



PCa-Clf: A Classifier of Prostate Cancer Patients into Patients with Indolent and Aggressive Tumors Using Machine Learning

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
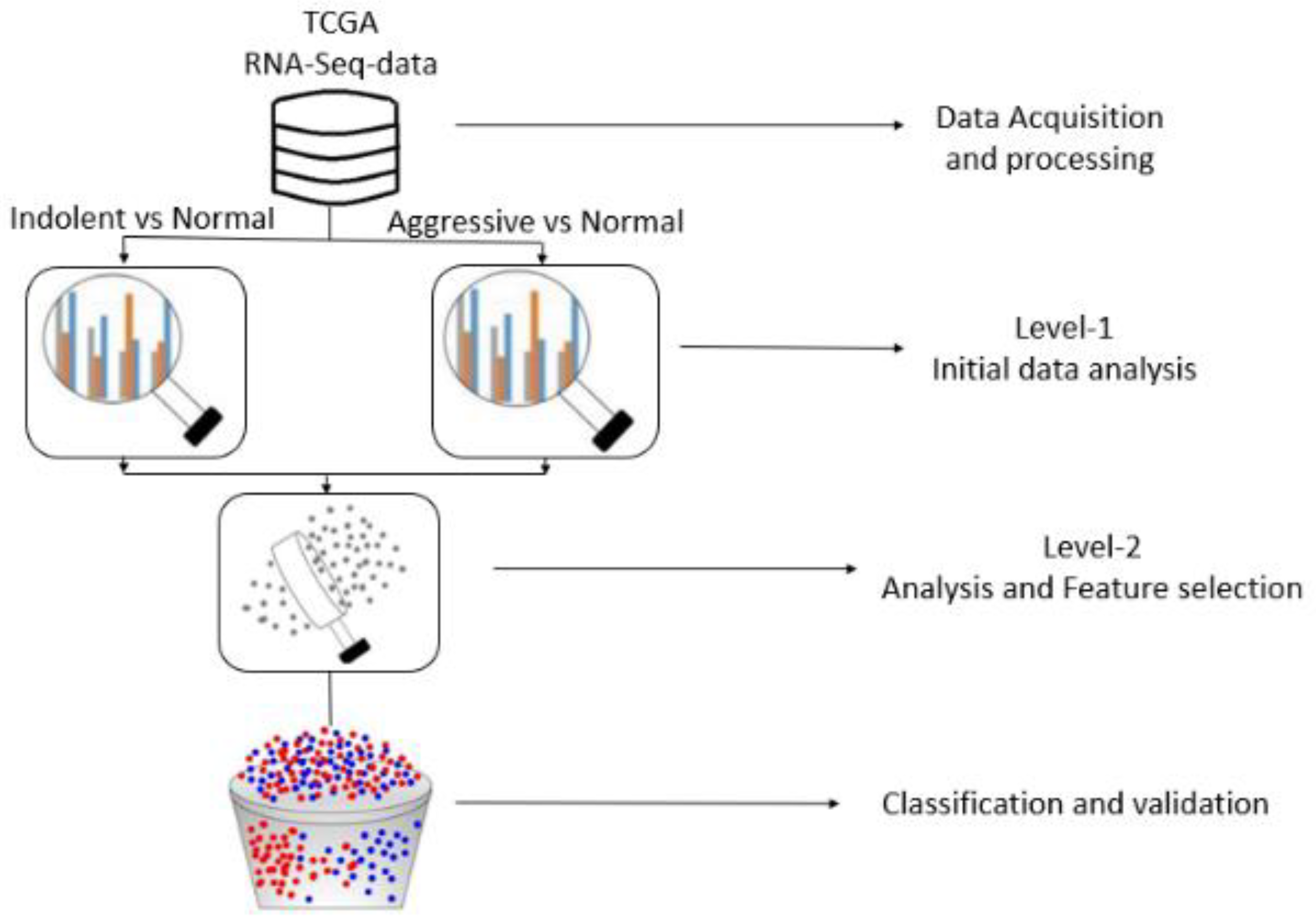
2. Experimental Materials and Methods
2.1. Source of Transcriptome and Clinical Datasets
2.2. Data Processing and Analysis for Gene Selection
2.2.1. Level 1 Analysis
2.2.2. Level 2 Analysis
2.2.3. Feature Selection and Implementation of ML and Genetic Algorithms
- (1)
- Support Vector Machine (SVM).
- (2)
- Logistic Regression (LR).
- (3)
- Random Decision Forest (RF).
- (4)
- Extra Tree Classifier (ETC).
- (5)
- Gradient Boosting Classifier (GBC).
- (6)
- K-Nearest Neighbors (KNNs).
- (7)
- eXtreme Gradient Boosting (XGB).
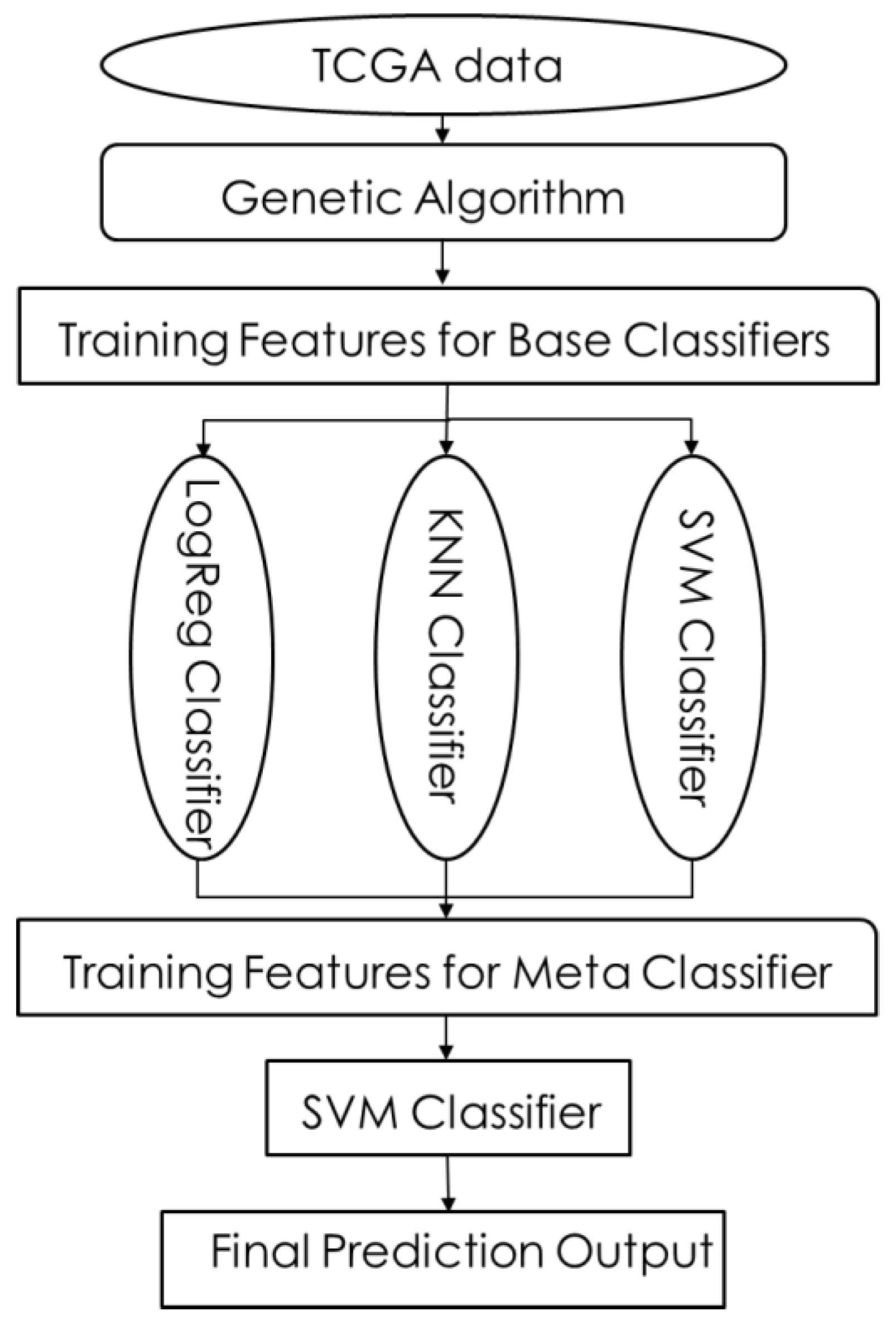
2.2.4. Stacking
- (1)
- SM-1: LR, KNNs, SVM as base classifiers; SVM as meta-classifier.
