For WSi at T = 300 K.
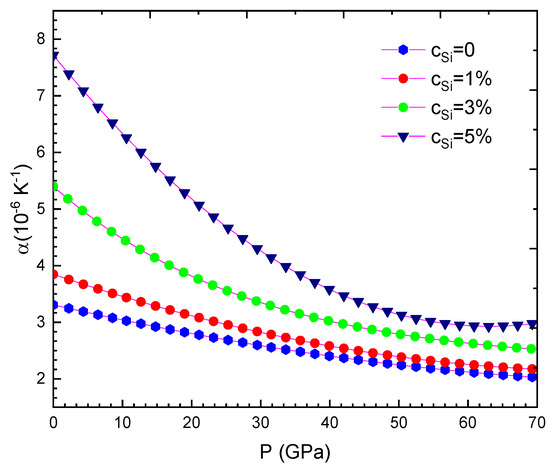
Figure 3.
For WSi at T = 300 K.
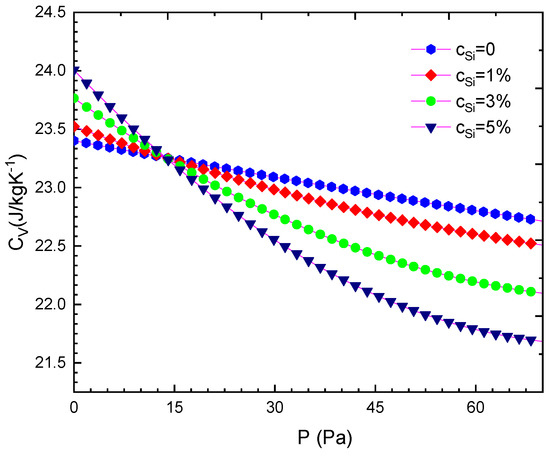
Figure 4.
For WSi at T = 300 K.
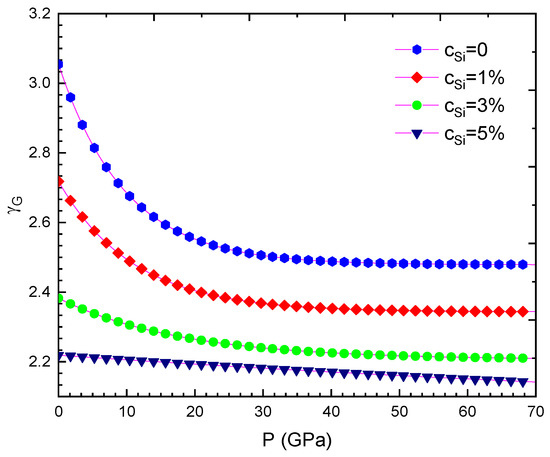
Figure 5.
For WSi at T = 300 K.
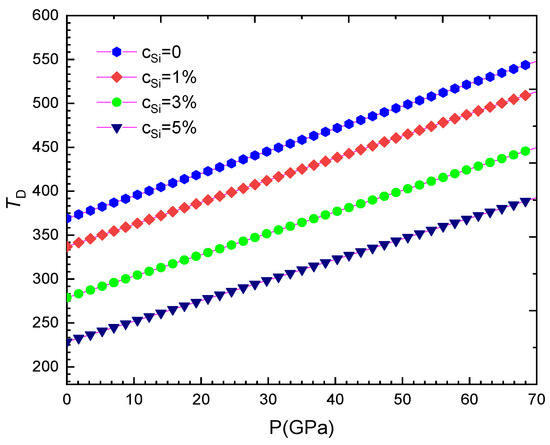
Figure 6.
For WSi at T = 300 K.
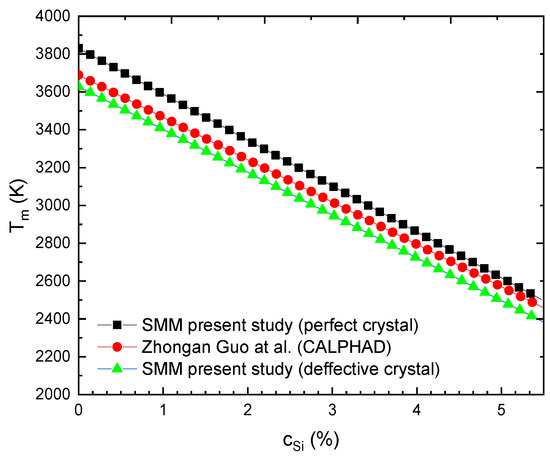
Figure 7.
For WSi at P = 0 obtained from the SMM for the perfect model [3], the SMM for the defective model [3] and CALPHAD [40].
