Microfluidic Devices for Drug Assays


Abstract
:1. Introduction
1.1. Microfluidics
1.2. The Physics of Microfluidics
1.3. Manufacturing Microfluidic Devices
1.4. Drug Discovery and Drug Assays
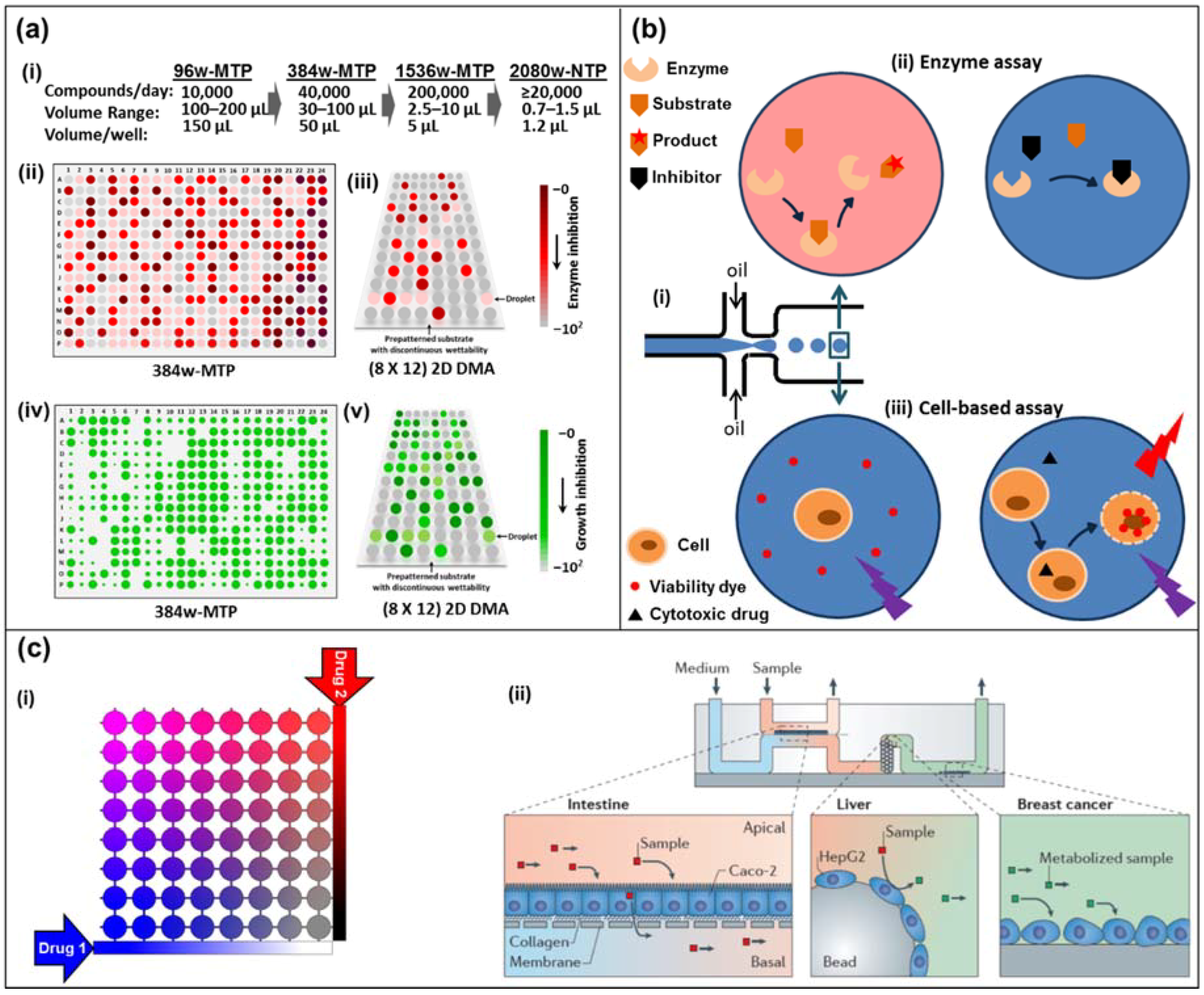
1.5. Definition: High-Throughput Screening (Well Plate vs. Droplet vs. Continuous Flow Microfluidics)
- (a)
- Traditionally, high-throughput drug assays are carried out in microtiter well plates, where the drug solutions are added to either target cells or target molecules. The entire well plates are incubated for days before all the wells are read out in parallel. The high level of parallelization and the usage of mere microliters of drugs gave rise to the microfluidic high-throughput label.
- (b)
- With the emerging field of microfluidics advancing rapidly, drug assays have been developed in two approaches. The first approach is based on droplet microfluidics, where each droplet’s contents correspond to a well from a microtiter plate. Hundreds of these droplets can be generated every second, stored for the incubation time, and read out automatically at similar speeds.
