
Electronic Characteristics of Layered Heterostructures Based on Graphene and Two-Dimensional Perovskites: First-Principle Study

 , , ,
, , ,  ,
,  and
and 
Abstract
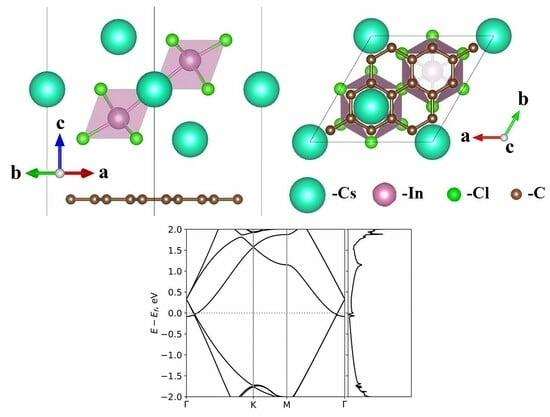
:
1. Introduction
2. Methods of Calculation
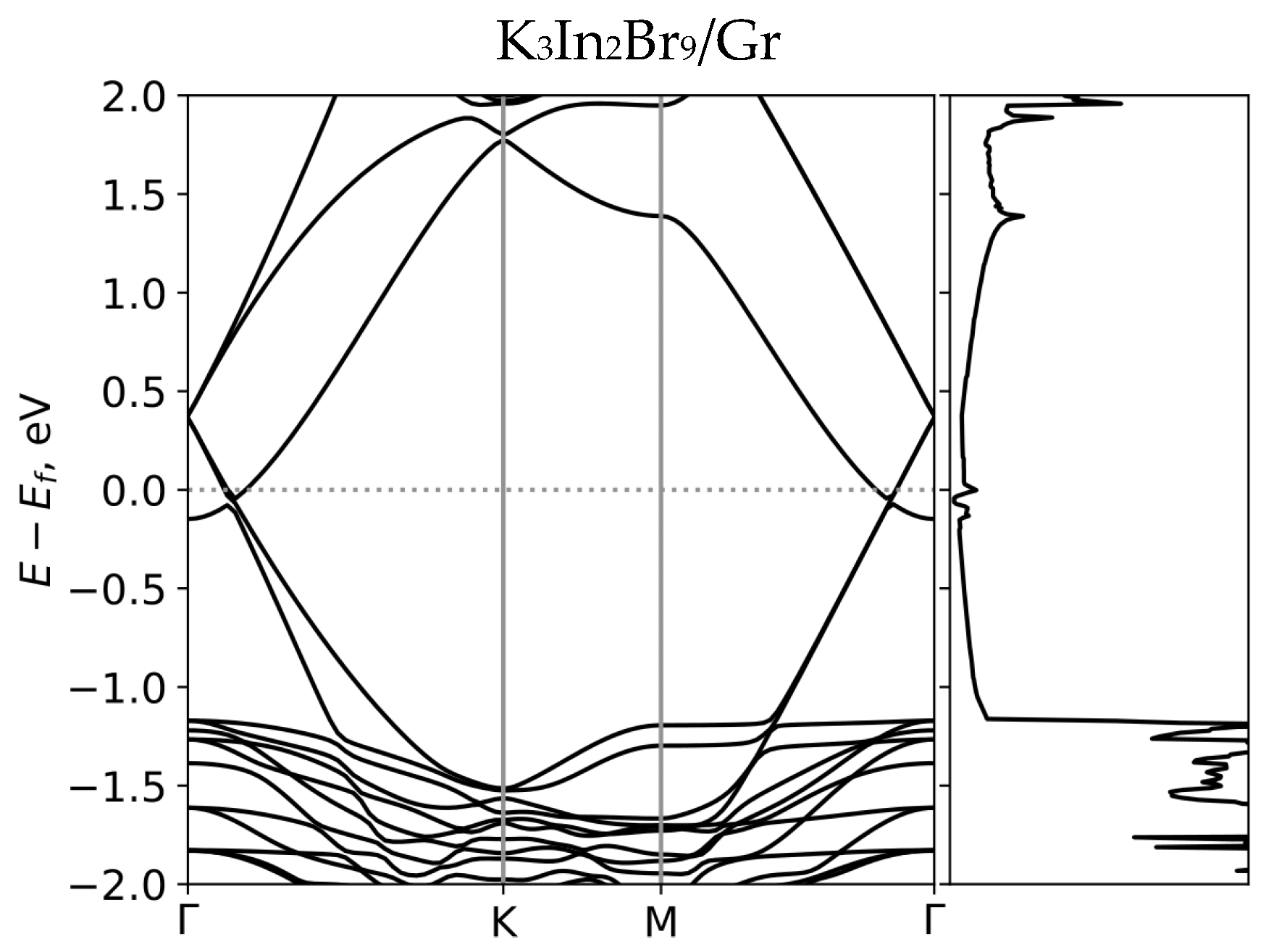
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Gao, X.; Zhang, X.; Shen, X.; Lu, M.; Wu, J.; Shi, Z.; Colvin, V.L.; Hu, J.; Bai, X.; et al. Lead-Free Halide Perovskites for Light Emission: Recent Advances and Perspectives. Adv. Sci. 2021, 8, 2003334. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Su, L.; Lu, Y.; Fang, L.; Shu, H.; Cao, D.; Chen, X. Computational Screening of 2D All-Inorganic Lead-Free Halide Perovskites A3B2X9 for Photovoltaic and Photocatalytic Applications. Adv. Theory Simul. 2024, 7, 2300988. [Google Scholar] [CrossRef]
- Zhao, S.; Lan, C.; Li, H.; Zhang, C.; Ma, T. Aurivillius Halide Perovskite: A New Family of Two-Dimensional Materials for Optoelectronic Applications. J. Phys. Chem. C 2020, 124, 1788–1793. [Google Scholar] [CrossRef]
- Shao, M.; Bie, T.; Yang, L.; Gao, Y.; Jin, X.; He, F.; Zheng, N.; Yu, Y.; Zhang, X. Over 21% Efficiency Stable 2D Perovskite Solar Cells. Adv. Mater. 2022, 34, 2107211. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, G.; Hu, T.; Chen, Y.; Cao, S.; Bellaiche, L.; Ren, W. Ferromagnetism, Jahn-Teller effect, and orbital order in the two-dimensional monolayer perovskite Rb2CuCl4. Phys. Rev. B 2021, 104, L241105. Available online: https://link.springer.com/article/10.1007/s12596-024-01819-9#citeas (accessed on 1 April 2025).
- Yang, Y.; Lou, F.; Xiang, H. Cooperative Nature of Ferroelectricity in Two-Dimensional Hybrid Organic-Inorganic Perovskites. Nano Lett. 2021, 21, 3170–3176. [Google Scholar] [CrossRef]
- Ma, K.; Li, X.; Yang, F.; Liu, H. Lead Leakage of Pb-Based Perovskite Solar Cells. Coatings 2023, 13, 1009. [Google Scholar] [CrossRef]
- Su, P.; Liu, Y.; Zhang, J.; Chen, C.; Yang, B.; Zhang, C.; Zhao, X. Pb-Based Perovskite Solar Cells and the Underlying Pollution behind Clean Energy: Dynamic Leaching of Toxic Substances from Discarded Perovskite Solar Cells. J. Phys. Chem. Lett. 2020, 11, 2812–2817. [Google Scholar] [CrossRef]
