
Progress in Studies of Disentangled Polymers and Composites



Abstract
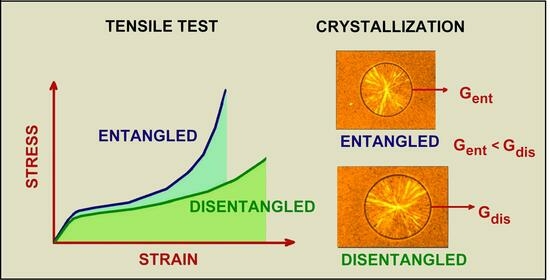
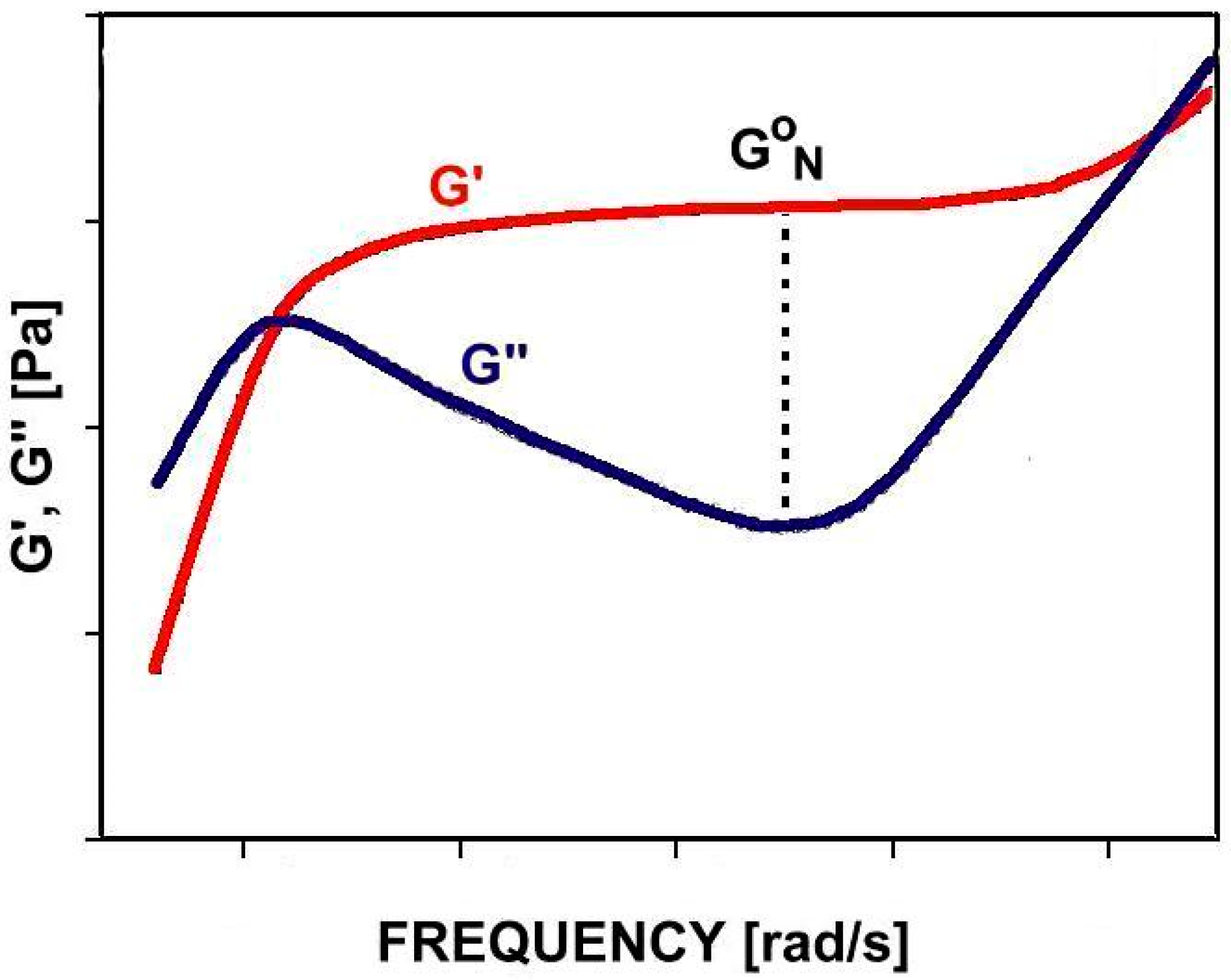
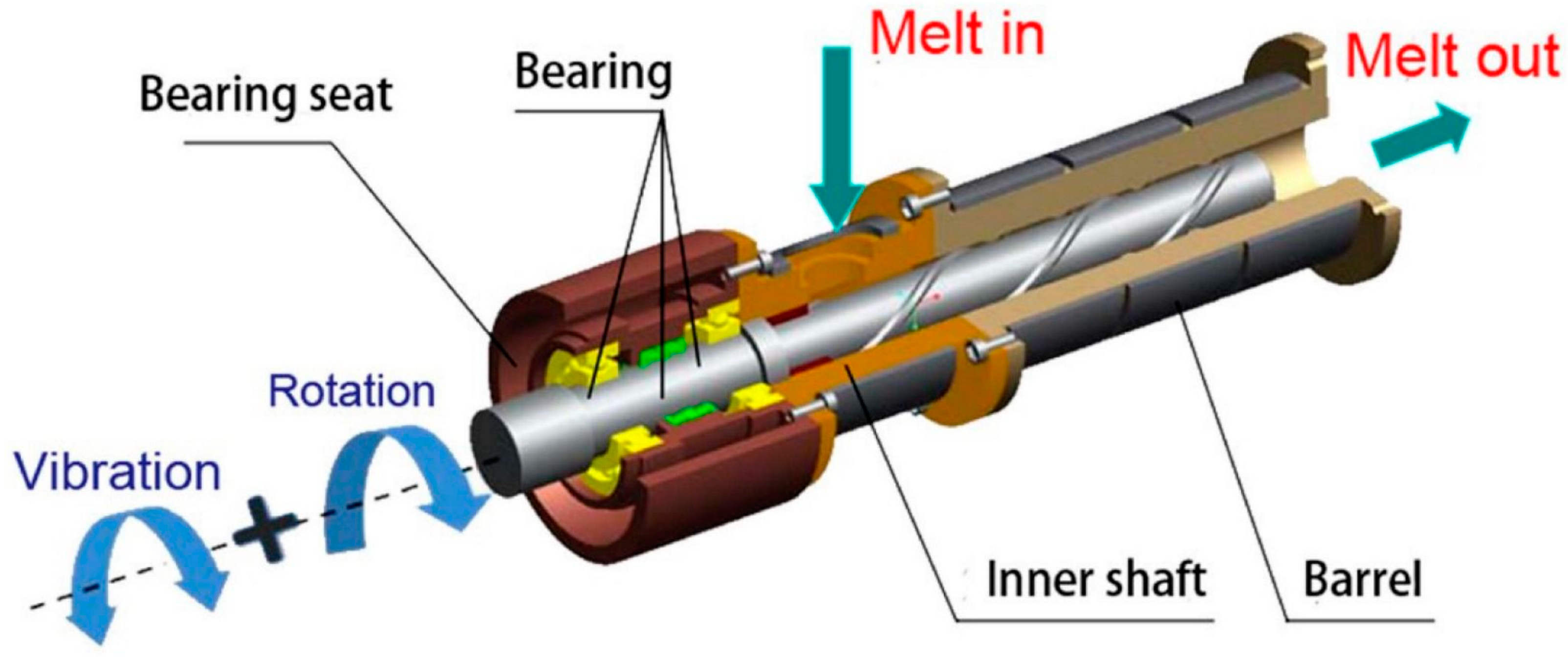
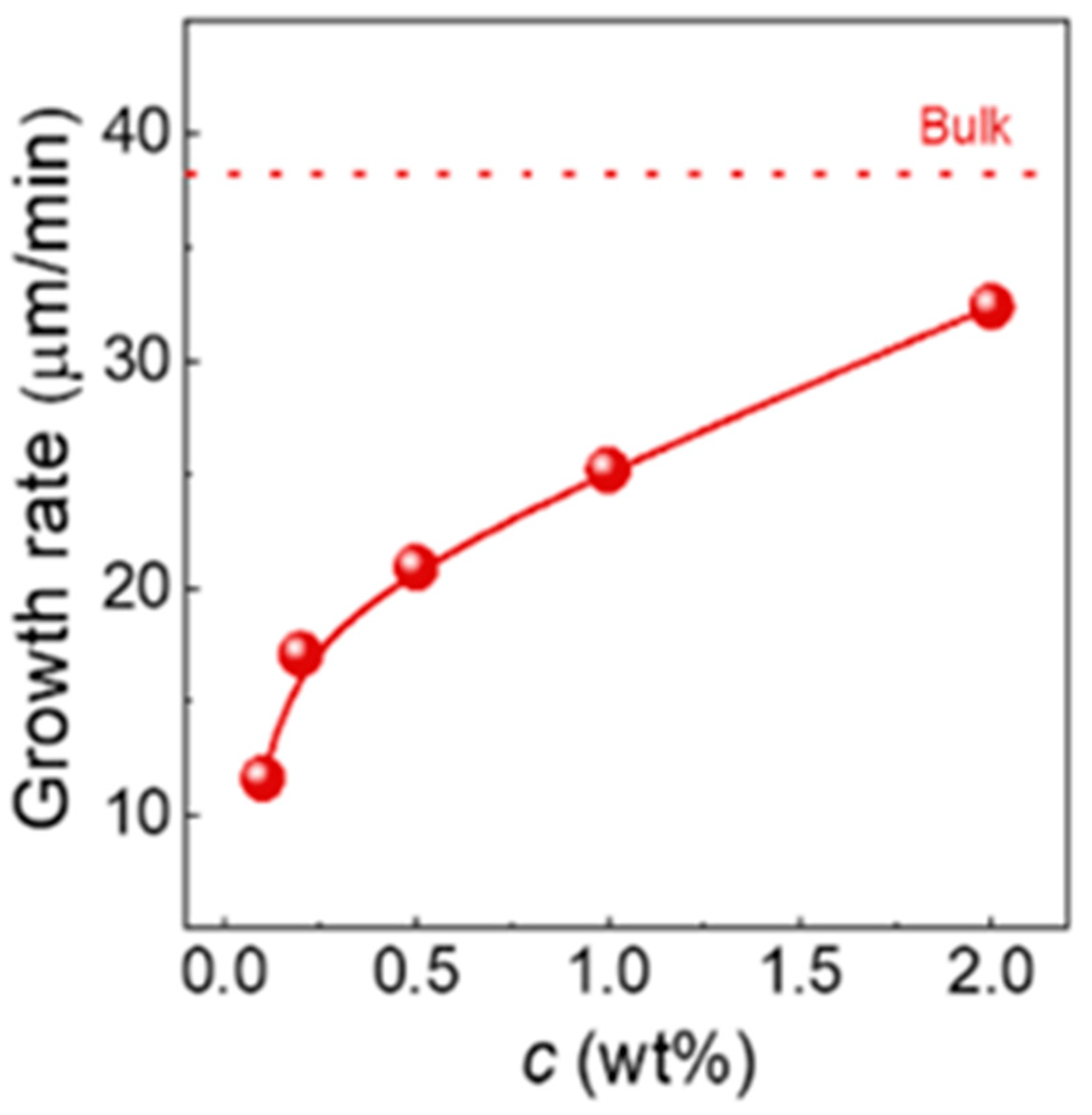
:
1. Entanglements in Polymers
2. Disentangling of Polymers
3. Change in Properties Due to Polymer Disentangling
4. Special Case: Ultra-High-Molecular-Weight Polyethylene
5. Re-Entangling Macromolecules
6. Composites Created Using Disentangled Polymers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Matrix | Filler | Contents of Filler (wt.%) | Method of Dispersion | Disentangling of Matrix | Reference |
|---|---|---|---|---|---|
| PMMA | Grafted SiO2, 11 nm | 2 | Solution | During mixing | [143] |
| PS | Grafted polyoxometalate, 0.5–6 nm | 1–5 | Solution | During mixing | [140] |
| UHMWPE | POSS | 0.2–3 | During polymerization | Previously disentangled | [63] |
| UHMWPE | POSS, 0.8 nm | 0.1–1 | Melt mixing | During mixing | [106] |
| UHMWPE | TiO2 | 0.1–0.5 | Solution | During mixing | [145] |
| UHMWPE | Gold, nano | 1 | Solution | Previously disentangled | [149] |
| PP | Graphene | 0.1–4 | Shear in melt | During mixing | [147] |
| PP | Boron nitride, 1, 5, 27 µm | 35 | Steady-state shear | During shear | [148] |
| PP | Al2O3, 78 nm | 1 | Melt mixing | Previously disentangled | [151] |
| Waterborne acrylic coatings | TiO2 | 1–3 | Solution | During mixing | [146] |
| PLA | MWCNT | 0.1–1 | Melt mixing | Previously disentangled | [152] |
7. Concludings Remarks and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Watanabe, H. Viscoelasticity and dynamics of entangled polymers. Prog. Polym. Sci. 1999, 24, 1253–1403. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, C.; Zhang, Y.; Zhao, J. Study on the chain entanglement of polyvinyl alcohol fiber during the dry-jet wet spinning process. Fibers Polym. 2015, 16, 345–353. [Google Scholar] [CrossRef]
- Wool, R.P. Polymer entanglements. Macromolecules 1993, 26, 1564–1569. [Google Scholar] [CrossRef]
- Pearson, D.S.; Ver Strate, G.; Von Meerwall, E.; Schilling, F.C. Viscosity and self-diffusion coefficient of linear polyethylene. Macromolecules 1987, 20, 1133–1141. [Google Scholar] [CrossRef]
- Berry, G.C.; Fox, T.G. The viscosity of polymers and their concentrated solutions. Adv. Polym. Sci. 1968, 5, 261–357. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity, 3rd ed.; Clarendon Press: Oxford, UK, 1975; pp. 64–65. [Google Scholar]
- Ward, M.; Sweeney, J. Mechanical Properties of Solid Polymers; John Wiley & Sons Ltd.: Chichester, UK, 2013; p. 72. [Google Scholar]
- Haward, R.N. Strain hardening of thermoplastics. Macromolecules 1993, 26, 5860–5869. [Google Scholar] [CrossRef]
- Deplancke, T.; Lame, O.; Rousset, F.; Seguela, F.; Vigier, G. Mechanisms of Chain Reentanglement During the Sintering of UHMWPE Nascent Powder: Effect of Molecular Weight. Macromolecules 2015, 48, 5328–5338. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; Wiley: New York, NY, USA, 1980; p. 372. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon: Oxford, UK, 1986. [Google Scholar]
- Liu, C.; He, J.; van Ruymbeke, E.; Keunings, R.; Bailly, C. Evaluation of different methods for the determination of the plateau modulus and the entanglement molecular weight. Polymer 2006, 47, 4461–4479. [Google Scholar] [CrossRef]
- Eckstein, A.; Suhm, J.; Friedrich, C.; Maier, R.; Sassmannshausen, J.; Bochmann, M.; Mulhaupt, R. Determination of Plateau Moduli and Entanglement Molecular Weights of Isotactic, Syndiotactic, and Atactic Polypropylenes Synthesized with Metallocene Catalysts. Macromolecules 1998, 31, 1335–1340. [Google Scholar] [CrossRef]
- Kong, D.-H.; Yang, M.-H.; Zhang, X.-S.; Du, Z.-C.; Fu, Q.; Gao, X.-Q.; Gong, J.W. Control of Polymer Properties by Entanglement: A Review. Macromol. Mater. Eng. 2021, 306, 2100536. [Google Scholar] [CrossRef]
- Wu, S. Chain structure an entanglement. J. Polym. Sci. Polym. Phys. 1989, 27, 723–741. [Google Scholar] [CrossRef]
- Wu, S.; Beckerbauer, R. Effect of Tacticity on Chain Entanglement in Poly(methyl methacrylate). Polym. J. 1992, 24, 1437–1442. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. The connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Colby, R.H. Physical Properties of Polymers Handbook, 2nd ed.; Mark, J.E., Ed.; Springer Science: New York, NY, USA, 2007; pp. 447–454. [Google Scholar]