- (2)
- SM-2: LR, SVM, KNNs, XGB as base classifiers; XGB as meta-classifier.
- (3)
- SM-3: LR, KNNs, SVM as base classifiers; XGB as meta-classifier.
- (4)
- SM-4: RDF, LR, KNNs as base classifiers; GBC as meta-classifier.
- (5)
- SM-5: RDF, LR, GBC as base classifiers; KNNs as meta-classifier.
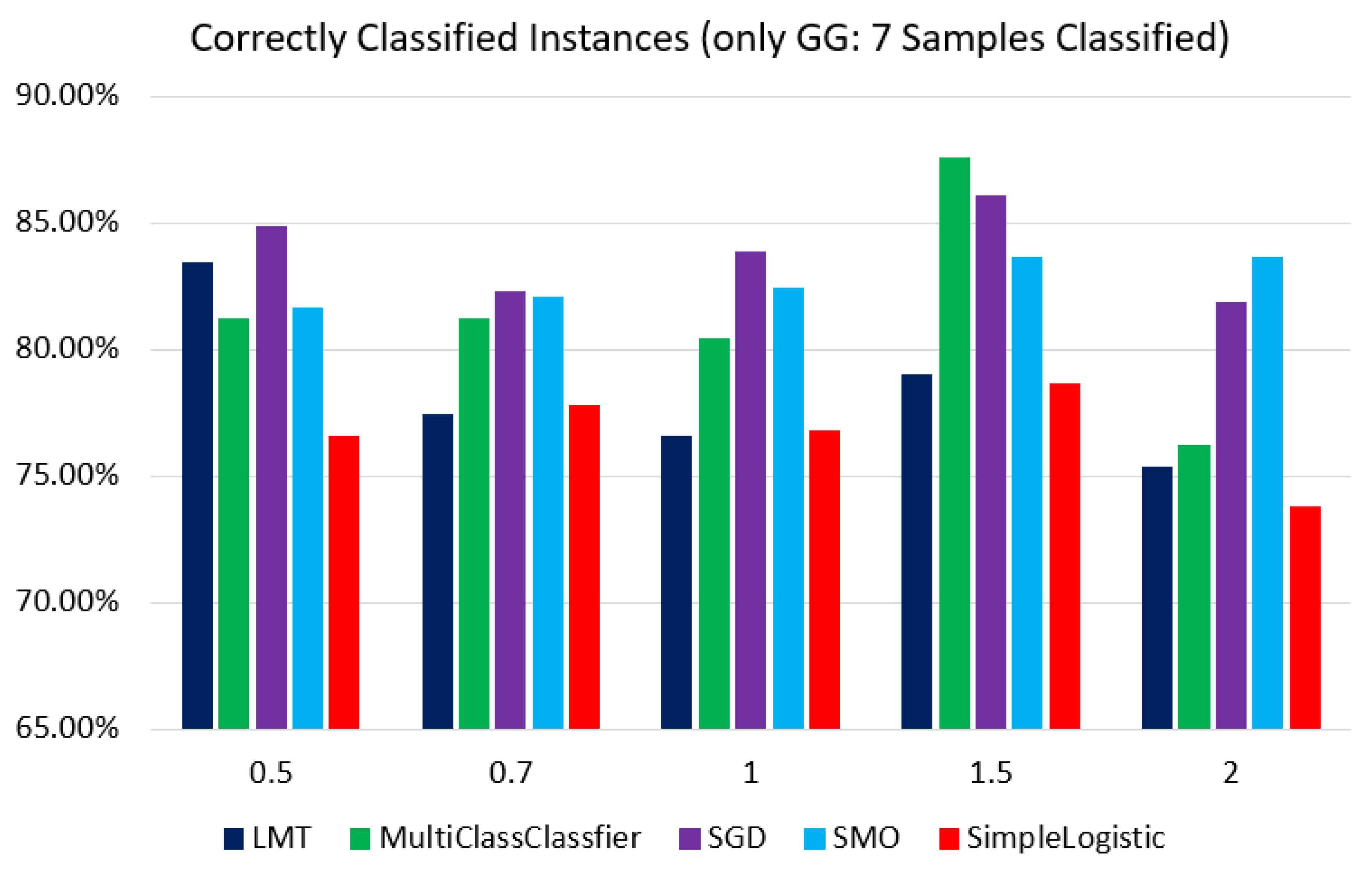
2.3. Model Selection and Validation by Correlating ML Algorithm with GGs
2.4. Performance Evaluation
3. Results
3.1. Discovery Genes Associated with Indolent and Aggressive PCas
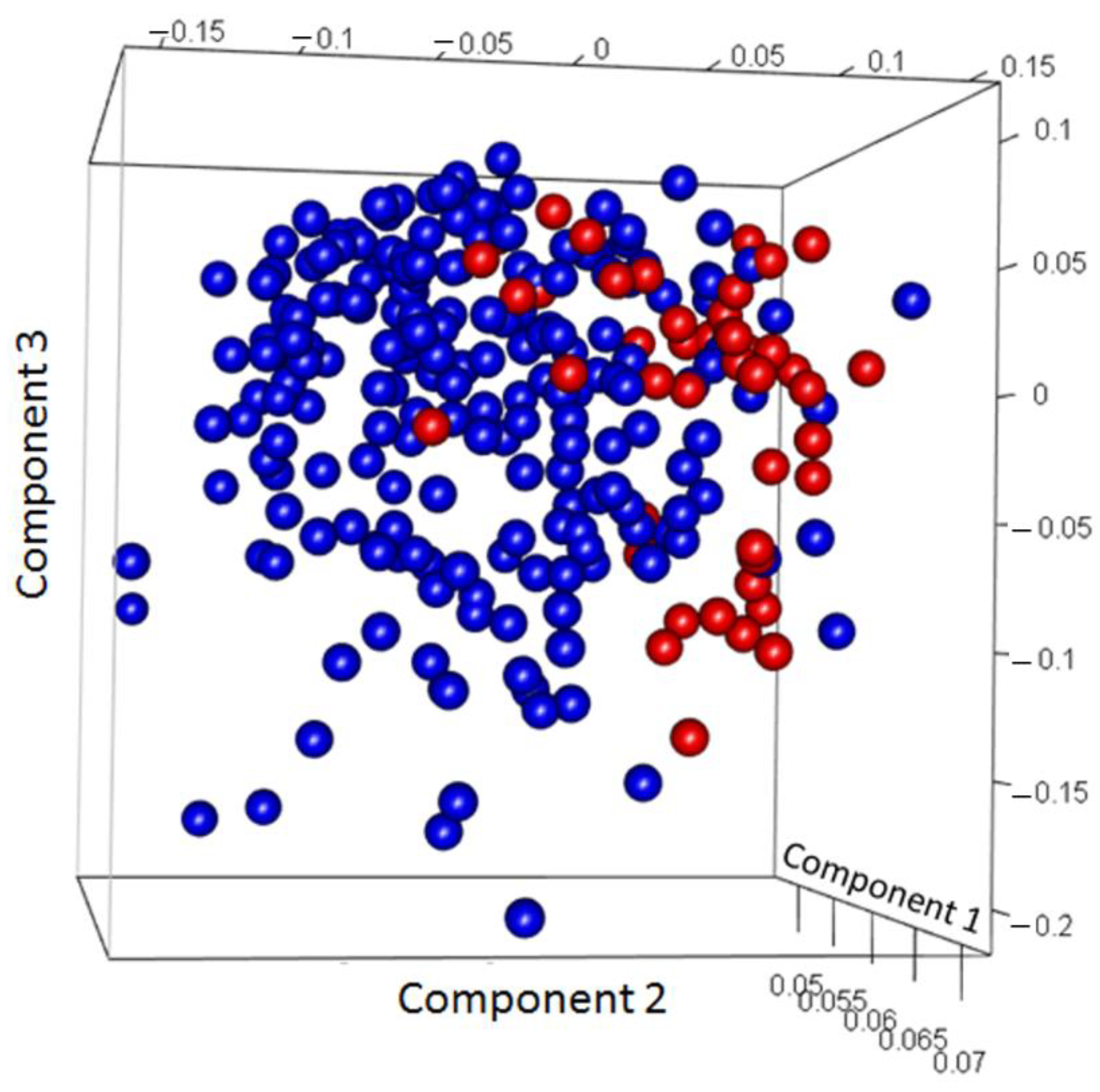
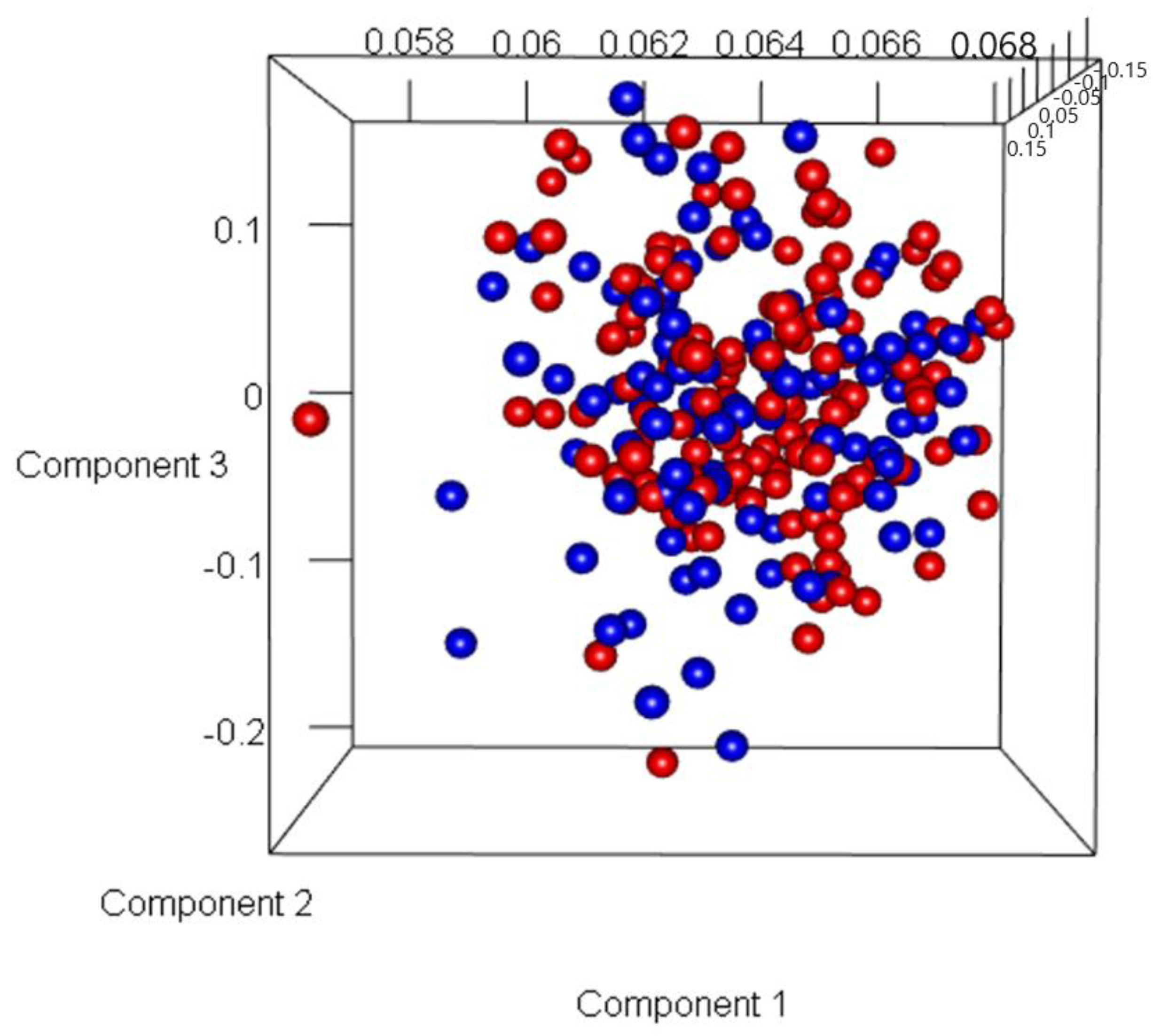
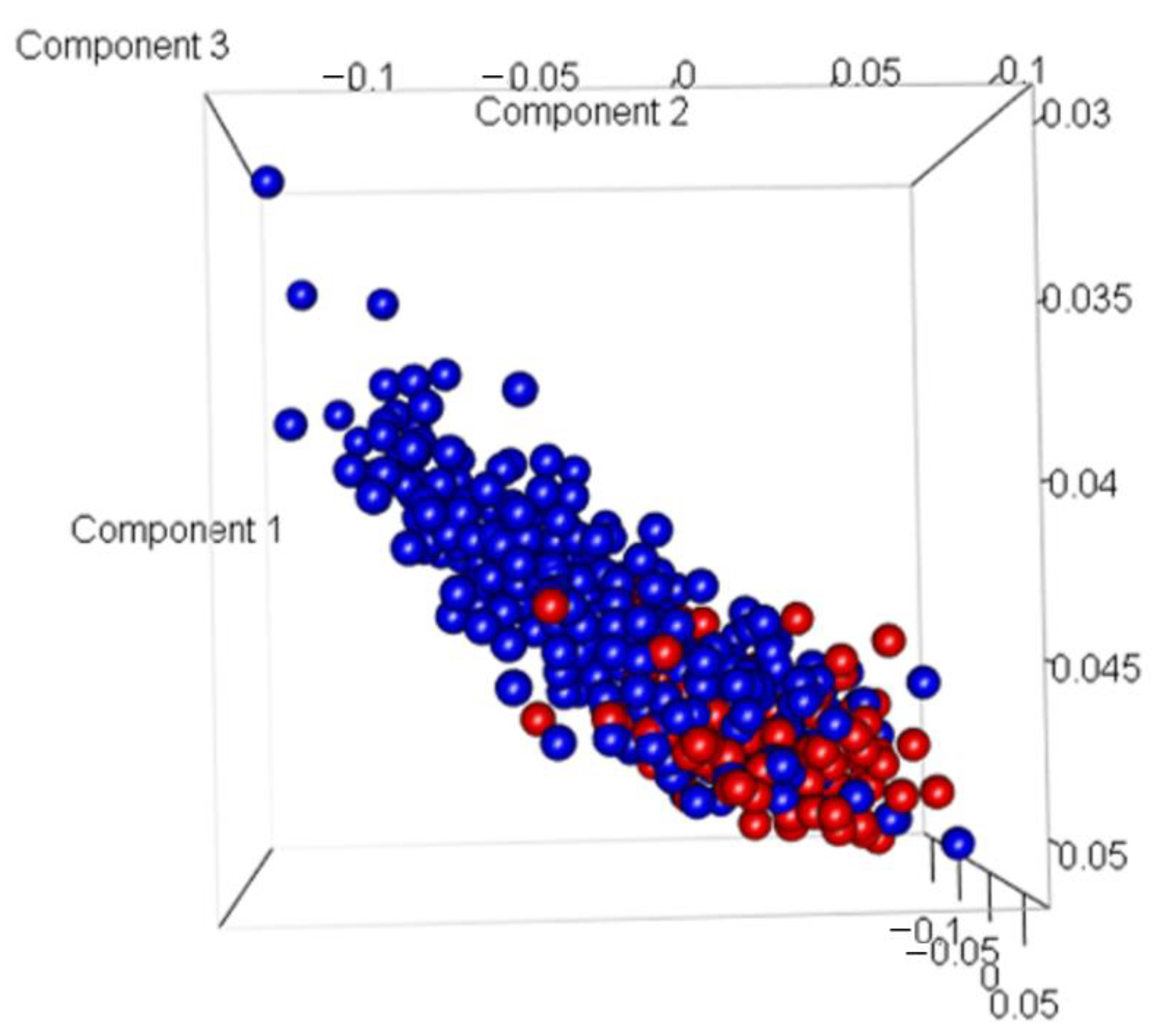
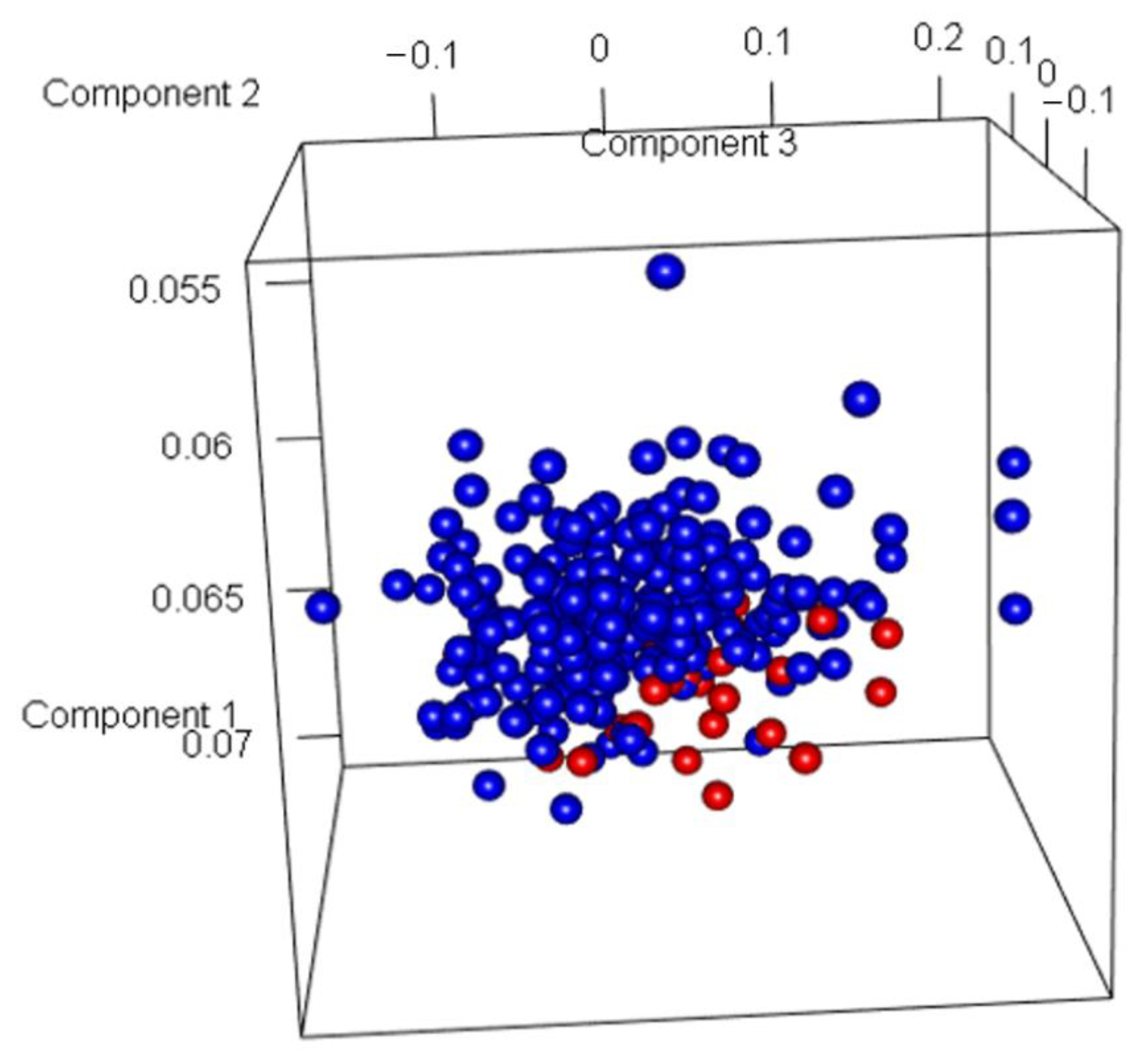
3.2. Discovery of Genes or Features Associated with the Two Types of PCa Used in ML Algorithms
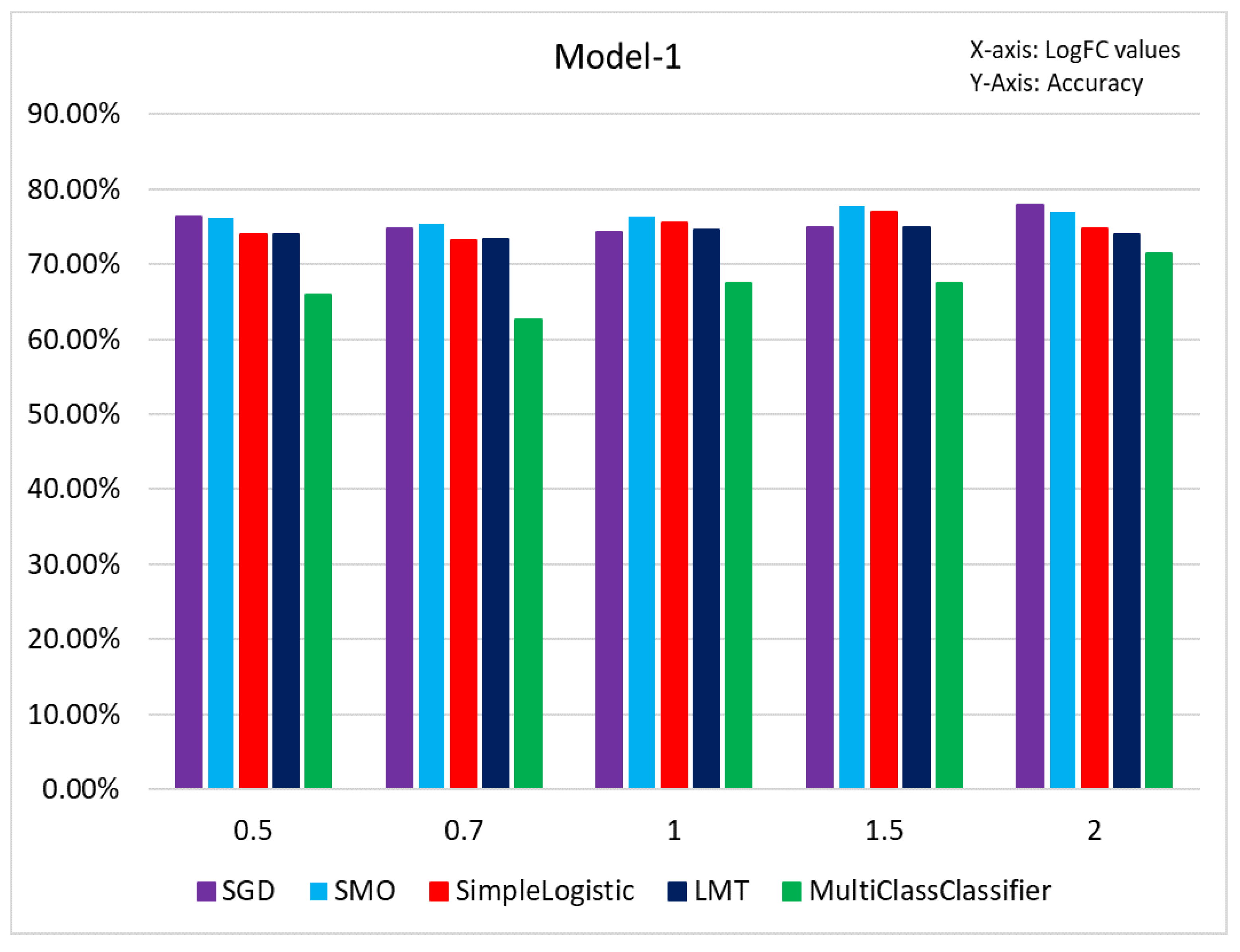
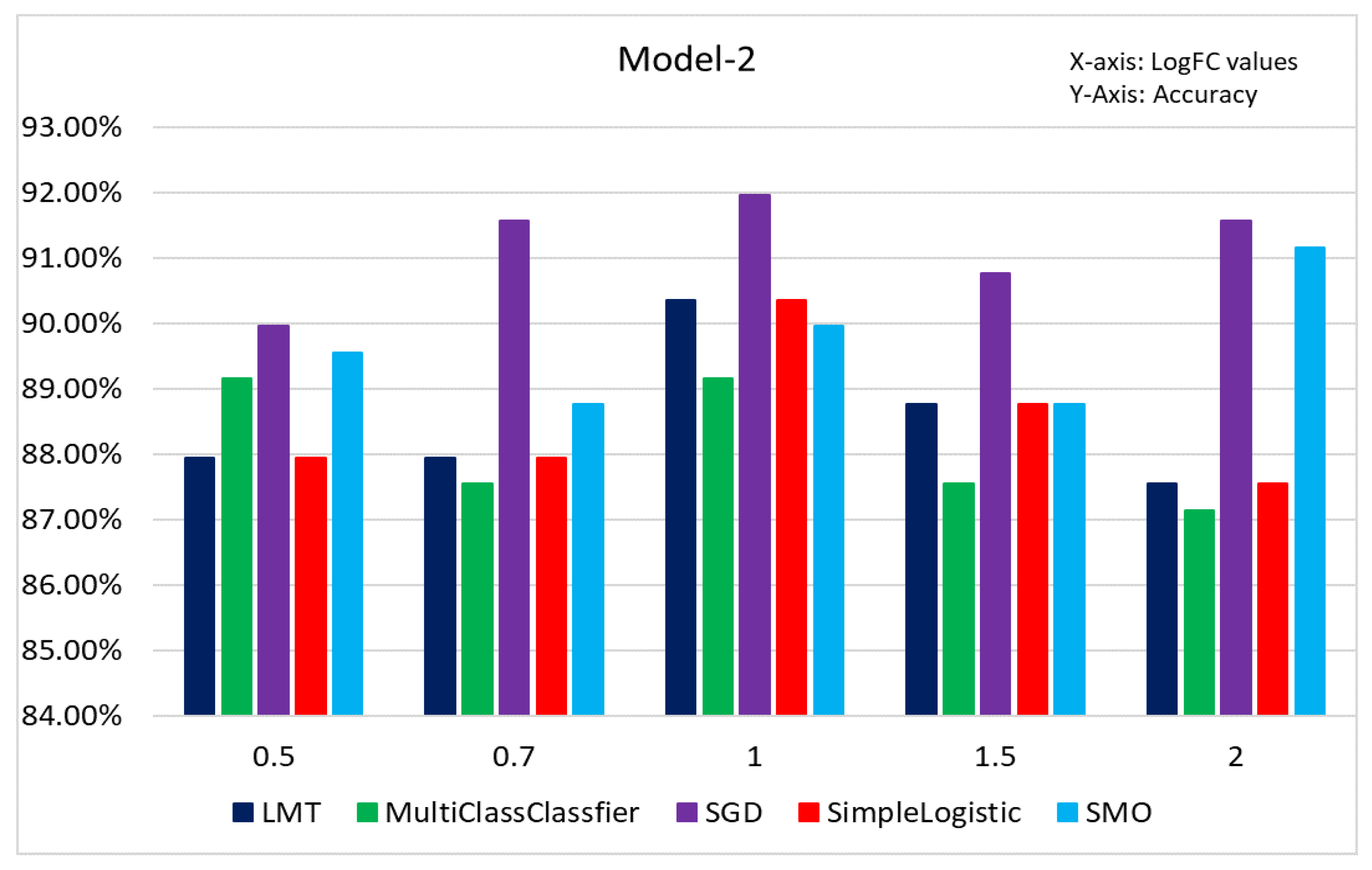