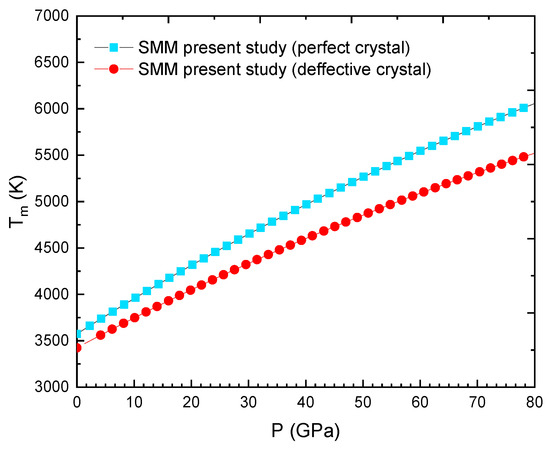
Figure 8.
Tm(P) for WSi at cSi = 1% obtained from the SMM for the perfect model and the SMM for the defective model [3].
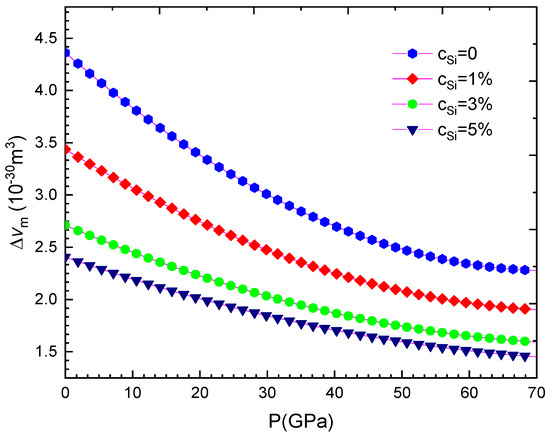
Figure 9.
For WSi at T = 300 K.
According to Figure 7 [3], when cSi increases from 0 to 5.5%, the Tm of WSi decreases (from 3810 to T = 2459 K) from the SMM for the perfect model, (from T = 3609 K to T = 2366 K) from the SMM for the defective model and from T = 3695 K to T = 2460 K from CALPHAD [40]. The melting slope for WSi at zero pressure is 245.6 K/% from the SMM for the perfect alloy, 226 K/% from the SMM for the defective alloy and 224.5K/% from CALPHAD [40]. The SMM calculations for the melting slope of the defective alloy are in good agreement with the CALPHAD calculations.
Figure 8 shows the melting curve of WSi at cSi = 1% obtained from the SMM for the perfect model and the SMM for the defective model [3]. In the range of pressure from P = 0 GPa to P = 80 GPa, the of the perfect W0.99Si0.01 increases from T = 3564 K to T = 6088 K and the of the defective W0.99Si0.01 increases from T = 3383 K to T = 5534 K.
3. Conclusions
The analytic expressions for structural and thermodynamic quantities such as the alloy parameters, the mean nearest neighbor distance, the melting temperature, the Helmholtz free energy, the equilibrium vacancy concentration, the cohesive energy, enthalpy and entropy, the isothermal compressibility, the limiting temperature of absolute stability for the crystalline state, the thermal expansion coefficient, the jumps of volume, the heat capacity at constant volume, the Gruneisen parameter and the Debye temperature for the defective and perfect binary interstitial alloy with a BCC structure are derived by combining the limiting condition of the absolute stability of the crystalline state with the statistical moment method, the Clapeyron–Clausius equation, the Debye model and the Gruneisen equation. Our numerical calculations of the melting curve and the relation are carried out for alloy WSi under a pressure of up to P = 80 GPa. Our calculated melting curve and relation between the melting temperature and the silicon concentration for WSi are in good agreement with other calculations. Our calculations for the melting curve, the jumping of volume, enthalpy and entropy, and the Debye temperature for WSi predict and orient experimental results in the future.