- (c)
- Another approach stems from microfluidic devices operating with a single (aqueous) phase, where drug concentrations are diffusion-controlled, and can be changed periodically and rapidly to obtain individual results within minutes instead of days. This also allows us to test drugs on entire tissues and organs-on-a-chip at the lower frequencies.
1.6. Cell-Based vs. Molecular Assays
2. Well Plate Setups
2.1. The 96-Well Microtiter Plate as an Industrial Standard
2.2. Miniaturization
2.3. Ultra-High-Throughput Screening and Low Volumes
2.4. Challenges and (Well-Less) Alternatives
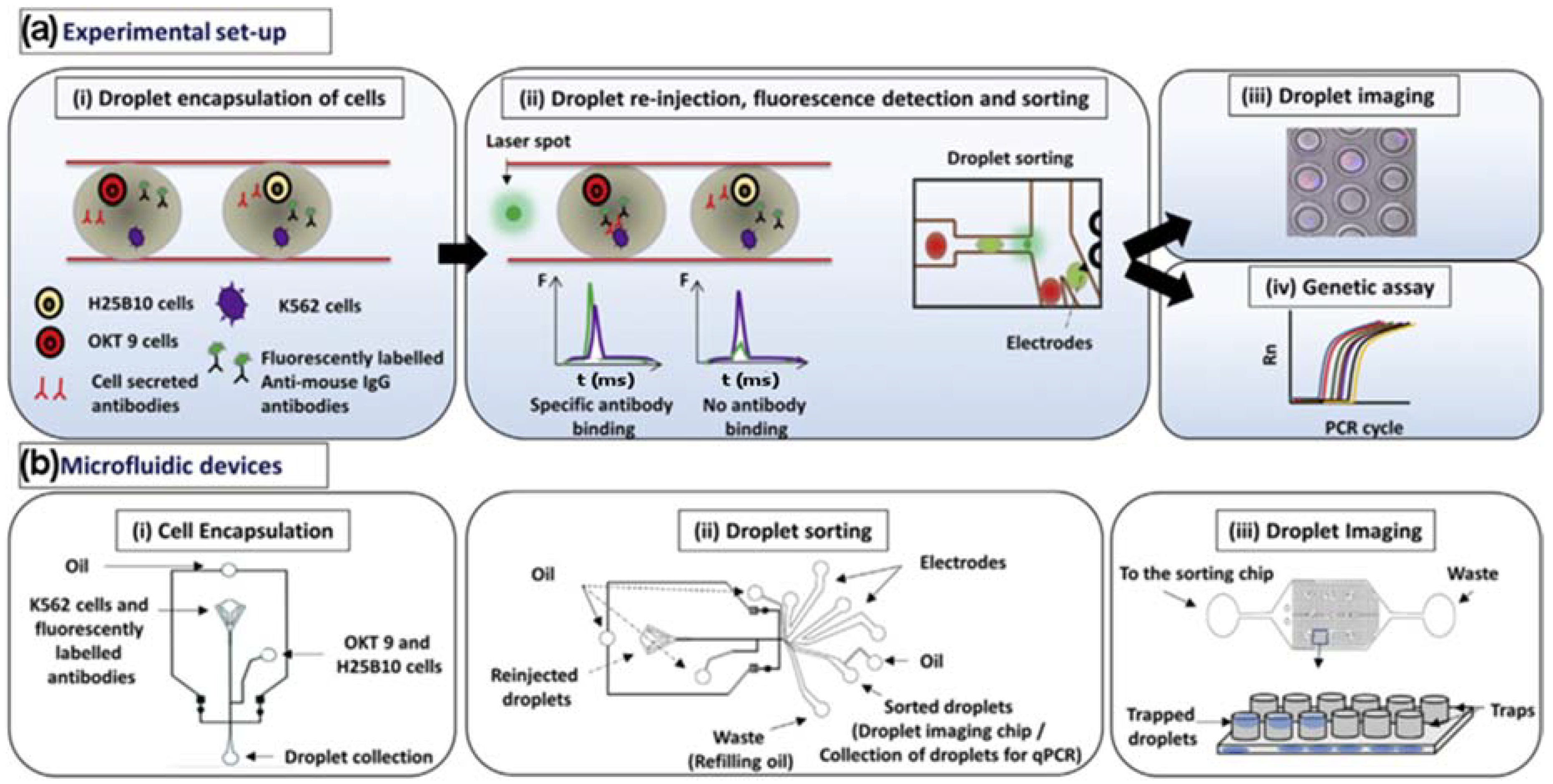
3. Droplet Microfluidics-Based Assays
Droplet Cytotoxicity Assays & Encapsulated Monoclonal Antibody Screening
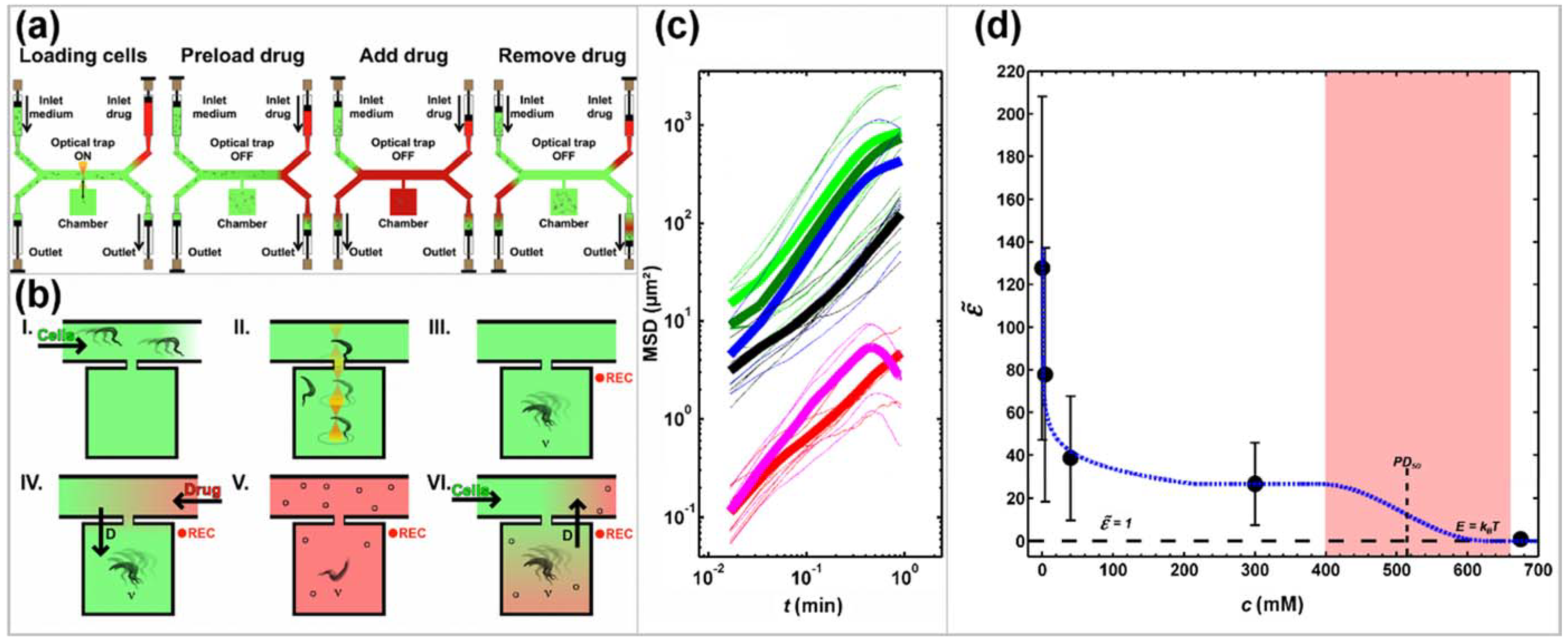
4. Continuous Flow, Diffusion and Concentration Gradients-Based Microfluidic Drug Assays
Further Notable Approaches
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Purcell, E.M. Life at low Reynolds number. Am. J. Phys. 1977, 45, 3. [Google Scholar] [CrossRef]