- Pawlak, A. The Entanglements of Macromolecules and Their Influence on the Properties of Polymers. Macromol. Chem. Phys. 2019, 220, 1900043. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, Z.; Lin, X.; Zhang, C.; Tanaka, K.; Zuo, B.; Zhang, W.; Wang, X. Suppressed Chain Entanglement Induced by Thickness of Ultrathin Polystyrene Films. Macromolecules 2021, 54, 3735–3743. [Google Scholar] [CrossRef]
- Doi, M. Viscoelastic and rheological properties. In Materials Science and Technology. A Comprehensive Treatment. Volume 12. Structure and Properties of Polymers; Cahn, R.W., Haasen, P., Kramer, E.J., Thomas, E.L., Eds.; VCH Verlag: Weinheim, 1993; pp. 389–425. [Google Scholar]
- Zulli, F.; Giordano, M.; Andreozzi, L. Onset of entanglement and reptation in melts of linear homopolymers: Consistent rheological simulations of experiments from oligomers to high polymers. Rheol. Acta 2015, 54, 185–205. [Google Scholar] [CrossRef]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. 4. Rheological properties. J. Chem. Soc. Faraday Trans. II 1979, 75, 38–54. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. Dynamics of concentrated polymer systems. Part 1—Brownian motion in the equilibrium state. J. Chem. Soc. Faraday Trans. 2 1978, 74, 1789–1801. [Google Scholar] [CrossRef]
- Talebi, S. Disentangled Polyethylene with Sharp Molar Mass Distribution: Implications for Sintering. Ph.D. Thesis, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2008. [Google Scholar]
- Robertson, C.G.; Warren, S.; Plazek, D.J.; Roland, C.M. Reentanglement kinetics in sheared polybutadiene solutions. Macromolecules 2004, 37, 10018–10022. [Google Scholar] [CrossRef]
- Costanzo, S.; Huang, Q.; Ianniruberto, G.; Marrucci, G.; Hassager, O.; Vlassopoulos, D. Shear and extensional rheology of polystyrene melts and solutions with the same number of entanglements. Macromolecules 2016, 49, 3925–3935. [Google Scholar] [CrossRef]
- Ransom, T.C.; Debjani, R.; Puskas, J.E.; Kaszas, G.; Roland, M.C. Molecular Weight Dependence of the Viscosity of Highly Entangled Polyisobutylene. Macromolecules 2019, 52, 5177–5182. [Google Scholar] [CrossRef]
- Hoy, R.; Robbins, M. Strain hardening of polymer glasses: Effect of entanglement density, temperature, and rate. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 3487–3500. [Google Scholar] [CrossRef]
- Hiss, R.; Hobeika, S.; Lynn, C.; Strobl, G. Network stretching, slip processes, and fragmentation of crystallites during uniaxial drawing of polyethylene and related copolymers. A comparative study. Macromolecules 1999, 32, 4390–4403. [Google Scholar] [CrossRef]
- Zuo, F.; Keum, J.K.; Chen, X.; Hsiao, B.S.; Chen, H.; Lai, S.Y.; Wevers, R.; Li, J. The role of interlamellar chain entanglement in deformation-induced structure changes during uniaxial stretching of isotactic polypropylene. Polymer 2007, 48, 6867–6880. [Google Scholar] [CrossRef]
- Haward, R.N. Strain Hardening of High Density Polyethylene. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 1090–1099. [Google Scholar] [CrossRef]
- van Melick, H.G.H.; Govaert, L.E.; Meijer, H.E.H. On the origin of strain hardening in glassy polymers. Polymer 2003, 44, 2493–2502. [Google Scholar] [CrossRef]
- Kennedy, M.A.; Peacock, A.J.; Mandelkern, L. Tensile properties of crystalline polymers: Linear polyethylene. Macromolecules 1994, 27, 5297–5310. [Google Scholar] [CrossRef]
- Bartczak, Z.; Kozanecki, M. Influence of molecular parameters on high-strain deformation of polyethylene in the plane-strain compression. Part I. Stress-strain behavior. Polymer 2005, 46, 8210–8221. [Google Scholar] [CrossRef]
- Schrauwen, B.A.G.; Janssen, R.P.M.; Govaert, L.E.; Meijer, H.E.H. Intrinsic deformation behavior of semicrystalline polymers. Macromolecules 2004, 37, 6069–6078. [Google Scholar] [CrossRef]
- Bartczak, Z.; Grala, M.; Richaud, E.; Gadzinowska, K. Erosion of the molecular network in the amorphous layers of polyethylene upon high-strain deformation. Polymer 2016, 99, 552–565. [Google Scholar] [CrossRef]
- Donald, A.M.; Kramer, E.J. Effects of molecular entanglements on craze microstructure in glassy polymers. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 899–909. [Google Scholar] [CrossRef]
- Donald, A.M.; Kramer, E.J. The competition between shear deformation and crazing in glassy polymers. J. Mater. Sci. 1982, 17, 1871–1879. [Google Scholar] [CrossRef]
- Garcıa-Franco, C.A.; Harrington, B.A.; Lohse, D.J. On the rheology of ethylene–octene copolymers. Rheol. Acta. 2005, 44, 591–599. [Google Scholar] [CrossRef]