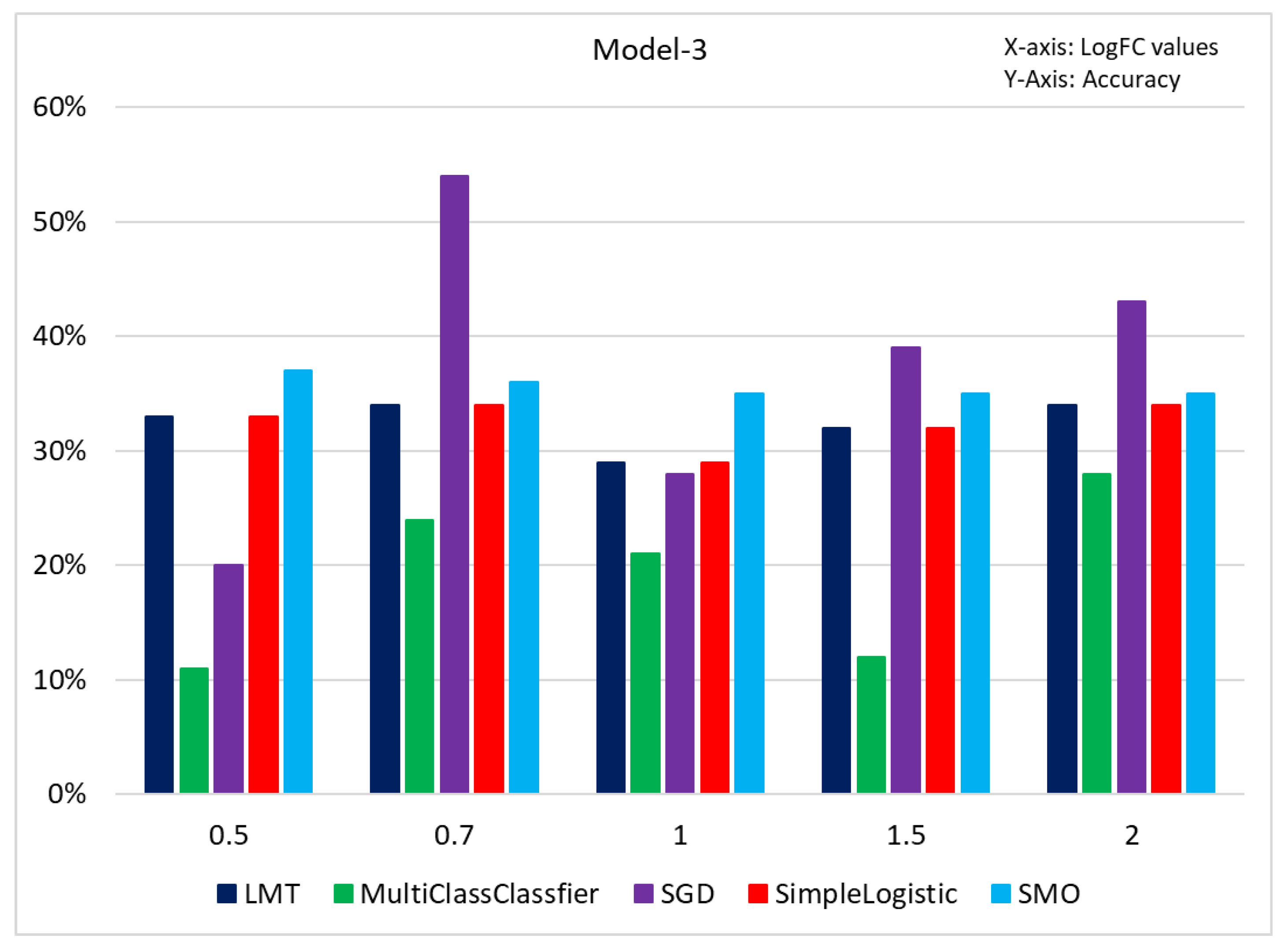
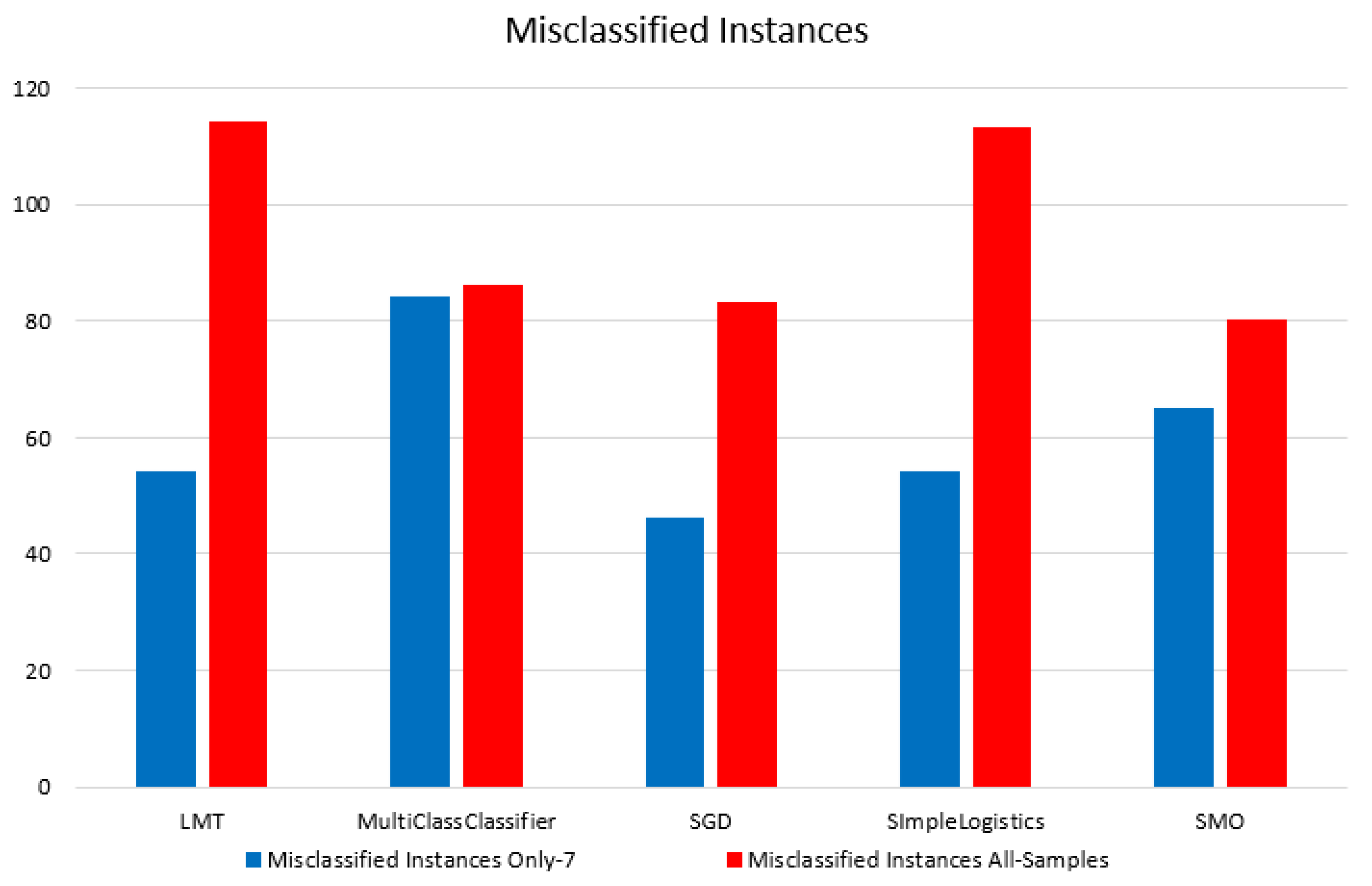
3.3. Results of Classification Based on Different Models
3.4. Stacking Results
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodney, S.; Shah, T.T.; Patel, H.R.; Arya, M. Key papers in prostate cancer. Expert Rev. Anticancer Ther. 2014, 14, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.J.; George, A.K.; Maruf, M.; Frye, T.P.; Muthigi, A.; Kongnyuy, M.; Valayil, S.G.; Pinto, P.A. Risk stratification of prostate cancer: Integrating multiparametric MRI, nomograms and biomarkers. Future Oncol. 2016, 12, 2417–2430. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Allsbrook, W.C., Jr.; Amin, M.B.; Egevad, L.L.; ISUP Grading Committee. The 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 2005, 29, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Allsbrook, W.C., Jr.; Amin, M.B.; Egevad, L.L.; ISUP Grading Committee. The 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma: Definition of Grading Patterns and Proposal for a New Grading System. Am. J. Surg. Pathol. 2016, 40, 244–252. [Google Scholar] [CrossRef]
- Lavi, A.; Cohen, M. Prostate cancer early detection using psacurrent trends and recent updates. Harefuah 2017, 156, 185–188. [Google Scholar]
- Moyer, V.A.; U.S. Preventive Services Task Force. Screening for prostate cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2012, 157, 120–134. [Google Scholar] [CrossRef]
- Lin, J.S.; O’Connor, E.A.; Evans, C.V.; Senger, C.A.; Rowland, M.G.; Groom, H.C. US Preventive Services Task Force evidence syntheses, formerly systematic evidence reviews. In Screening for Colorectal Cancer: A Systematic Review for the U.S. Preventive Services Task Force; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2016. [Google Scholar]
- Yang, L.; Wang, S.; Zhou, M.; Chen, X.; Jiang, W.; Zuo, Y.; Lv, Y. Molecular classification of prostate adenocarcinoma by the integrated somatic mutation profiles and molecular network. Sci. Rep. 2017, 7, 738. [Google Scholar] [CrossRef]
- Danaee, P.; Ghaeini, R.; Hendrix, D.A. A deep learning approach for cancer detection and relevant gene identification. In Pacific Symposium on Biocomputing 2017; World Scientific: Singapore, 2017; pp. 219–229. [Google Scholar]
- Takeuchi, T.; Hattori-Kato, M.; Okuno, Y.; Iwai, S.; Mikami, K. Prediction of prostate cancer by deep learning with multilayer artificial neural network. Can. Urol. Assoc. J. 2019, 13, E145–E150. [Google Scholar] [CrossRef]
