
Real-World Data in Children with Spinal Muscular Atrophy Type 1 on Long-Term Ventilation Receiving Gene Therapy: A Prospective Cohort Study



{kind=link}
{kind=link}
Abstract
Highlights
- Gene therapy at a late age was safe and effective in resolving paradoxical breathing, improving cough ability, reducing airway secretions, and enhancing CHOP-INTEND scores over a short-term period in patients with SMA-1 ventilated via tracheostomy.
- The clinical assessment and management implemented pre-gene therapy were effective in safely weaning patients for at least 8 h off ventilator per day.
- Older patients with SMA-1 still have a chance for improvement if presented late for gene therapy.
- In conjunction with gene therapy, high-quality clinical care is beneficial and should be paired with gene therapy management.
Abstract
1. Introduction
1.1. Definition
1.2. Clinical Features
1.3. Gene Therapy
1.4. Aims
2. Materials and Methods
2.1. Design and Setting
2.2. Study Size
2.3. Assessment and Management
2.4. Gene Therapy Infusion
2.5. Data Collection and Analysis
2.6. Follow-Up Period
2.7. Ethical Approval
3. Results
3.1. Demographics and Characteristics
3.2. Gene Therapy Safety
3.3. Work of Breathing
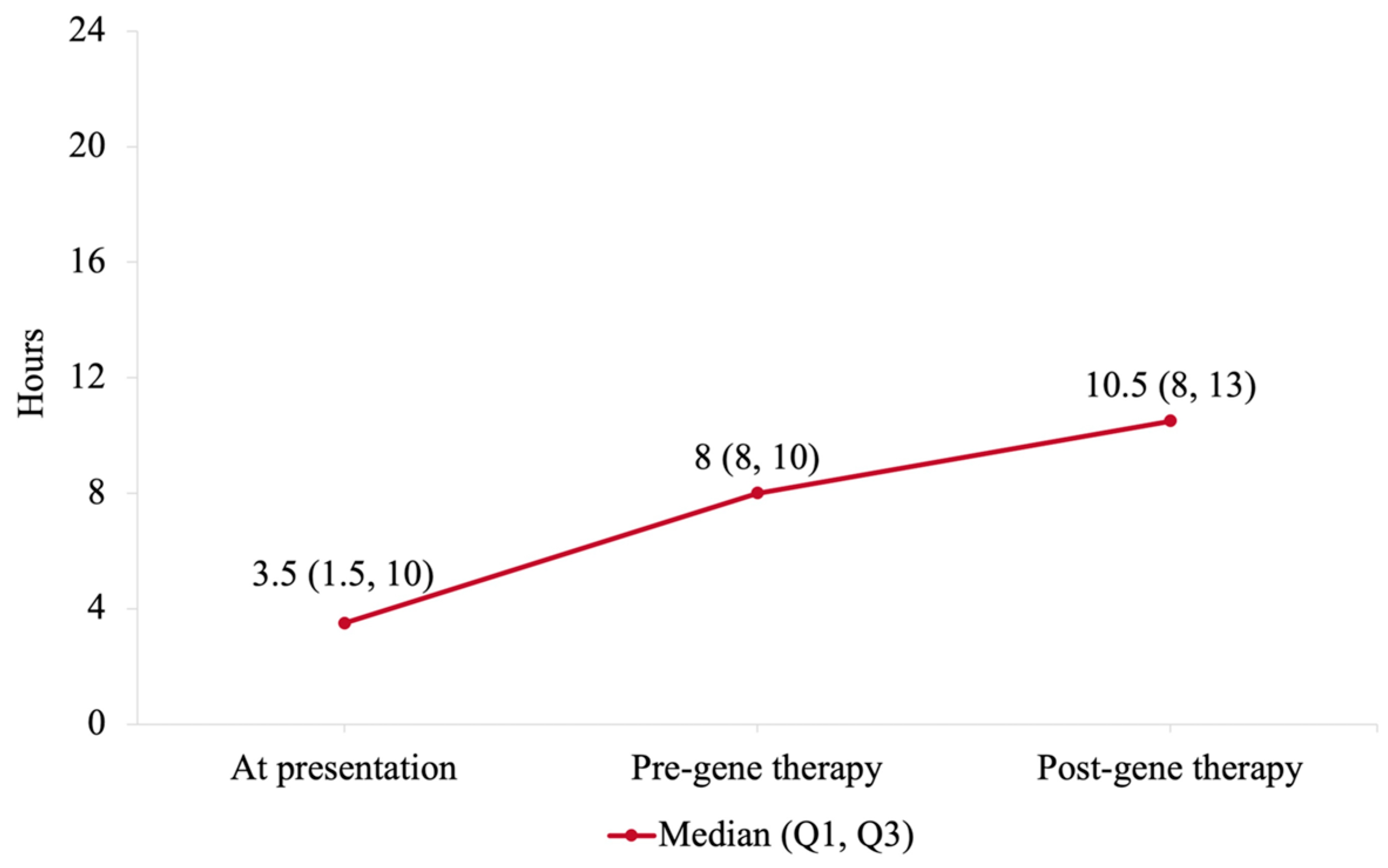
3.4. Time off Ventilator
3.5. Ventilator Settings
3.6. Polysomnography Assessment
3.7. Cough Ability and Augmentation
3.8. Airway Secretions
3.9. Bacterial Colonization and Infections
3.10. Swallowing Ability
3.11. Motor Assessment
4. Discussion
4.1. Key Findings
4.2. Results Comparison
4.3. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Farrar, M.A.; Park, S.B.; Vucic, S.; Carey, K.A.; Turner, B.J.; Gillingwater, T.H.; Swoboda, K.J.; Kiernan, M.C. Emerging therapies and challenges in spinal muscular atrophy. Ann. Neurol. 2017, 81, 355–368. [Google Scholar] [CrossRef]
- Panagiotou, P.; Kanaka-Gantenbein, C.; Kaditis, A.G. Changes in Ventilatory Support Requirements of Spinal Muscular Atrophy (SMA) Patients Post Gene-Based Therapies. Children 2022, 9, 1207. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.S.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef]
- Fauroux, B.; Griffon, L.; Amaddeo, A.; Stremler, N.; Mazenq, J.; Khirani, S.; Baravalle-Einaudi, M. Respiratory management of children with spinal muscular atrophy (SMA). Arch. Pediatr. 2020, 27, 7S29–7S34. [Google Scholar] [CrossRef]
- Ali, H.G.; Ibrahim, K.; Elsaid, M.F.; Mohamed, R.B.; Abeidah, M.I.A.; Al Rawwas, A.O.; Elshafey, K.; Almulla, H.; El-Akouri, K.; Almulla, M.; et al. Gene therapy for spinal muscular atrophy: The Qatari experience. Gene Ther. 2021, 28, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Bitetti, I.; Lanzara, V.; Margiotta, G.; Varone, A. Onasemnogene abeparvovec gene replacement therapy for the treatment of spinal muscular atrophy: A real-world observational study. Gene Ther. 2023, 30, 592–597. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Mercuri, E.; Sumner, C.J.; Muntoni, F.; Darras, B.T.; Finkel, R.S. Spinal muscular atrophy. Nat. Rev. Dis. Primers 2022, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Erdos, J.; Wild, C. Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: A systematic review of real-world study data. Eur. J. Paediatr. Neurol. 2022, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muni-Lofra, R.; Murphy, L.B.; Adcock, K.; Farrugia, M.E.; Irwin, J.; Lilleker, J.B.; McConville, J.; Merrison, A.; Parton, M.; Ryburn, L.; et al. Real-World Data on Access to Standards of Care for People With Spinal Muscular Atrophy in the UK. Front. Neurol. 2022, 13, 866243. [Google Scholar] [CrossRef] [PubMed]
- AlNaimi, A.; Hamad, S.G.; Mohamed, R.B.A.; Ben-Omran, T.; Ibrahim, K.; Osman, M.F.E.; Abu-Hasan, M. A breakthrough effect of gene replacement therapy on respiratory outcomes in children with spinal muscular atrophy. Pediatr. Pulmonol. 2023, 58, 1004–1011. [Google Scholar] [CrossRef]
- Ropars, J.; Barnerias, C.; Hully, M.; Chabalier, D.; Peudenier, S.; Barzic, A.; Cros, P.; Desguerre, I. Thoracic circumference: A new outcome measure in spinal muscular atrophy type 1? Neuromuscul. Disord. 2019, 29, 415–421. [Google Scholar] [CrossRef]
- Morley, S.L. Non-invasive ventilation in paediatric critical care. Paediatr. Respir. Rev. 2016, 20, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Khoshnood, M.; O’Rourke, D.; Zaveri, H.; Davis, L.; Strober, J.; Konersman, C.; Ramos-Platt, L. Onasemnogene Abeparvovec (Zolgensma) Decreases Ventilator/Bi-Pap Support in 3 Infants with SMA 1. Ann. Case Rep. 2021, 6, 583. [Google Scholar] [CrossRef]
- Al-Zaidy, S.; Pickard, A.S.; Kotha, K.; Alfano, L.N.; Lowes, L.; Paul, G.; Church, K.; Lehman, K.; Sproule, D.M.; Dabbous, O.; et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr. Pulmonol. 2019, 54, 179–185. [Google Scholar] [CrossRef]
- Finkel, R.S.; McDermott, M.P.; Kaufmann, P.; Darras, B.T.; Chung, W.K.; Sproule, D.M.; Kang, P.B.; Foley, A.R.; Yang, M.L.; Martens, W.B.; et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology 2014, 83, 810–817. [Google Scholar] [CrossRef]
- Han, Y.J.; Park, J.D.; Lee, B.; Choi, Y.H.; Suh, D.I.; Lim, B.C.; Chae, J. Home mechanical ventilation in childhood-onset hereditary neuromuscular diseases: 13 years’ experience at a single center in Korea. PLoS ONE 2015, 10, e0122346. [Google Scholar] [CrossRef]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: The Phase III SPR1NT trial. Nat. Med. 2022, 28, 1390–1397. [Google Scholar] [CrossRef]
- Strauss, K.A.; Farrar, M.A.; Muntoni, F.; Saito, K.; Mendell, J.R.; Servais, L.; McMillan, H.J.; Finkel, R.S.; Swoboda, K.J.; Kwon, J.M.; et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: The Phase III SPR1NT trial. Nat. Med. 2022, 28, 1381–1389. [Google Scholar] [CrossRef]