- Yamazaki, S.; Hikosaka, M.; Gu, F.; Ghosh, S.K.; Arakaki, M.; Toda, A. Effect of entanglement on nucleation rate of polyethylene. Polymer J. 2001, 33, 906–908. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Zhong, L.W.; Yang, S.; Liang, D.H.; Chen, E.Q. Memory effect on solution crystallization of high molecular weight poly(ethylene oxide). Polymer 2012, 53, 3621–3628. [Google Scholar] [CrossRef]
- Robelin-Souffache, E.; Rault, J. Origin of the long period and crystallinity in quenched semicrystalline polymers. Macromoelcules 1989, 22, 3581–3594. [Google Scholar] [CrossRef]
- Flory, P.J.; Yoon, D.Y. Molecular morphology in semicrystalline polymers. Nature 1978, 272, 226–229. [Google Scholar] [CrossRef]
- Jani, F.; Sepahi, A.; Afzali, S.K.; Moyad, S.H. Experimental study on the effect of molecular weight and chemical composition distribution on the mechanical response of high-density polyethylene. Polym. Eng. Sci. 2023, 63, 176–188. [Google Scholar] [CrossRef]
- Peters, G.W.M.; Balzano, L.; Steenbakkers, R.J.A. Flow-induced Crystallization. In Handbook of Polymer Crystallization; Piorkowska, E., Rutledge, G.C., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 399–431, Chapter 14. [Google Scholar]
- Martins, J.A.; Zhang, W.; Brito, A.M. Origin of the melt memory effect in polymer crystallization. Polymer 2010, 51, 4185–4194. [Google Scholar] [CrossRef]
- Song, L. Effect of Entanglement Density on Mechanical Properties and Deformation Behavior of Rubber-Modified PVC/α-MSAN Blends. Ind. Eng. Chem. Res. 2013, 52, 12567–12573. [Google Scholar] [CrossRef]
- Prest, W.M.; Porter, R.S. Rheological properties of poly(2, 6-dimethylphenylene oxide)-polystyrene blends. J. Polym. Sci. (A-2) 1972, 10, 1639–1655. [Google Scholar] [CrossRef]
- Hao, X.; Kaschta, J.; Liu, X.; Pan, Y.; Schubert, D.W. Entanglement network formed in miscible PLA/PMMA blends and its role in rheological and thermo-mechanical properties of the blends. Polymer 2015, 80, 38–45. [Google Scholar] [CrossRef]
- Song, L.; Zhang, Y.; Ren, J.; Li, Y.; Yang, B.; Xing, E.; Wang, Y.; Shi, Y. Effect of Entanglement Density on Mechanical Properties and the Deformation Mechanism of Rubber-Modified PPO/PS Blends. Macromol. Mater. Eng. 2022, 307, 2200325. [Google Scholar] [CrossRef]
- Xie, M.; Li, M. Viscosity reduction and disentanglement in ultrahigh molecular weight polyethylene melt: Effect of blending with polypropylene and poly(ethylene glycol). Eur. Polym. J. 2007, 43, 3480–3487. [Google Scholar] [CrossRef]
- Huang, Y.F.; Xu, J.Z.; Zhang, Z.C.; Xu, L.; Li, L.B.; Li, J.F.; Li, Z.M. Melt processing and structural manipulation of highly linear disentangled ultrahigh molecular weight polyethylene. Chem. Eng. J. 2017, 315, 132–141. [Google Scholar] [CrossRef]
- Pandey, A.; Champouret, Y.; Rastogi, S. Heterogeneity in the Distribution of Entanglement Density during Polymerization in Disentangled Ultrahigh Molecular Weight Polyethylene. Macromolecules 2011, 44, 4952–4960. [Google Scholar] [CrossRef]
- Rastogi, S.; Kurelec, L.; Cuijpers, J.; Lippits, D.; Wimmer, M.; Lemstra, P.J. Disentangled state in polymer melts; a route to ultimate physical and mechanical properties. Macromol. Mater. Eng. 2003, 288, 964–970. [Google Scholar] [CrossRef]
- Westfahl, H., Jr.; Cardoso, M.B. Accessing the hidden lamellar nanostructure of semi-crystalline nascent polymers by small-angle X-ray scattering contrast variation. J. Appl. Crystall. 2011, 44, 1123–1126. [Google Scholar] [CrossRef]
- Yamazaki, S.; Gu, F.; Watanabe, K.; Okada, K.; Toda, A.; Hikosaka, M. Two-step formation of entanglement from disentangled polymer melt detected by using nucleation rate. Polymer 2006, 47, 6422–6428. [Google Scholar] [CrossRef]
- Wang, B.; Cavallo, D.; Zhang, X.; Zhang, B.; Chen, J. Evolution of chain entanglements under large amplitude oscillatory shear flow and its effect on crystallization of isotactic polypropylene. Polymer 2020, 186, 121899. [Google Scholar] [CrossRef]
- Bu, H.S.; Gu, F.M.; Bao, L.; Chen, M. Influence of entanglements on crystallization of macromolecules. Macromolecules 1998, 31, 7108–7110. [Google Scholar] [CrossRef]
- Hao, H.; Liu, R.J.; Zhao, Y.L. Concentration Dependence of Crystalline Poly(L-lactide) Prepared by Freeze-drying Solutions. Polym. Polym. Compos. 2009, 17, 31–35. [Google Scholar] [CrossRef]