Author Contributions
H.N.Q.: conceptualization, methodology, validation, investigation, writing—original draft preparation, data curation. H.N.D.: writing—original draft preparation. D.N.T.: writing—original draft preparation, writing—review and editing. V.C.L.: writing—original draft preparation. Ş.Ţ.: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hoc, N.Q.; Tinh, B.D.; Hien, N.D. Elastic moduli and elastic constants of interstitial alloy AuCuSi with FCC structure under pressure. High Temp. Mater. Proc. 2019, 38, 264–272. [Google Scholar] [CrossRef]
- Tinh, B.D.; Hoc, N.Q.; Vinh, D.Q.; Cuong, T.D.; Hien, N.D. Thermodynamic and elastic properties of interstitial alloy FeC with BCC structure at zero pressure. Adv. Mater. Sci. Eng. 2018, 5251741. [Google Scholar] [CrossRef]
- Hoc, N.Q.; Cuong, T.D.; Tinh, B.D.; Viet, L.H. Study on the melting of defective interstitial alloys TaSi and WSi with BCC structure. J. Korean Phys. Soc. 2019, 71, 801–805. [Google Scholar] [CrossRef]
- Tuyen, L.T.C.; Học, N.Q.; Tinh, B.D.; Vinh, D.Q.; Cuong, T.D. Study on the melting of interstitial alloys FeH and FeC with BCC structure under pressure. Chin. J. Phys. 2019, 59, 1–9. [Google Scholar] [CrossRef]
- Cuong, T.D.; Hoc, N.Q.; Phan, A.D. Application of the statistical moment method to melting properties of ternary alloys with FCC structure. J. Appl. Phys. 2019, 125, 215306. [Google Scholar] [CrossRef]
- Hoc, N.Q.; Viet, L.H.; Dung, N.T. On the melting of defective FCC interstitial alloy FeC under pressure up to 100 GPa. J. Electron. Mater. 2020, 49, 910–916. [Google Scholar] [CrossRef]
- Hoc, N.Q.; Tinh, B.D.; Hien, N.D. Influence of temperature and pressure on the electrical resistivity of gold and copper up to 1350K and 100GPa. Mater. Res. Bull. 2020, 128, 110874. [Google Scholar] [CrossRef]
- Hoc, N.Q.; Tinh, B.D.; Hien, N.D. Stress-strain curve of FCC interstitial alloy AuSi under pressure. Rom. J. Phys. 2020, 65, 608. [Google Scholar]
- Hoc, N.Q.; Tinh, B.D.; Coman, G.; Hien, N.D. On the melting of alloys FeX (X = Ni, Ta, Nb, Cr) under pressure up to 5 GPa. J. Phys. Soc. Jpn. 2020, 89, 114602. [Google Scholar] [CrossRef]
- Dung, N.T. Influence of impurity concentration, atomic number, temperature and tempering time on microstructure and phase transformation of Ni1-xFex (x = 0:1, 0.3, 0.5) nanoparticles. Mod. Phys. Lett. B 2018, 32, 1850204. [Google Scholar] [CrossRef]
- Tuan, T.Q.; Dung, N.T. Effect of heating rate, impurity concentration of Cu, atomic number, temperatures, time annealing temperature on the structure, crystallization temperature and crystallization process of Ni1-xCux bulk; x = 0.1, 0.3, 0.5, 0.7. Int. J. Mod. Phys. B 2018, 32, 1830009. [Google Scholar] [CrossRef]
- Nguyen-Trong, D.; Pham-Huu, K.; Nguyen-Tri, P. Simulation on the factors affecting the crystallization process of FeNi alloy by molecular dynamics. ACS Omega 2019, 4, 14605–14612. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Trong, D.; Nguyen-Chinh, C.; Duong-Quoc, V. Study on the effect of doping on lattice constant and electronic structure of bulk AuCu by the density functional theory. J. Multiscale Model. 2020, 11, 2030001. [Google Scholar] [CrossRef]
- Nguyen-Trong, D.; Nguyen-Tri, P. Factors affecting the structure, phase transition and crystallization process of AlNi nanoparticles. J. Alloy. Compd. 2020, 812, 152133. [Google Scholar] [CrossRef]
- Nguyen-Trong, D.; Nguyen-Tri, P. Molecular dynamic study on factors influencing the structure, phase transition and crystallization process of NiCu6912 nanoparticle. Mater. Chem. Phys. 2020, 250, 123075. [Google Scholar] [CrossRef]
- Long, V.C.; Quoc, V.D.; Trong, D.N. Ab initio calculations on the structural and electronic properties of AgAu alloys. ACS Omega 2020, 5, 31391–31397. [Google Scholar] [CrossRef] [PubMed]
- Trong, D.N.; Long, V.C.; Ţălu, Ş. The structure and crystallizing process of NiAu alloy: A molecular dynamics simulation method. J. Compos. Sci. 2021, 5, 18. [Google Scholar] [CrossRef]
- Nguyen-Trong, D. Z-AXIS deformation method to investigate the influence of system size, structure phase transition on mechanical properties of bulk nickel. Mater. Chem. Phys. 2020, 252, 123275. [Google Scholar] [CrossRef]