- Malone, D.C.; Dean, R.; Arjunji, R.; Jensen, I.; Cyr, P.; Miller, B.; Maru, B.; Sproule, D.M.; Feltner, D.E.; Dabbous, O. Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J. Mark. Access Health Policy 2019, 7, 1601484. [Google Scholar] [CrossRef] [PubMed]
- Novartis. 2021 Global Managed Access Program Community Update. Available online: https://www.novartis.com/news/2021-global-managed-access-program-community-update (accessed on 27 June 2024).
- SMA Newsroom. EMA Grants Positive Recommendation for Zolgensma. Available online: https://www.sma-europe.eu/news/european-medicines-agency-grants-positive-recommendation-for-conditional-marketing-authorisation-of-gene-therapy-product-onasemnogene-abeparvovec-zolgensma (accessed on 27 June 2024).
- Friese, J.; Geitmann, S.; Holzwarth, D.; Müller, N.; Sassen, R.; Baur, U.; Adler, K.; Kirschner, J. Safety Monitoring of Gene Therapy for Spinal Muscular Atrophy with Onasemnogene Abeparvovec–A Single Centre Experience. J. Neuromuscul. Dis. 2021, 8, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Muntoni, F.; Baranello, G.; Masson, R.; Boespflug-Tanguy, O.; Bruno, C.; Corti, S.; Daron, A.; Deconinck, N.; Servais, L.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Finkel, R.S.; Chiriboga, C.A.; Connolly, A.M.; Crawford, T.O.; Darras, B.T.; Iannaccone, S.T.; Kuntz, N.L.; Peña, L.D.M.; Shieh, P.B.; et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): An open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021, 20, 284–293. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Published by MDPI on behalf of the Polish Respiratory Society. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alajjuri, M.A.; Abusamra, R.; Mundada, V.; Narayan, O. Real-World Data in Children with Spinal Muscular Atrophy Type 1 on Long-Term Ventilation Receiving Gene Therapy: A Prospective Cohort Study. Adv. Respir. Med. 2024, 92, 338-347. https://doi.org/10.3390/arm92050032
Alajjuri MA, Abusamra R, Mundada V, Narayan O. Real-World Data in Children with Spinal Muscular Atrophy Type 1 on Long-Term Ventilation Receiving Gene Therapy: A Prospective Cohort Study. Advances in Respiratory Medicine. 2024; 92(5):338-347. https://doi.org/10.3390/arm92050032
Chicago/Turabian StyleAlajjuri, Mohammad Ala’, Rania Abusamra, Vivek Mundada, and Omendra Narayan. 2024. "Real-World Data in Children with Spinal Muscular Atrophy Type 1 on Long-Term Ventilation Receiving Gene Therapy: A Prospective Cohort Study" Advances in Respiratory Medicine 92, no. 5: 338-347. https://doi.org/10.3390/arm92050032
APA StyleAlajjuri, M. A., Abusamra, R., Mundada, V., & Narayan, O. (2024). Real-World Data in Children with Spinal Muscular Atrophy Type 1 on Long-Term Ventilation Receiving Gene Therapy: A Prospective Cohort Study. Advances in Respiratory Medicine, 92(5), 338-347. https://doi.org/10.3390/arm92050032