- Guo, W.H.; Shi, J.-J.; Zhu, Y.-H.; Wu, M.; Du, J.; Cen, Y.-L.; Liu, S.-M.; Han, S.-P. Two-Dimensional 111-Type in -Based Halide Perovskite Cs3In2X9 (X=Cl, Br, I) with Optimal Band Gap for Photovoltaics and Defect-Insensitive Blue Emission. Phys. Rev. Appl. 2020, 13, 024031. [Google Scholar] [CrossRef]
- Pradhan, A.; Jena, M.K.; Samal, S.L. Understanding of the Band Gap Transition in Cs3Sb2Cl9- xBrx: Anion Site Preference-Induced Structural Distortion. ACS Appl. Energy Mater. 2022, 5, 6952–6961. [Google Scholar] [CrossRef]
- Pradhan, A.; Sahoo, S.C.; Sahu, A.K.; Samal, S.L. Effect of Bi Substitution on Cs3Sb2Cl9: Structural Phase Transition and Band Gap Engineering. Cryst. Growth Des. 2020, 20, 3386–3395. [Google Scholar] [CrossRef]
- Wang, X.D.; Miao, N.-H.; Liao, J.-F.; Li, W.-Q.; Xie, Y.; Chen, J.; Sun, Z.-M.; Chen, H.-Y.; Kuang, D.-B. The top-down synthesis of single-layered Cs4CuSb2Cl12 halide perovskite nanocrystals for photoelectrochemical application. Nanoscale 2019, 11, 5180–5187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Deng, H.; Farooq, U.; Yang, X.; Khan, J.; Tang, J.; Song, H. High Quantum Yield Blue Emission from Lead-Free Inorganic Antimony Halide Perovskite Colloidal Quantum Dots. ACS Nano 2017, 11, 9294–9302. [Google Scholar] [CrossRef] [PubMed]
- Kalesaki, E.; Boneschanscher, M.P.; Geuchies, J.; Delerue, C.; Delerue, C.; Smith, C.M.; Evers, W.; Allan, G.; Altantzis, T.; Bals, S.; et al. Preparation and Study of 2-D Semiconductors with Dirac Type Bands due to the Honeycomb Nanogeometry, in Physics. In Simulation, and Photonic Engineering of Photovoltaic Devices III, Proceedings of the SPIE Photonics West, San Francisco, CA, USA, 1–6 February 2014; SPIE: Bellingham, WC, USA, 2014; p. 898107. [Google Scholar] [CrossRef]
- Louie, S.G.; Park, C.H. Making massless dirac fermions from a patterned two-dimensional electron gas. Nano Lett. 2009, 9, 1793–1797. [Google Scholar] [CrossRef]