- Gu, F.M.; Bu, H.S.; Zhang, Z. A unique morphology of freeze-dried poly(ethylene oxide) and its transformation. Polymer 2000, 41, 7605–7609. [Google Scholar] [CrossRef]
- Li, W.; Guan, C.; Xu, J.; Mu, J.; Gong, D.; Chen, Z.R.; Zhou, Q. Disentangled UHMWPE/POSS nanocomposites prepared by ethylene in situ polymerization. Polymer 2014, 55, 1792–1798. [Google Scholar] [CrossRef]
- Huang, B.; Ito, M.; Kanamoto, T. Deformation mechanism of amorphous poly(ethylene terephthalate) as function of molecular weight and entanglements. Polymer 1994, 35, 1210–1216. [Google Scholar] [CrossRef]
- Sun, Q.; Fu, Q.; Xue, G.; Chen, W. Crystallization Behavior of Syndiotactic Poly(propylene) Freeze-Dried from Toluene at very Dilute Concentration. Macromol. Rapid Comm. 2001, 22, 1182–1185. [Google Scholar] [CrossRef]
- Xue, G.; Wang, Y.; Liu, S.; Liao, Y.T. FT-IR Study of Concentration Dependence for Crystallization of Isotactic Polystyrene Arising from Freeze-Drying Dilute Solutions. Macromolecules 1995, 28, 4344–4346. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ohta, T. The influence of chain entanglement density on ultra-drawing behavior of ultra-high-molecular-weight polypropylene in the gel-casting method. Polymer 2008, 49, 621–627. [Google Scholar] [CrossRef]
- Fu, J.; Wang, Y.; Shen, K.; Fu, Q.; Zhang, J. Insight into Shear-Induced Modification for Improving Processability of Polymers: Effect of Shear Rate on the Evolution of Entanglement State. J. Polym. Sci. B Polym. Phys. 2019, 57, 598–606. [Google Scholar] [CrossRef]
- Wang, B.; Cavallo, D.; Chen, J. Delay of re-entanglement kinetics by shear-induced nucleation precursors in isotactic polypropylene melt. Polymer 2020, 210, 123000. [Google Scholar] [CrossRef]
- Liu, M.; Chen, J.; Luo, J.; Min, J.; Fu, Q.; Zhang, J. Investigating the disentanglement of long chain branched polypropylene under different shear fields. J. Appl. Polym. Sci. 2022, 139, 51642. [Google Scholar] [CrossRef]
- Kamkar, M.; Salehiyan, R.; Goudoulas, T.B.; Abbasi, M.; Saengow, C.; Erfanian, E.; Sadeghi, S.; Natale, G.; Rogers, S.A.; Giacomini, A.J.; et al. Large amplitude oscillatory shear flow: Microstructural assessment of polymeric systems. Prog. Polym. Sci. 2022, 132, 101580. [Google Scholar] [CrossRef]
- Wang, S.-Q.; Ravindranath, S.; Wang, Y.; Boukany, P. New theoretical considerations in polymer rheology: Elastic breakdown of chain entanglement network. J. Chem. Phys. 2007, 127, 064903. [Google Scholar] [CrossRef] [PubMed]
- Ibar, J.P. Processing Polymer Melts under Rheo-Fluidification Flow Conditions, Part 1: Boosting Shear-Thinning by Adding Low Frequency Nonlinear Vibration to Induce Strain Softening. J. Macromol. Sci. B Phys. 2013, 52, 407–441. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Zhou, N.-Q.; Liu, B.; Jin, G. Improved Mechanical Propertes and Structure of Polypropylene Pipe Prepared Under Vibration Force Field. J. Appl. Polym. Sci. 2009, 114, 3612–3620. [Google Scholar] [CrossRef]
- An, F.Z.; Gao, X.Q.; Lei, J.; Deng, C.; Li, Z.M.; Shen, K.Z. Vibration assisted extrusion of polypropylene. Chin. J. Polym. Sci. 2015, 33, 688–696. [Google Scholar] [CrossRef]
- Isayev, A.I.; Wong, C.M.; Zeng, X. Effect of oscillations during extrusion on rheology and mechanical properties of polymers. Adv. Polym. Technol. 1990, 10, 31–45. [Google Scholar] [CrossRef]
- Lin, W.; Yang, Z.T.; Qu, J.-P. Short-time fabrication of well-mixed high-density polyethylene/ultrahigh-molecular-weight polyethylene blends under elongational flow: Morphology, mechanical properties and mechanism. Polym. Int. 2019, 68, 904–914. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Chen, J.; Luo, J.; Min, J.; Fu, Q.; Zhang, J. Efficient disentanglement of polycarbonate melts under complex shear field. Polymer 2020, 201, 122610. [Google Scholar] [CrossRef]
- Hu, H.; Chen, J.; Yang, T.; Wang, P.; Min, J.; Fu, Q.; Zhang, J. Regulation of Entanglement Networks under Different Shear Fields and Its Effect on the Properties of Poly(L-lactide). Ind. Eng. Chem. Res. 2023, 62, 7434–7446. [Google Scholar] [CrossRef]
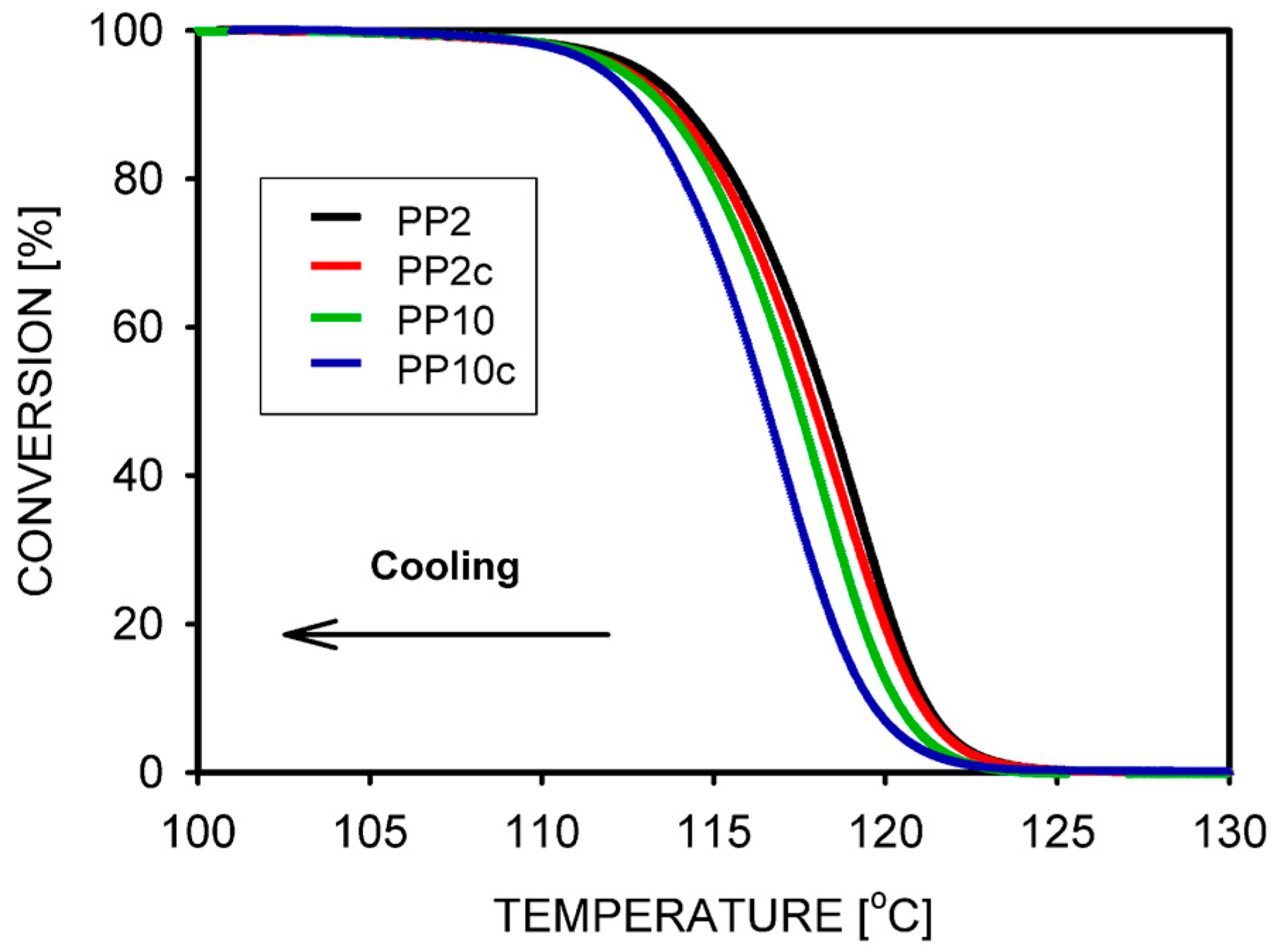
- Wang, X.H.; Liu, R.; Wu, M.; Wang, Z.; Huang, Y. Effect of chain disentanglement on melt crystallization behavior of isotactic polypropylene. Polymer 2009, 50, 5824–5827. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. The crystallization of polypropylene with reduced density of entanglements. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 748–756. [Google Scholar] [CrossRef]