- Wulczyn, E.; Nagpal, K.; Symonds, M.; Moran, M.; Plass, M.; Reihs, R.; Nader, F.; Tan, F.; Cai, Y.; Brown, T.; et al. Predicting prostate cancer specific-mortality with artificial intelligence-based Gleason grading. Commun. Med. 2021, 1, 1–8. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. The Cancer Genome Atlas Research Network. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef]
- The Genomics Data Commons. Available online: https://portal.gdc.cancer.gov/ (accessed on 26 September 2023).
- Bekelman, J.E.; Rumble, R.B.; Chen, R.C.; Pisansky, T.M.; Finelli, A.; Feifer, A.; Nguyen, P.L.; Loblaw, D.A.; Tagawa, S.T.; Gillessen, S.; et al. Clinically Localized Prostate Cancer: ASCO Clinical Practice Guideline Endorsement of an American Urological Association/American Society for Radiation Oncology/Society of Urologic Oncology Guideline. J. Clin. Oncol. 2018, 36, 3251–3258. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Witten, D.M.; Johnstone, I.M.; Tibshirani, R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics 2012, 13, 523–538. [Google Scholar] [CrossRef]
- Mamidi, T.K.K.; Wu, J.; Hicks, C. Interactions between Germline and Somatic Mutated Genes in Aggressive Prostate Cancer. Prostate Cancer 2019, 2019, 4047680. [Google Scholar] [CrossRef]
- Doyle, M.; Phipson, B.; Ritchie, M.; Doyle, M.; Dashnow, H.; Law, C. RNA-Seq Analysis in R. Available online: http://combine-australia.github.io/2016-05-11-RNAseq/ (accessed on 26 September 2023).
- Brownlee, J. How to Run Your First Classifier in Weka. Mach. Learn. Mastery. 2020. Available online: https://machinelearningmastery.com/how-to-run-your-first-classifier-in-weka/ (accessed on 26 September 2023).