- Li, Z.; Han, Y.; Qiao, Z. Large-Gap Quantum Anomalous Hall Effect in Monolayer Halide Perovskite. Phys. Rev. B 2021, 104, 205401. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Shen, D. A simple rule for finding Dirac cones in bilayered perovskites. Chin. Phys. B 2019, 28, 077106. [Google Scholar] [CrossRef]
- Guo, S.; Bu, K.; Li, J.; Hu, Q.; Luo, H.; He, Y.; Wu, Y.; Zhang, D.; Zhao, Y.; Yang, W.; et al. Enhanced Photocurrent of All-Inorganic Two-Dimensional Perovskite Cs2PbI2Cl2 via Pressure-Regulated Excitonic Features. J. Am. Chem. Soc. 2021, 143, 2545–2551. [Google Scholar] [CrossRef]
- Saffari, M.; Soleimani, H.R.; Tagani, M.B. Quantum confinement and strain effects on the low-dimensional all-inorganic halide Cs2XI2Cl2 (X= Pb, Sn) perovskites: A theoretical approach for modulating electronic and optical properties. Phys. E Low-Dimens. Syst. Nanostructures 2020, 124, 114226. [Google Scholar] [CrossRef]
- Hwang, J.; Feng, Z.; Charles, N.; Wang, X.R.; Lee, D.; Stoerzinger, K.A.; Muy, S.; Rao, R.R.; Lee, D.; Jacobs, R.; et al. Tuning perovskite oxides by strain: Electronic structure, properties, and functions in (electro)catalysis and ferroelectricity. Mater. Today 2019, 31, 100–118. [Google Scholar] [CrossRef]
- Torma, A.J.; Li, W.; Zhang, H.; Tu, Q.; Klepov, V.V.; Brennan, M.C.; McCleese, C.L.; Krzyaniak, M.D.; Wasielewski, M.R.; Katan, C.; et al. Interstitial Nature of Mn2+ Doping in 2D Perovskites. ACS Nano 2021, 15, 20550–20561. [Google Scholar] [CrossRef]
- Liu, G.; Kong, L.; Guo, P.; Stoumpos, C.C.; Hu, Q.; Liu, Z.; Cai, Z.; Gosztola, D.J.; Mao, H.-K.; Kanatzidis, M.G.; et al. Two Regimes of Bandgap Red Shift and Partial Ambient Retention in Pressure-Treated Two-Dimensional Perovskites. ACS Energy Lett. 2017, 2, 2518–2524. [Google Scholar] [CrossRef]
- Tan, R.; Zou, C.; Pan, K.; Zou, D.; Liu, Y. Enhancing thermoelectric properties of BiCuSeO via uniaxial compressive strain: First-principles calculations. J. Alloys Compd. 2018, 743, 610–617. [Google Scholar] [CrossRef]
- Wang, M.; Hou, T.-P.; Wu, K.-M.; Lin, H.-F. Graphene/Cs2PbI2Cl2 van der Waals heterostructure with tunable Schottky barriers and contact types. J. Appl. Phys. 2022, 132, 165101. [Google Scholar] [CrossRef]