- Xiao, Z.G.; Sun, Q.; Xue, G.; Yuan, Z.; Dai, Q.; Hu, Y.L. Thermal behavior of isotactic polypropylene freeze-extracted from solutions with varying concentrations. Europ. Polym. J. 2003, 39, 927–931. [Google Scholar] [CrossRef]
- Krajenta, J.; Pawlak, A.; Galeski, A. Deformation of disentangled polypropylene crystalline grains into nanofibers. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 1983–1994. [Google Scholar] [CrossRef]
- Pandey, A.; Toda, A.; Rastogi, S. Influence of Amorphous Component on Melting of Semicrystalline Polymers. Macromolecules 2011, 44, 8042–8055. [Google Scholar] [CrossRef]
- Ji, G.; Xue, G.; Ma, J.; Dong, C.; Gu, X. Concentration dependence of crystallinity of polycarbonate by shock-cooling and subsequent freeze-drying of its various solutions. Polymer 1996, 37, 3255–3258. [Google Scholar] [CrossRef]
- Xie, Z.P.; Liu, D.; Zhu, P.P.; Yang, H.Y. Crystallization behavior of chain-disentangled poly(ethylene terephthalate). Acta Polym. Sinica 2010, 5, 522–529. [Google Scholar] [CrossRef]
- Romano, D.; Tops, N.; Andablo-Reyes, E.; Ronca, S.; Rastogi, S. Influence of Polymerization Conditions on Melting Kinetics of Low Entangled UHMWPE and Its Implications on Mechanical Properties. Macromolecules 2014, 47, 4750–4760. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, J.; Liu, M.; Fu, Q.; Zhang, J. Understanding the effect of chain entanglement state on melt crystallization of the polymer freeze-extracted from solution: The role of critical overlap concentration. Polymer 2019, 178, 121588. [Google Scholar] [CrossRef]
- Gaonkar, A.A.; Murudkar, V.V.; Deshpande, V.D. Comparison of crystallization kinetics of polyethylene terephthalate (PET) and reorganized PET. Thermochim. Acta 2020, 683, 178472. [Google Scholar] [CrossRef]
- Ni, L.; Xu, S.; Sun, C.; Qin, Y.; Zheng, Y.; Zhou, J.; Yu, C.; Pan, P. Retarded Crystallization and Promoted Phase Transition of Freeze-Dried Polybutene-1: Direct Evidence for the Critical Role of Chain Entanglement. ACS Macro Lett. 2022, 11, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, W. Weak Shear-Induced Slowdown in Crystallization of Less-Entangled Poly(ε-caprolactone). Macromolecules 2021, 54, 3347–3357. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Fu, Q.; Zhang, J. Novel Strategy to Improve the Performance of Poly(L-lactide): The Synergistic Effect of Disentanglement and Strong Shear Field. ACS Sustain. Chem. Eng. 2023, 11, 9630–9642. [Google Scholar] [CrossRef]
- Sun, C.; Zheng, Y.; Xu, S.; Ni, L.; Li, X.; Shan, G.; Bao, Y.; Pan, P. Role of Chain Entanglements in the Stereocomplex Crystallization between Poly(lactic acid) Enantiomers. ACS Macro. Lett. 2021, 10, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Krajenta, J.; Safandowska, M.; Pawlak, A. The re-entangling of macromolecules in polypropylene. Polymer 2019, 175, 215–226. [Google Scholar] [CrossRef]
- Krajenta, J.; Safandowska, M.; Pawlak, A.; Galeski, A. All-polymer composites—A new approach with the use of disentangled semi-crystalline polymers. Part 1. Disentangling and properties of disentangled polylactide. Polimery 2020, 65, 167–173. [Google Scholar] [CrossRef]
- Krajenta, J.; Polinska, M.; Lapienis, G.; Pawlak, A. The crystallization of poly(ethylene oxide) with limited density of macromolecular entanglements. Polymer 2020, 197, 122500. [Google Scholar] [CrossRef]
- Zhai, Z.; Fusco, C.; Morthomas, J.; Perez, M.; Lame, O. Disentangling and Lamellar Thickening of Linear Polymers during Crystallization: Simulation of Bimodal and Unimodal Molecular Weight Distribution Systems. ACS Nano. 2019, 13, 11310–11319. [Google Scholar] [CrossRef]
- Peng, F.; Nie, C.; Xu, T.Y.; Sheng, J.F.; Chen, W.; Yu, W.C.; Li, L.B. Entanglement on nucleation barrier of polymer crystal. Chin. J. Polym. Sci. 2022, 40, 1640–1650. [Google Scholar] [CrossRef]
- Dai, Q.; Lu, Y.; Xue, G.; Liao, Y.T. Glass transition of atactic polystyrene with less chain entanglements. Polym. Bull. 1995, 35, 209–214. [Google Scholar] [CrossRef]
- Sasaki, T.; Yamauchi, N.; Irie, S.; Sakurai, K. Differential scanning calorimetry study on thermal behaviors of freeze-dried poly(L-lactide) from dilute solutions. J. Polym. Sci. B Polym. Phys. 2005, 43, 115–124. [Google Scholar] [CrossRef]
- Huang, D.H.; Yang, Y.; Zhuang, G.; Li, B. Influence of Entanglements on the Glass Transition and Structural Relaxation Behaviors of Macromolecules. 1. Polycarbonate. Macromolecules 1999, 32, 6675–6678. [Google Scholar] [CrossRef]