- Kuchi, A.; Hoque, M.T.; Abdelguerfi, M.; Flanagin, M.C. Machine learning applications in detecting sand boils from images. Array 2019, 3–4, 100012. [Google Scholar] [CrossRef]
- Vapnik, V.N. An overview of statistical learning theory. IEEE Trans. Neural Netw. 1999, 10, 988–999. [Google Scholar] [CrossRef]
- Szilágyi, A.; Skolnick, J. Efficient Prediction of Nucleic Acid Binding Function from Low-resolution Protein Structures. J. Mol. Biol. 2006, 358, 922–933. [Google Scholar] [CrossRef]
- Ho, T.K. Random decision forests. In Proceedings of the Proceedings of 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 271, pp. 278–282. [Google Scholar]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Altman, N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am. Stat. 1992, 46, 175–185. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016. [Google Scholar]
- Gattani, S.; Mishra, A.; Hoque, T. StackCBPred: A stacking based prediction of protein-carbohydrate binding sites from sequence. Carbohydr. Res. 2019, 486, 107857. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Merchante, C.; Stepanova, A.N.; Alonso, J.M.; Heber, S. A Stacking-Based Approach to Identify Translated Upstream Open Reading Frames in Arabidopsis Thaliana. In Book A Stacking-Based Approach to Identify Translated Upstream Open Reading Frames in Arabidopsis Thaliana; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; pp. 138–149. [Google Scholar]
- Iqbal, S.; Hoque, M. PBRpredict-Suite: A Suite of Models to Predict Peptide Recognition Domain Residues from Protein Sequence. Bioinformatics 2018, 34, 3289–3299. [Google Scholar] [CrossRef]
- Mishra, A.; Pokhrel, P.; Hoque, T. StackDPPred: A stacking based prediction of DNA-binding protein from sequence. Bioinformatics 2018, 35, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Flot, M.; Mishra, A.; Kuchi, A.S.; Hoque, T. StackSSSPred: A Stacking-Based Prediction of Supersecondary Structure from Sequence. Protein Supersecondary Struct. Methods Protoc. 2019, 1958, 101–122. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar]
- Casey, M.; Chen, B.; Zhou, J.; Zhou, N. A machine learning approach to prostate cancer risk classification through use of RNA sequencing data. In International Conference on Big Data; Springer International Publishing: Cham, Switzerland, 2019; pp. 65–79. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SVM | LR | RF | ETC | GBC | KNNs | XGB |
|---|---|---|---|---|---|---|
| C = 1.0 | penalty = “l2” | n_estimators = 100 | n_estimators = 100 | n_estimators = 100 | n_neighbors = 5 | n_estimatorst = 100 |
| kernel = “rbf” | tol = 1 × 10−4 | criterion = “gini” | criterion = “gini” | loss = “log_loss” | weights = “uniform” | learning_rate = 0.3 |
| gamma = “scale” | C = 1.0 | max_depth = None | max_depth = None | learning_rate = 0.1 | algorithm = “ball_tree” | max_deptht = 10 |
| Name of Metric | Definition |
|---|---|
| True positive (TP) | Correctly predicted positive samples |
| True negative (TN) | Correctly predicted negative samples |
| False positive (FP) | Incorrectly predicted positive samples |
| False negative (FN) | Incorrectly predicted negative samples |
| Recall/sensitivity/true positive rate (TPR) | |
| Specificity/true negative rate (TNR) | |
| Fall-out rate/false positive rate (FPR) | |
| Miss rate/false negative rate (FNR) | |
| Accuracy (ACC) | |
| Balanced accuracy (Bal_ACC) | |
| Precision | |
| F1 score (harmonic mean of precision and recall) | |
| Mathews correlation coefficient (MCC) |
| LogFC Cutoff | No. of Genes | ||
|---|---|---|---|
| Model 1 | Model 2 | Model 3 | |
| 0.5 | 2074 | 3513 | 513 |
| 0.7 | 821 | 2028 | 381 |
| 1 | 213 | 836 | 174 |
| 1.5 | 24 | 186 | 25 |
| 2 | 3 | 52 | 5 |
| Metric/ Method | LR | ETC | KNNs | SVM | GBC | RF | XGB |
|---|---|---|---|---|---|---|---|
| Sensitivity | 0.85 | 0.93 | 0.86 | 0.91 | 0.91 | 0.92 | 0.90 |
| Specificity | 0.67 | 0.49 | 0.59 | 0.69 | 0.54 | 0.51 | 0.67 |
| Bal. acc. | 0.76 | 0.72 | 0.72 | 0.90 | 0.72 | 0.71 | 0.80 |
| Accuracy | 0.80 | 0.82 | 0.79 | 0.86 | 0.82 | 0.82 | 0.85 |
| Precision | 0.88 | 0.84 | 0.86 | 0.90 | 0.86 | 0.85 | 0.89 |
| F1 score | 0.87 | 0.88 | 0.86 | 0.90 | 0.88 | 0.88 | 0.89 |
| MCC | 0.50 | 0.84 | 0.45 | 0.61 | 0.49 | 0.49 | 0.61 |
| Method/ Metric | Sensitivity | Specificity | Accuracy | Precision | F1 Score | MCC | Balanced Accuracy |
|---|---|---|---|---|---|---|---|
| SM-1 | 0.99 | 0.85 | 0.96 | 0.95 | 0.97 | 0.88 | 0.92 |
| SM-2 | 0.96 | 0.83 | 0.93 | 0.95 | 0.95 | 0.81 | 0.87 |
| SM-3 | 0.97 | 0.84 | 0.93 | 0.95 | 0.96 | 0.84 | 0.91 |
| SM-4 | 0.98 | 0.68 | 0.91 | 0.90 | 0.94 | 0.74 | 0.83 |
| SM-5 | 0.94 | 0.81 | 0.91 | 0.94 | 0.94 | 0.74 | 0.87 |
| Method/ Metric | Sensitivity | Specificity | Accuracy | Precision | F1 Score | MCC | Balanced Accuracy |
|---|---|---|---|---|---|---|---|
| SM-1 | 0.98 | 0.90 | 0.97 | 0.99 | 0.98 | 0.87 | 0.94 |
| SM-2 | 0.95 | 0.79 | 0.94 | 0.97 | 0.96 | 0.72 | 0.91 |
| SM-3 | 0.96 | 0.79 | 0.95 | 0.97 | 0.97 | 0.72 | 0.92 |
| SM-4 | 0.98 | 0.62 | 0.94 | 0.95 | 0.96 | 0.66 | 0.80 |
| SM-5 | 0.98 | 0.59 | 0.93 | 0.95 | 0.96 | 0.64 | 0.78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamidi, Y.K.K.; Mamidi, T.K.K.; Kabir, M.W.U.; Wu, J.; Hoque, M.T.; Hicks, C. PCa-Clf: A Classifier of Prostate Cancer Patients into Patients with Indolent and Aggressive Tumors Using Machine Learning. Mach. Learn. Knowl. Extr. 2023, 5, 1302-1319. https://doi.org/10.3390/make5040066
Mamidi YKK, Mamidi TKK, Kabir MWU, Wu J, Hoque MT, Hicks C. PCa-Clf: A Classifier of Prostate Cancer Patients into Patients with Indolent and Aggressive Tumors Using Machine Learning. Machine Learning and Knowledge Extraction. 2023; 5(4):1302-1319. https://doi.org/10.3390/make5040066
Chicago/Turabian StyleMamidi, Yashwanth Karthik Kumar, Tarun Karthik Kumar Mamidi, Md Wasi Ul Kabir, Jiande Wu, Md Tamjidul Hoque, and Chindo Hicks. 2023. "PCa-Clf: A Classifier of Prostate Cancer Patients into Patients with Indolent and Aggressive Tumors Using Machine Learning" Machine Learning and Knowledge Extraction 5, no. 4: 1302-1319. https://doi.org/10.3390/make5040066
APA StyleMamidi, Y. K. K., Mamidi, T. K. K., Kabir, M. W. U., Wu, J., Hoque, M. T., & Hicks, C. (2023). PCa-Clf: A Classifier of Prostate Cancer Patients into Patients with Indolent and Aggressive Tumors Using Machine Learning. Machine Learning and Knowledge Extraction, 5(4), 1302-1319. https://doi.org/10.3390/make5040066