- Sun, Y.; Morice, C.; Garrot, D.; Weil, R.; Watanabe, K.; Taniguchi, T.; Monteverde, M.; Chepelianskii, A.D. Quantum Transport and Spectroscopy of 2D Perovskite/Graphene Heterostructures. Adv. Elect. Mater. 2024, 10, 2400211. [Google Scholar] [CrossRef]
- Hu, J.; Ji, G.; Ma, X.; He, H.; Huang, C. Probing interfacial electronic properties of graphene/CH3NH3PbI3 heterojunctions: A theoretical study. Appl. Surf. Sci. 2018, 440, 35–41. [Google Scholar] [CrossRef]
- Zibouche, N.; Volonakis, G.; Giustino, F. Graphene Oxide/Perovskite Interfaces for Photovoltaics. J. Phys. Chem. C 2018, 122, 16715–16726. [Google Scholar] [CrossRef]
- Agresti, A.; Pescetelli, S.; Taheri, B.; Del Rio Castillo, A.E.; Cinà, L.; Bonaccorso, F.; Di Carlo, A. Graphene–Perovskite Solar Cells Exceed 18% Efficiency: A Stability Study. ChemSusChem 2016, 9, 2609–2619. [Google Scholar] [CrossRef]
- Zhao, L.; Tian, H.; Silver, S.H.; Kahn, A.; Ren, T.-L.; Rand, B.P. Ultrasensitive Heterojunctions of Graphene and 2D Perovskites Reveal Spontaneous Iodide Loss. Joule 2018, 2, 2133–2144. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Revised values of effective ionic radii. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 1046–1048. [Google Scholar] [CrossRef]
- Tiwari, D.; Hutter, O.S.; Longo, G. Chalcogenide perovskites for photovoltaics: Current status and prospects. J. Phys. Energy 2021, 3, 034010. [Google Scholar] [CrossRef]
- Jacobsson, T.J.; Pazoki, M.; Hagfeldt, A.; Edvinsson, T. Goldschmidt’s Rules and Strontium Replacement in Lead Halogen Perovskite Solar Cells: Theory and Preliminary Experiments on CH3NH3SrI3. J. Phys. Chem. C 2015, 119, 25673–25683. [Google Scholar] [CrossRef]
- Xia, M.; Yuan, J.-H.; Niu, G.; Du, X.; Yin, L.; Pan, W.; Luo, J.; Li, Z.; Zhao, H.; Xue, K.-H.; et al. Unveiling the Structural Descriptor of A3B2X9 Perovskite Derivatives toward X-Ray Detectors with Low Detection Limit and High Stability. Adv. Funct. Mater. 2020, 30, 1910648. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Yun, Y.; Shin, S.; Lee, J.H.; Heo, Y.-W.; Lee, S. Wide range tuning of band gap energy of A3B2X9 perovskite-like halides. Scr. Mater. 2019, 166, 107–111. [Google Scholar] [CrossRef]