- Bernazzani, P.; Simon, S.L.; Plazek, D.J.; Ngai, K.L. Effects of entanglement concentration on Tg and local segmental motions. Europ. Phys. J. E 2002, 8, 201–207. [Google Scholar] [CrossRef]
- Rong, W.; Fan, Z.; Yu, Y.; Bu, H.; Wang, M. Influence of entanglements on glass transition of atactic polystyrene. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2243–2251. [Google Scholar] [CrossRef]
- Huang, D.H.; Yang, Y.; Zhuang, G.; Li, B. Influence of Intermolecular Entanglements on the Glass Transition and Structural Relaxation Behaviors of Macromolecules. 2. Polystyrene and Phenolphthalein Poly(ether sulfone). Macromolecules 2000, 33, 461–464. [Google Scholar] [CrossRef]
- Singh, M.K.; Hu, M.; Cang, Y.; Hsu, H.P.; Therien-Aubin, H.; Koynov, K.; Fytas, G.; Landfester, K.; Kremer, K. Glass Transition of Disentangled and Entangled Polymer Melts: Single-Chain-Nanoparticles Approach. Macromolecules 2020, 53, 7312–7321. [Google Scholar] [CrossRef]
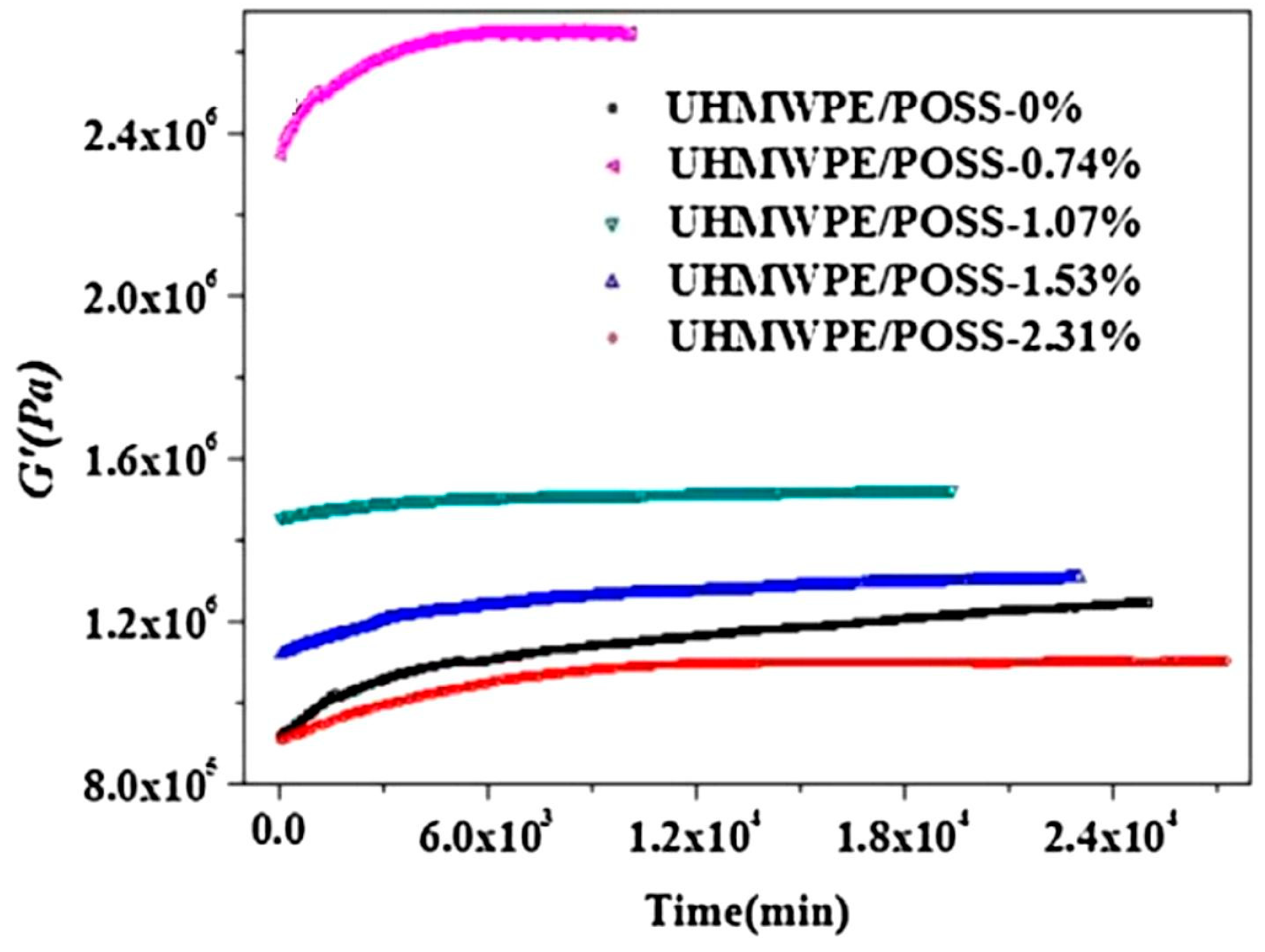
- Zhang, X.; Zhao, S.; Xin, Z. The chain dis-entanglement effect of polyhedral oligomeric silsesquioxanes (POSS) on ultra-high molecular weight polyethylene (UHMWPE). Polymer 2020, 202, 122631. [Google Scholar] [CrossRef]
- Lu, X.L.; Xue, G.; Mi, Y.L. Understanding the effect of chain entanglement on the glass transition of a hydrophilic polymer. J. Appl. Polym. Sci. 2011, 119, 2310–2317. [Google Scholar] [CrossRef]
- Li, N.; Yang, Q.; Huang, Y.; Zhang, Q.; Zhao, W. Entropy reduction phenomenon in the non-equilibrium state of freeze-dried polymethyl methacrylate samples. J. Polym. Res. 2014, 21, 392. [Google Scholar] [CrossRef]
- Zheng, W.; Simon, S.L. Polystyrene freeze-dried from dilute solution: Tg depression and residual solvent effects. Polymer 2006, 47, 3520–3527. [Google Scholar] [CrossRef]
- Yue, Z.; Wang, N.; Cao, Y.; Li, W.; Dong, C.-D. Reduced Entanglement Density of Ultrahigh-Molecular-Weight Polyethylene Favored by the Isolated Immobilization on the MgCl2 (110) Plane. Ind. Eng. Chem. Res. 2020, 59, 3351–3358. [Google Scholar] [CrossRef]
- Shan, W.; Xiao, K.; Thomas, E.L. Influence of Entanglements on Ultrahigh Strain Rate Deformation of Polystyrene Micro projectiles. Macromolecules 2022, 55, 9594–9600. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, S.; Yu, X.; Xin, Z.; Ye, C.; Li, Z.; Xia, J. Nascent Particle Sizes and Degrees of Entanglement Are Responsible for the Significant Differences in Impact Strength of Ultrahigh Molecular Weight Polyethylene. J. Polym. Sci. B Polym. Phys. 2019, 57, 632–641. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. Cavitation phenomenon and mechanical properties of partially disentangled polypropylene. Polymer 2018, 151, 15–26. [Google Scholar] [CrossRef]
- Logunova, M.A.; Orekhov, N.D. The Role of Intermolecular Entanglements in the Formation of Nanosized Pores during Deformation of Polyethylene: Atomistic Modeling. Polym. Sci. Series A 2021, 63, 591–599. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Psarras, G.C.; Forte, G.; Martin-Fabiani, I.; Ronca, S. Entanglement dynamics in ultra-high molecular weight polyethylene as revealed by dielectric spectroscopy. Polymer 2018, 50, 35–43. [Google Scholar] [CrossRef]
- Liu, G.; Larson, R.G.; Li, L.; Luo, H.; He, X.; Niu, Y.; Li, G. Influence of Chain Entanglement on Rheological and Mechanical Behaviors of Polymerized Ionic Liquids. Macromolecules 2023, 56, 2719–2728. [Google Scholar] [CrossRef]
- Wu, B.; Cai, Y.; Zhao, X.; Ye, L. Polymer Testing, Fabrication of well-miscible and highly enhanced polyethylene/ultrahigh molecular weight polyethylene blends by facile construction of interfacial intermolecular entanglement. Polym. Test. 2021, 93, 106973. [Google Scholar] [CrossRef]
- Tanaka, H.; Saijo, S.; Kakiage, M.; Yamanobe, T.; Uehara, H. In-situ analysis for melt-drawing behavior of ultra-high molecular weight polyethylene/normal molecular weight polyethylene blend films. Polymer 2021, 213, 123213. [Google Scholar] [CrossRef]
- Takazawa, A.; Kakiage, M.; Yamanobe, T.; Uehara, H.; Shimizu, Y.; Ohnishi, T.; Wakabayashi, Y.; Inatomi, K.; Abe, S.; Aoyama, K. Effect of blending small amount of high-density polyethylene on molecular entanglements during melt-drawing of ultrahigh-molecular-weight polyethylene. Polymer 2022, 241, 124528. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, Y.; Li, W.; Wang, J.; Yang, Y. Melt spinning of polyethylene composites fibers containing disentangled ultra-high molecular weight polyethylene. In Proceedings of the World Textile Conference AUTEX 2022, Lodz, Poland, 7–10 June 2022. [Google Scholar] [CrossRef]