- Quoc, T.T.; Trong, D.N.; Ţălu, Ş. Study on the influence of factors on the structure and mechanical properties of amorphous aluminium by molecular dynamics method. Adv. Mater. Sci. Eng. 2021, 5564644, 1–10. [Google Scholar] [CrossRef]
- Kraftmakher, Y. Equilibrium vacancy and thermal property of metals. Phys. Rep. 1998, 299, 79–198. [Google Scholar] [CrossRef]
- Siegel, R.W. Vacancy concentrations in metals. J. Nucl. Mater. 1978, 69–70, 117–146. [Google Scholar] [CrossRef]
- Maier, K.; Peo, M.; Saile, B.; Schaefer, H.E.; Seeger, A. High-temperature positron annihilation and vacancy formation in refractory metals. Philos. Mag. A 1979, 40, 701. [Google Scholar] [CrossRef]
- Pamato, M.G.; Wood, I.G.; Dobson, D.P.; Hunt, S.A.; Vocadlo, L. The thermal expansion of gold: Point defect concentrations and pre-melting I a face-centered cubic metal. J. Appl. Crystallogr. 2018, 51, 470–480. [Google Scholar] [CrossRef]
- Hung, V.V.; Hai, N.T. Investigation of the thermodynamic properties of anharmonic crystals with defects and influence of anharmonicity in EXAFs by the moment method. Int. Mod. Phys. B 1998, 12, 191. [Google Scholar] [CrossRef]
- Vereshchagin, L.F.; Fateeva, N.S. Melting temperatures of refractory metals at high pressures. High Temp. High Press. 1977, 9, 619–628. [Google Scholar]
- Errandonea, D.; Schwager, B.; Ditz, R.; Gessmann, C.; Boehler, R.; Ross, M. Systematics of transition-metal melting. Phys. Rev. B 2001, 63, 132104. [Google Scholar] [CrossRef]
- Saxen, S.K.; Zhang, J. Thermodynamical and pressure-volume-temperature systematics of data on solids, examples: Tungsten and MgO. Phys. Chem. Miner. 1990, 17, 45–51. [Google Scholar] [CrossRef]
- Moriarty, J.A. Ultra-high pressure structural phase transitions in Cr, Mo and W. Phys. Rev. B 1992, 45, 2004–2014. [Google Scholar] [CrossRef]
- Tang, N.; Hung, V.V. Investigation of the thermodynamic properties of anharmonic crystals by the momentum method, (I) General results for FCC crystals. Phys. Stat. Sol. 1988, 149, 511. [Google Scholar] [CrossRef]
- Tang, N.; Hung, V.V. Investigation of the thermodynamic properties of anharmonic crystals by the momentum method, (II) Comparison of calculations with experiments for inert gas crystals. Phys. Stat. Sol. 1990, 161, 165. [Google Scholar] [CrossRef]
- Tang, N.; Hung, V.V. Investigation of the thermodynamic properties of anharmonic crystals by the momentum method, (III) Thermodynamic properties of the crystals at various pressures. Phys. Stat. Sol. 1990, 162, 371. [Google Scholar] [CrossRef]
- Tang, N.; Hung, V.V. Investigation of the thermodynamic properties of anharmonic crystals by the momentum method, (IV) The limiting of absolute stability and the melting temperature of crystals. Phys. Stat. Sol. 1990, 162, 379. [Google Scholar] [CrossRef]
- Hung, V.V. Statistical Moment Method in Studying Elastic and Thermodynamic Properties of Crystals; HNUE Publishing House: Hanoi, Vietnam, 2009. [Google Scholar]
- Cuong, T.D.; Anh, P.D. Modification of the statistical moment method for the high-pressure melting curve by the inclusion of thermal vacancies. Vacuum 2020, 179, 109444. [Google Scholar] [CrossRef]
- Hung, V.V. Investigation of the change in volume, entropy and specific heat for metals on melting. In Proceedings of the 22nd National Conference of Theoretical Physics, Do Son, Vietnam, 3–5 August 1997; pp. 199–203. [Google Scholar]
- Good, R.J.; Hope, C.J. New combining rule for intermolecular distances in intermolecular potential functions. J. Chem. Phys. 1970, 53, 540–543. [Google Scholar] [CrossRef]
- Graf, M.J.; Greeff, C.W.; Boettger, J.C. High-pressure Debye-Waller and Grüneisen parameters of gold and copper. AIP Conf. Proc. 2004, 706, 65–68. [Google Scholar]
- Girifalco, L.A. Statistical Physics of Materials; John Wiley & Sons: Hoboken, NJ, USA, 1973. [Google Scholar]
- Magomedov, M.N. On calculating the Debye temperature and the Gruneisen parameter. Z. Fiz. Khimii 1987, 61, 1003–1009. (In Russian) [Google Scholar]
- Guo, Z.; Yuan, W.; Sun, Y.; Cai, Z.; Qiao, Z. Thermodynamic assessment of the Si-Ta and Si-W systems. J. Phase Equilibria Diffus. 2000, 30, 564–570. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).