- Peresh, E.Y.; Sidei, V.I.; Zubaka, O.V.; Stercho, I.P. K2(Rb2,Cs2,Tl2)TeBr6(I6) and Rb3(Cs3)Sb2(Bi2)Br9(I9) perovskite compounds. Inorg. Mater. 2011, 47, 208–212. [Google Scholar] [CrossRef]
- Saparov, B.; Hong, F.; Sun, J.-P.; Duan, H.-S.; Meng, W.; Cameron, S.; Hill, I.G.; Yan, Y.; Mitzi, D.B. Thin-Film Preparation and Characterization of Cs3Sb2I9: A Lead-Free Layered Perovskite Semiconductor. Chem. Mater. 2015, 27, 5622–5632. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter. 2017, 29, 465901. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Itanze, D.S.; Aragon, A.G.; Ma, X.; Li, H.; Ucer, K.B.; Hewitt, C.; Carroll, D.L.; Williams, R.T.; Qiu, Y.; et al. Synthesis of lead-free Cs3Sb2Br9 perovskite alternative nanocrystals with enhanced photocatalytic CO2 reduction activity. Nanoscale 2020, 12, 2987–2991. [Google Scholar] [CrossRef]
- Long, Y.; Zhang, H.; Cheng, X. Stability, electronic structure, and optical properties of lead-free perovskite monolayer Cs3B2X9 (B = Sb, Bi; X = Cl, Br, I) and bilayer vertical heterostructure Cs3B2X9/Cs3B2’X9 (B,B’ = Sb, Bi; X = Cl, Br, I). Chin. Phys. B 2022, 31, 027102. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Yang, C.-L.; Wang, M.-S.; Ma, X.-G.; Yi, Y.-G. Theoretical insight into the optoelectronic properties of lead-free perovskite derivatives of Cs3Sb2X9 (X = Cl, Br, I). J. Mater. Sci. 2019, 54, 4732–4741. [Google Scholar] [CrossRef]
- Laref, A.; Ahmed, A.; Bin-Omran, S.; Luo, S.J. First-principle analysis of the electronic and optical properties of boron and nitrogen doped carbon mono-layer graphenes. Carbon 2015, 81, 179–192. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.K.; Sinha, S.R.P. Fermi-Level Modulation of Chemical Vapor Deposition-Grown Monolayer Graphene via Nanoparticles to Macromolecular Dopants. ACS Omega 2022, 7, 744–751. [Google Scholar] [CrossRef]
- Adhikari, S.; Perello, D.J.; Biswas, C.; Ghosh, A.; Van Luan, N.; Park, J.; Yao, F.; Rotkinac, S.V.; Lee, Y.H. Determining the Fermi level by absorption quenching of monolayer graphene by charge transfer doping. Nanoscale 2016, 8, 18710–18717. [Google Scholar] [CrossRef]