- Zhang, Y.; Di, Y.; Chunlin Ye, C.; Zhang, L.; Tang, X.; Shu, B.; Yan, X.; Li, W.; Wang, J.; Yang, Y. Morphology evolution and mechanical property enhancement of linear low-density polyethylene by adding disentangled ultra high molecular weight polyethylene. Polym. Adv. Technol. 2022, 33, 1047–1056. [Google Scholar] [CrossRef]
- Schirmeister, C.G.; Hees, T.; Dolynchuk, O.; Licht, E.H.; Thurn-Albrecht, T.; Muelhaupt, R. Digitally Tuned Multidirectional All-Polyethylene Composites via Controlled 1D Nanostructure Formation during Extrusion-Based 3D Printing. ACS Appl. Polym. Mater. 2021, 3, 1675–1686. [Google Scholar] [CrossRef]
- Tao, G.; Chen, Y.; Mu, J.; Zhang, L.; Ye, C.; Li, W. Exploring the entangled state and molecular weight of UHMWPE on the microstructure and mechanical properties of HDPE/UHMWPE blends. J. Appl. Polym. Sci. 2021, 138, 50741. [Google Scholar] [CrossRef]
- Tang, X.; Xing, J.; Yan, X.; Ye, C.; Zhang, L.; Zhang, Y.; Shu, B.; Mu, J.; Wei, L.; Wang, J.; et al. Metallocene Polyolefins Reinforced by Low-Entanglement UHMWPE through Interfacial Entanglements. Adv. Polym. Technol. 2022, 2022, 9344096. [Google Scholar] [CrossRef]
- Smith, P.; Lemstra, P.J. Ultra-drawing of high molecular weight polyethylene cast from solution. Coll. Polym. Sci. 1980, 258, 891–894. [Google Scholar] [CrossRef]
- Spronck, M.; Klein, A.; Blom, B.; Romano, D. Synthesis of Disentangled Ultra-High Molecular Weight Polyethylene using Vanadium(V)-Based Catalysts. Z. Anorg. Allg. Chem. 2018, 644, 993–998. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, P.; Yue, Z.; Li, W.; Dong, C.; Jiang, B.; Wang, J.; Yang, Y. Entanglement Formation Mechanism in the POSS Modified Heterogeneous Ziegler−Natta Catalysts. Macromolecules 2019, 52, 7593–7602. [Google Scholar] [CrossRef]
- Cao, Y.; Wu, Y.; Tang, X.; Zhou, Q.; Stapf, S.; Mattea, C.; Li, W. Long-term efficiency for reducing entanglements of nascent polyethylene by a polystyrene-modified Ziegler-Natta catalyst. J. Appl. Polym. Sci. 2022, 39, 51790. [Google Scholar] [CrossRef]
- Chammingkwan, P.; Bando, Y.; Mai, L.T.T.; Wada, T.; Thakur, A.; Terano, M.; Sinthusai, L.; Taniike, T. Less Entangled Ultrahigh-Molecular-Weight Polyethylene Produced by Nano-Dispersed Ziegler−Natta Catalyst. Ind. Eng. Chem. Res. 2021, 60, 2818–2827. [Google Scholar] [CrossRef]
- Chen, Y.; Li, W.; Zhang, L.; Ye, C.; Tao, G.; Ren, C.; Jiang, B.; Wang, J.; Yang, Y. In Situ Synthesized Self-Reinforced HDPE/UHMWPE Composites with High Content of Less Entangled UHMWPE and High Gradient- Distributed Oriented Structures. ACS Appl. Polym. Mater. 2023, 5, 88–98. [Google Scholar] [CrossRef]
- Chen, Y.; Tao, G.; Li, W.; Zhou, Q.; Shu, B.; Jiang, B.; Wang, J.; Yang, Y. The in situ synthesis of weakly entangled ultrahigh-molecular weight polyethylene/polyethylene wax blends and its synergetic disentanglement effect on LLDPE reinforcement. Polym. Adv. Technol. 2022, 33, 3728–3739. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Forte, G.; Ronca, S. Relaxation Dynamics in Disentangled Ultrahigh Molecular Weight Polyethylene via Torsional Rheology. Ind. Eng. Chem. Res. 2020, 59, 4515–4523. [Google Scholar] [CrossRef]
- Christakopoulos, F.; Bersenev, E.; Grigorian, S.; Brem, A.; Ivanow, D.; Tervoort, T.O.; Litvinov, V. Melting-Induced Evolution of Morphology, Entanglement Density, and Ultradrawability of Solution-Crystallized Ultrahigh-Molecular-Weight Polyethylene. Macromolecules 2021, 54, 5683–5693. [Google Scholar] [CrossRef]
- Petrov, A.; Rudyak, V.Y.; Chertovich, A. Optimal. Entanglement of Polymers Promotes the Formation of Highly Oriented Fibers. Macromolecules 2022, 55, 6493–6504. [Google Scholar] [CrossRef]
- Li, W.; Guan, C.; Xu, J.; Chen, Z.; Jiang, B.; Wang, J.; Yang, Y. Bimodal/broad polyethylene prepared in a disentangled state. Industr. Eng. Chem. Res. 2014, 53, 1088–1096. [Google Scholar] [CrossRef]
- Lippits, D.R.; Rastogi, S.; Talebi, S.; Bailly, C. Formation of entanglements in initially disentangled polymer melts. Macromolecules 2006, 39, 8882–8885. [Google Scholar] [CrossRef]
- Talebi, S.; Duchateau, R.; Rastogi, S.; Kaschta, J.; Peters, G.; Lemstra, P.J. Molar mass and molecular weight distribution determination of UHMWPE synthesized using a living homogeneous catalyst. Macromolecules 2010, 43, 2780–2788. [Google Scholar] [CrossRef]