- Madito, M.J. Correlation of the Graphene Fermi-Level Shift and the Enhanced Electrochemical Performance of Graphene-Manganese Phosphate for Hybrid Supercapacitors: Raman Spectroscopy Analysis. ACS Appl. Mater. Interfaces 2021, 13, 37014–37026. [Google Scholar] [CrossRef]
- Hui, Z.; Wang, M.; Chen, J.; Yin, X.; Yue, Y.; Lu, J. Predicting photovoltaic parameters of perovskite solar cells using machine learning. J. Phys. Condens. Matter. 2024, 36, 355901. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.A.M.; Lee, S.; Kim, K.H.; Kim, J.; Jeong, K. Machine Learning Approach for Predicting the Hole Mobility of the Perovskite Solar Cells. Adv. Theory Sims 2024, 7, 2300978. [Google Scholar] [CrossRef]
- He, Z.; Peng, J.; Lei, C.; Xie, S.; Zou, D.; Liu, Y. Prediction of superior thermoelectric performance in unexplored doped-BiCuSeO via machine learning. Mater. Des. 2023, 229, 111868. [Google Scholar] [CrossRef]
- Mammeri, M.; Bencherif, H.; Dehimi, L.; Hajri, A.; Sasikumar, P.; Syed, A.; AL-Shwaiman, H.A. Stability forecasting of perovskite solar cells utilizing various machine learning and deep learning techniques. J. Opt. 2024. [Google Scholar] [CrossRef]



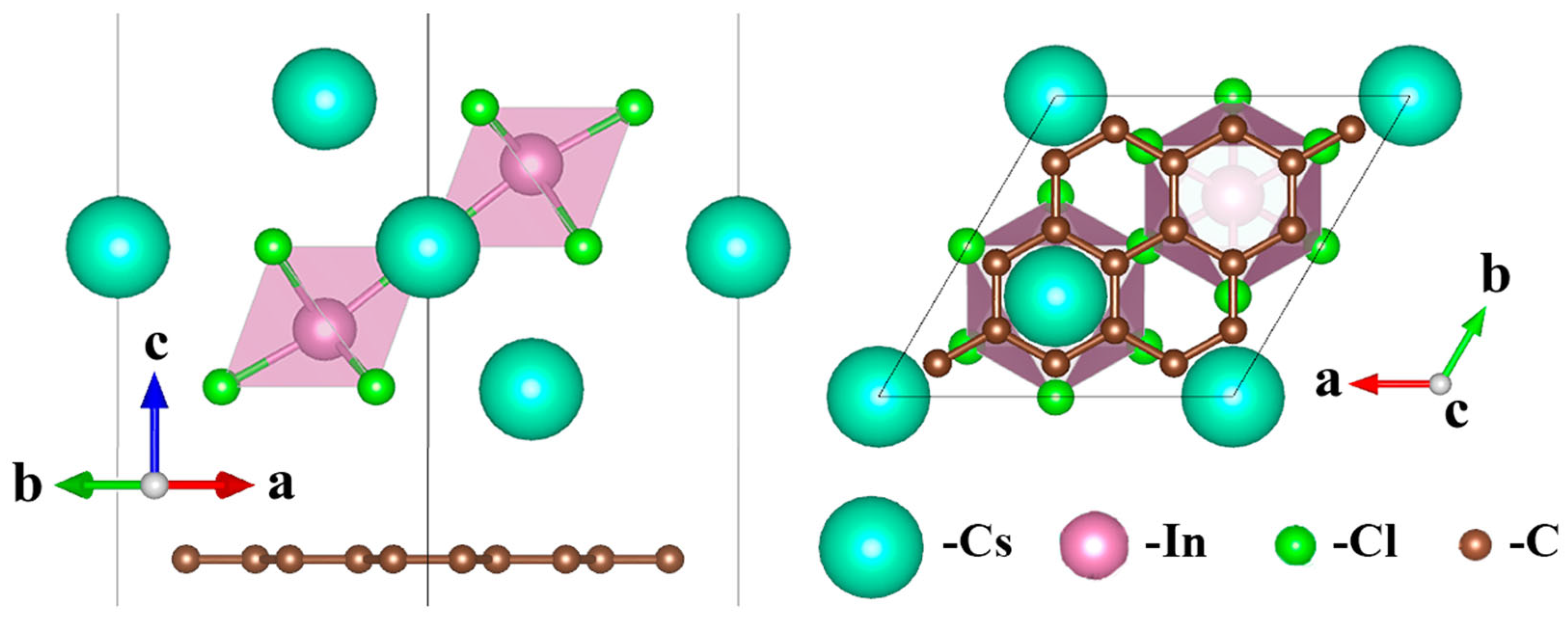

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | t | a = b, Å | c, Å | Band Gap, eV (Direct/Indirect) | B-X, Terminal Bond, Å | B-X, Bridging Bond, Å | |
|---|---|---|---|---|---|---|---|
| Cs3In2Cl9 | 0.442 | 0.973 | 7.457 (7.45 [9]) | 6.057 | 2.00 (D) | 2.448 | 2.770 |
| Cs3In2Br9 | 0.408 | 0.967 | 7.848 (7.86 [9]) | 6.272 | 1.18 (D) | 2.601 | 2.955 |
| Rb3In2Cl9 | 0.442 | 0.940 | 7.328 (7.327 [2]) | 5.819 | 2.04 (D) | 2.439 | 2.755 |
| Rb3In2Br9 | 0.408 | 0.927 | 7.744 (7.752 [2]) | 6.235 | 1.16 (D) | 2.591 | 2.945 |
| Cs3Sb2Cl9 | 0.420 | 0.997 | 7.712 (7.715 [46]) | 6.312 | 2.76 (D) | 2.519 | 2.869 |
| Cs3Sb2Br9 | 0.388 | 0.972 | 8.026 (7.935 [46]) | 6.693 | 2.32 (I) | 2.679 | 3.014 |
| Rb3Sb2Cl9 | 0.420 | 0.971 | 7.561 (7.561 [2]) | 6.320 | 2.73(I) | 2.513 | 2.859 |
| Rb3Sb2Br9 | 0.388 | 0.957 | 7.906 (7.848 [35]) | 6.692 | 2.27 (I) | 2.671 | 3.008 |
| K3In2Cl9 | 0.442 | 0.933 | 7.261 (7.264 [2]) | 5.775 | 2.05 (D) | 2.430 | 2.751 |