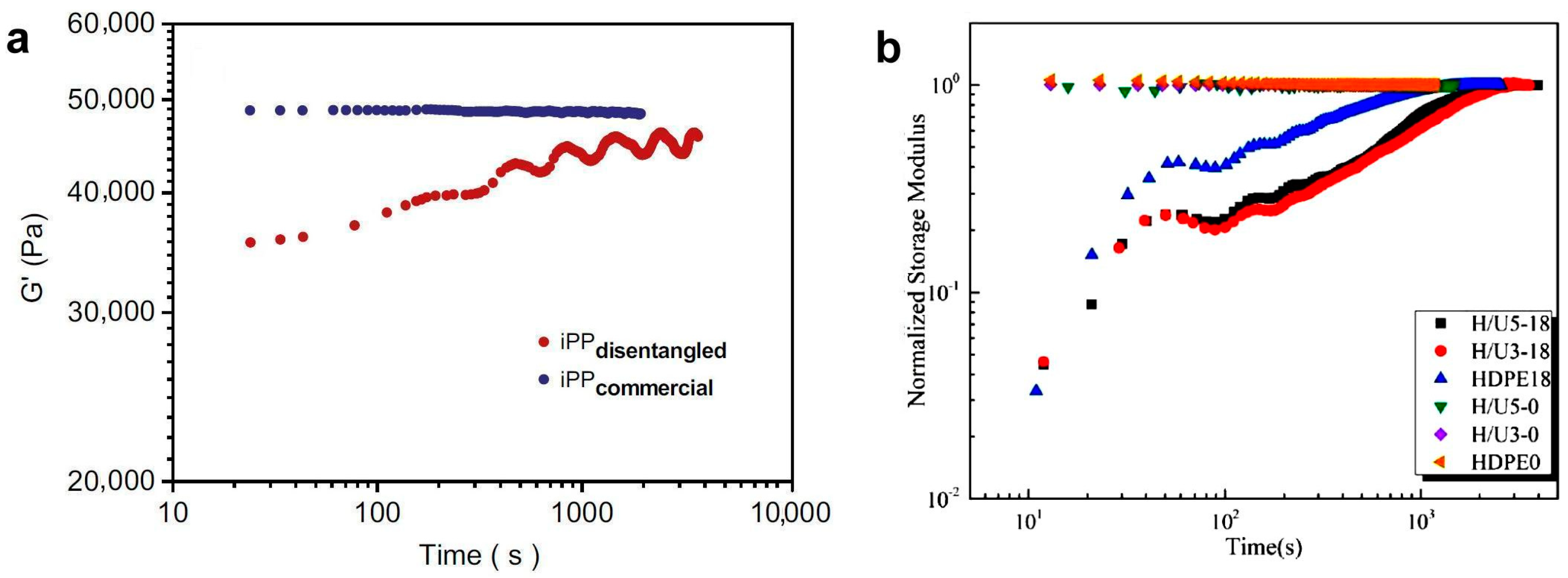
- Liu, M.; Wang, Y.; Chen, J.; Luo, J.; Fu, Q.; Zhang, J. The retarded recovery of disentangled state by blending HDPE with ultra-high molecular weight polyethylene. Polymer 2020, 192, 122329. [Google Scholar] [CrossRef]
- Barham, P.J.; Sadler, D.M. A neutron scattering study of the melting behavior of polyethylene single crystals. Polymer 1991, 32, 393–395. [Google Scholar] [CrossRef]
- Chai, S.-C.; Xu, T.-Y.; Cao, X.; Wang, G.; Chen, Q.; Li, H.-L. Ultrasmall Nanoparticles Diluted Chain Entanglement in Polymer Nanocomposites. Chin. J. Polym. Sci. 2019, 37, 797–805. [Google Scholar] [CrossRef]
- Mackay, M.E.; Dao, T.T.; Tuteja, A.; Ho, D.L.; Horn, B.V.; Kim, H.C.; Hawker, C.J. Nanoscale effects leading to non-Einstein-like decrease in viscosity. Nat. Mater. 2003, 2, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Nusser, K.; Schneider, G.J.; Pyckhout-Hintzen, W.; Richter, D. Viscosity decrease and reinforcement in polymer-silsesquioxane composites. Macromolecules 2011, 44, 7820–7830. [Google Scholar] [CrossRef]
- Mangal, R.; Srivastava, S.; Archer, L.A. Phase stability and dynamics of entangled polymer-nanoparticle composites. Nat. Commun. 2015, 6, 7198. [Google Scholar] [CrossRef] [PubMed]
- Senses, E.; Ansar, S.M.; Kitchens, C.L.; Mao, Y.; Narayanan, S.; Natarajan, B.; Faraone, A. Small Particle Driven Chain Disentanglements in Polymer Nanocomposites. Phys. Rev. Lett. 2017, 118, 147801. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Yui, Y.; Wei, P.; Cong, C.; Meng, X.; Ye, H.-M.; Zhou, Q. Nanoscale effects of TiO2 nanoparticles on the rheological behaviors of ultra-high molecular weight polyethylene (UHMWPE). Soft Matter 2023, 19, 5459–5467. [Google Scholar] [CrossRef]
- Romo-Uribe, A. Dispersion at Single Unit TiO2 Nanoparticles Reduced Tg, Induced Chain Disentanglement and Reduced Tensile Modulus in Waterborne Acrylic Coatings. Macromol. Mater. Eng. 2021, 306, 200059. [Google Scholar] [CrossRef]
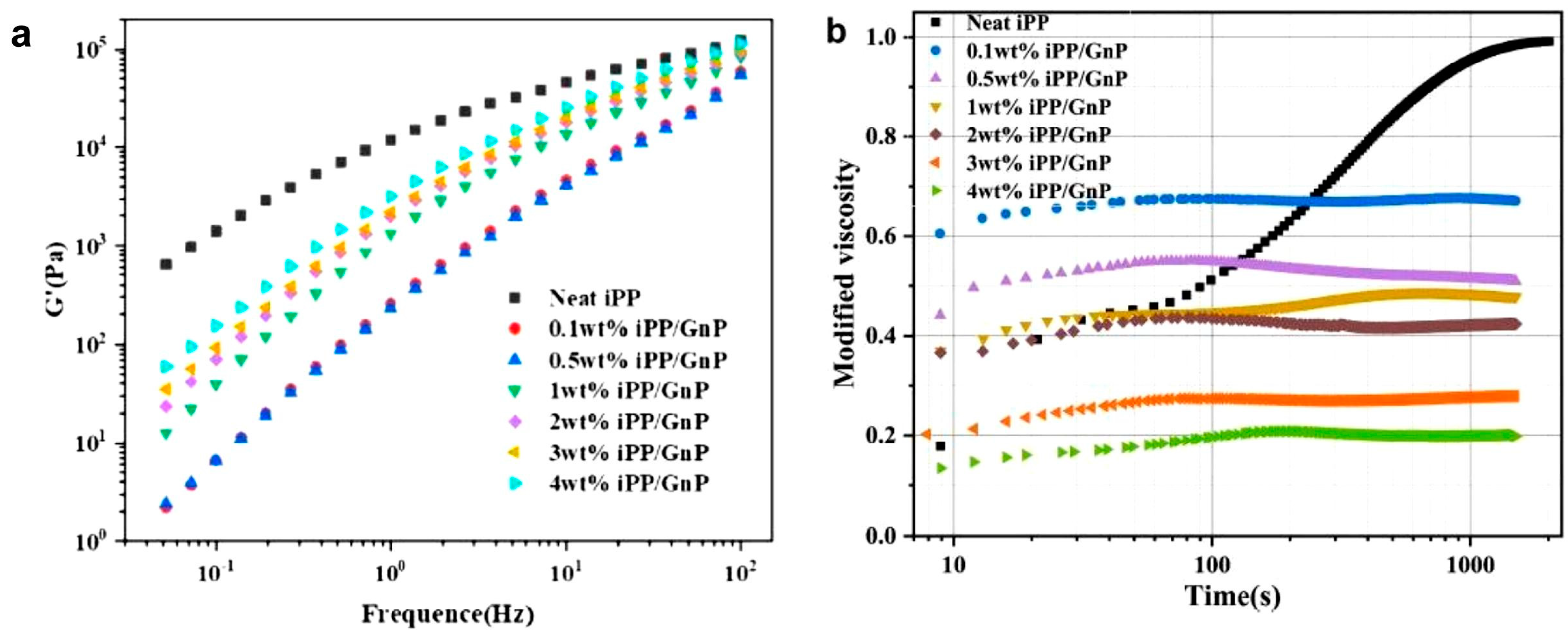
- Luo, J.; Liu, M.; Chen, J.; Min, J.; Fu, Q.; Zhang, J. Effectively maintaining the disentangled state of isotactic polypropylene in the presence of graphene nanoplatelet. Polymer 2021, 226, 23806. [Google Scholar] [CrossRef]
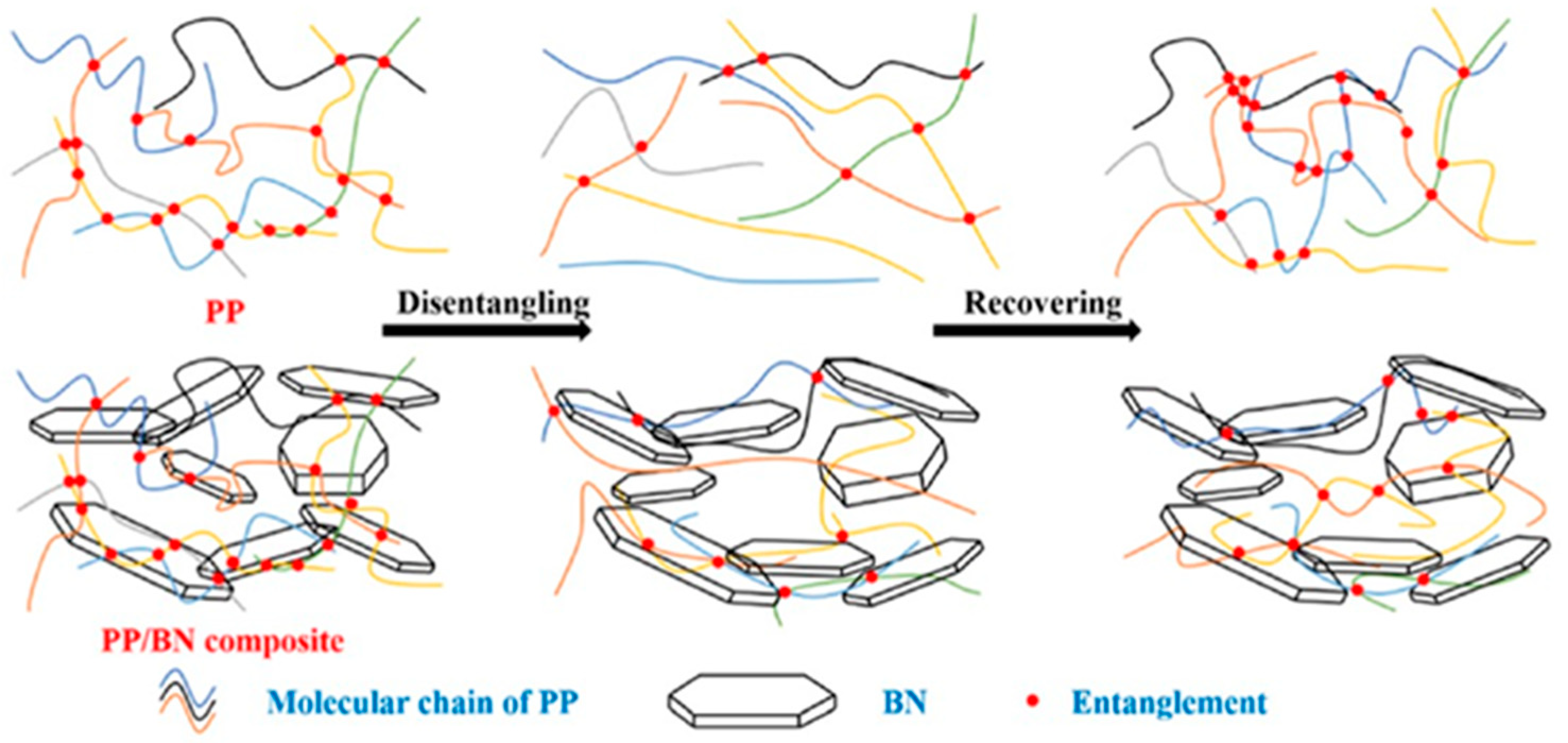
- Luo, J.; Chen, J.; Liu, M.; Min, J.; Fu, Q.; Zhang, J. Investigating the Influence of Incorporation of Boron Nitride on the Kinetics of Isotactic Polypropylene Entanglement Recovery. Ind. Eng. Chem. Res. 2021, 60, 12901–12910. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Tarallo, O.; Guan, L.; Martin-Fabiani, I.; Ronca, S. Nanocomposites of Au/Disentangled UHMWPE: A Combined Optical and Structural Study. Molecules 2020, 25, 3225. [Google Scholar] [CrossRef]
- Drakopoulos, S.X.; Psarras, G.C.; Ronca, S. Oriented ultra-high molecular weight polyethylene/gold nanocomposites: Electrical conductivity and chain entanglement dynamics. Express Polym. Lett. 2021, 15, 492–502. [Google Scholar] [CrossRef]
- Barangizi, H.; Pawlak, A. Crystallization of partially disentangled polypropylene in nanocomposites with aluminum oxide. Polymer 2022, 254, 125049. [Google Scholar] [CrossRef]
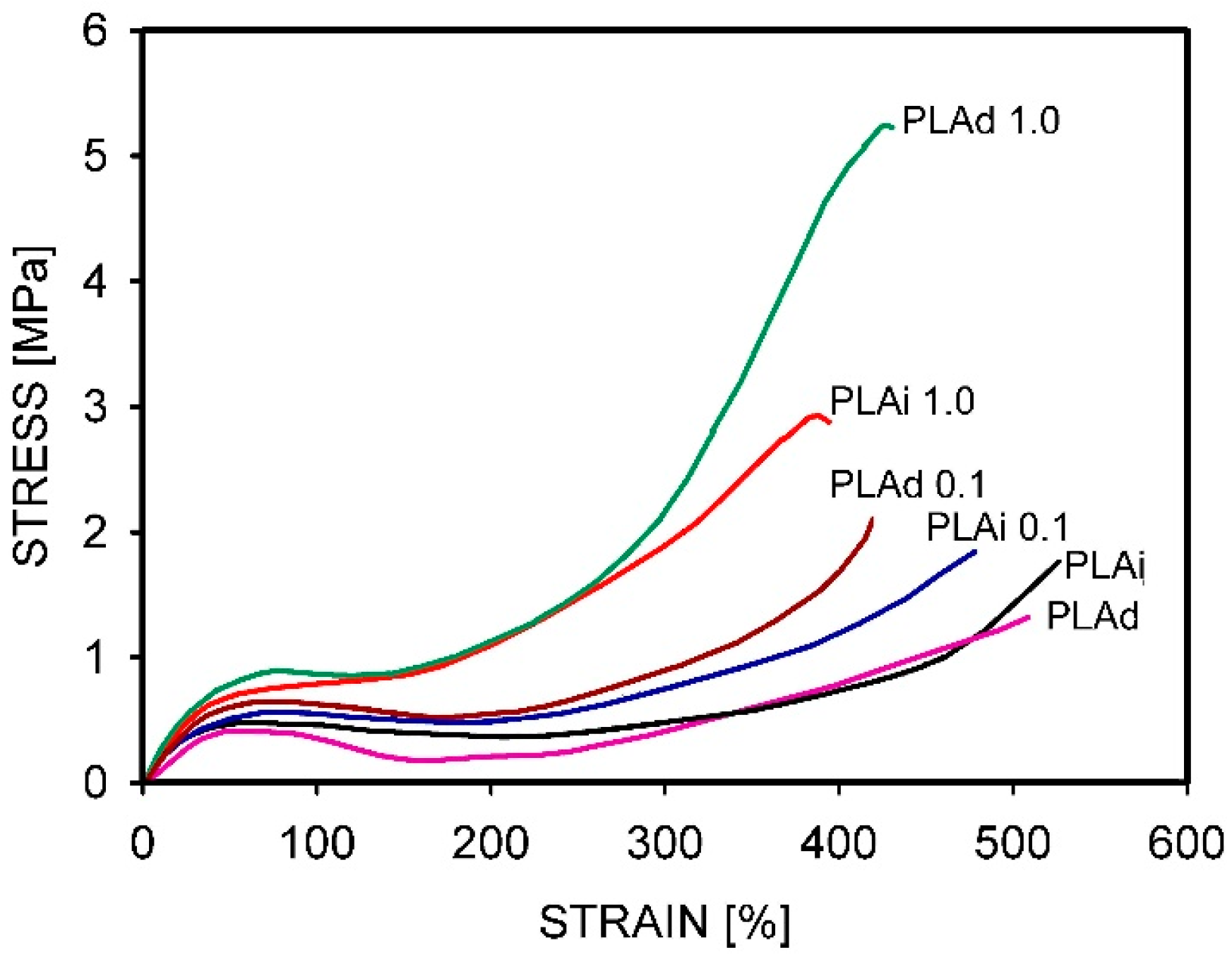
- Barangizi, H.; Krajenta, J.; Pawlak, A. The influence of entanglements of macromolecules on the mechanical and thermal properties of polylactide composites with carbon nanotubes. Express Polym. Lett. 2023, 17, 738–758. [Google Scholar] [CrossRef]
- Jurczuk, K.; Galeski, A.; Piorkowska, E. All-polymer nanocomposites with nanofibrillar inclusions generated in situ during compounding. Polymer 2013, 54, 4617. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlak, A.; Krajenta, J. Progress in Studies of Disentangled Polymers and Composites. J. Compos. Sci. 2023, 7, 521. https://doi.org/10.3390/jcs7120521
Pawlak A, Krajenta J. Progress in Studies of Disentangled Polymers and Composites. Journal of Composites Science. 2023; 7(12):521. https://doi.org/10.3390/jcs7120521
Chicago/Turabian StylePawlak, Andrzej, and Justyna Krajenta. 2023. "Progress in Studies of Disentangled Polymers and Composites" Journal of Composites Science 7, no. 12: 521. https://doi.org/10.3390/jcs7120521
APA StylePawlak, A., & Krajenta, J. (2023). Progress in Studies of Disentangled Polymers and Composites. Journal of Composites Science, 7(12), 521. https://doi.org/10.3390/jcs7120521