| K3In2Br9 | 0.408 | 0.907 | 7.693 (7.693 [2]) | 6.192 | 1.16 (D) | 2.582 | 2.946 |
| Compound | a = b, Å | c, Å | d, Å | Downshifting of the Fermi Level, eV | B-X, Terminal Bond, Å | B-X, Bridging Bond, Å | Compound |
|---|---|---|---|---|---|---|---|
| Cs3In2Cl9 | 7.403 | 6.071 | 3.610 | 0.326 | 2.450 | 2.755 | Cs3In2Cl9 |
| Cs3In2Br9 | 7.444 | 6.563 | 3.660 | 0.328 | 2.592 | 2.877 | Cs3In2Br9 |
| Rb3In2Cl9 | 7.390 | 5.793 | 3.548 | 0.253 | 2.445 | 2.753 | Rb3In2Cl9 |
| Rb3In2Br9 | 7.429 | 6.393 | 3.578 | 0.341 | 2.583 | 2.871 | Rb3In2Br9 |
| Cs3Sb2Cl9 | 7.418 | 6.404 | 3.579 | - | 2.522 | 2.823 | Cs3Sb2Cl9 |
| Cs3Sb2Br9 | 7.452 | 6.965 | 3.636 | - | 2.664 | 2.941 | Cs3Sb2Br9 |
| Rb3Sb2Cl9 | 7.405 | 6.296 | 3.488 | - | 2.519 | 2.814 | Rb3Sb2Cl9 |
| Rb3Sb2Br9 | 7.437 | 6.877 | 3.560 | - | 2.657 | 2.931 | Rb3Sb2Br9 |
| K3In2Cl9 | 7.382 | 5.693 | 3.459 | 0.279 | 2.440 | 2.754 | K3In2Cl9 |
| K3In2Br9 | 7.419 | 6.316 | 3.545 | 0.364 | 2.575 | 2.871 | K3In2Br9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubkov, L.; Kulyamin, P.; Grishakov, K.; Kaya, S.; Katin, K.; Maslov, M. Electronic Characteristics of Layered Heterostructures Based on Graphene and Two-Dimensional Perovskites: First-Principle Study. Colloids Interfaces 2025, 9, 23. https://doi.org/10.3390/colloids9020023
Zubkov L, Kulyamin P, Grishakov K, Kaya S, Katin K, Maslov M. Electronic Characteristics of Layered Heterostructures Based on Graphene and Two-Dimensional Perovskites: First-Principle Study. Colloids and Interfaces. 2025; 9(2):23. https://doi.org/10.3390/colloids9020023
Chicago/Turabian StyleZubkov, Lev, Pavel Kulyamin, Konstantin Grishakov, Savaş Kaya, Konstantin Katin, and Mikhail Maslov. 2025. "Electronic Characteristics of Layered Heterostructures Based on Graphene and Two-Dimensional Perovskites: First-Principle Study" Colloids and Interfaces 9, no. 2: 23. https://doi.org/10.3390/colloids9020023
APA StyleZubkov, L., Kulyamin, P., Grishakov, K., Kaya, S., Katin, K., & Maslov, M. (2025). Electronic Characteristics of Layered Heterostructures Based on Graphene and Two-Dimensional Perovskites: First-Principle Study. Colloids and Interfaces, 9(2), 23. https://doi.org/10.3390/colloids9020023