- Shembekar, N.; Chaipan, C.; Utharala, R.; Merten, C.A. Droplet-based microfluidics in drug discovery, transcriptomics and high-throughput molecular genetics. Lab Chip 2016, 16, 1314–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, F.; Ferreira, M.P.A.; Correia, A.; Hirvonen, J.; Santos, H.A. Microfluidics as a cutting-edge technique for drug delivery applications. J. Drug Deliv. Sci. Technol. 2016, 34, 76–87. [Google Scholar] [CrossRef]
- Tsui, J.H.; Lee, W.; Pun, S.H.; Kim, J.; Kim, D.-H. Microfluidics-assisted in vitro drug screening and carrier production. Adv. Drug Deliv. Rev. 2013, 65, 1575–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riahi, R.; Tamayol, A.; Shaegh, S.A.M.; Ghaemmaghami, A.M.; Dokmeci, M.R.; Khademhosseini, A. Microfluidics for advanced drug delivery systems. Curr. Opin. Chem. Eng. 2015, 7, 101–112. [Google Scholar] [CrossRef]
- Du, G.; Fang, Q.; den Toonder, J.M.J. Microfluidics for cell-based high throughput screening platforms-A review. Anal. Chim. Acta 2016, 903, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Junkin, M.; Tay, S. Microfluidic single-cell analysis for systems immunology. Lab Chip 2014, 14, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Clausell-Tormos, J.; Lieber, D.; Baret, J.-C.; El-Harrak, A.; Miller, O.J.; Frenz, L.; Blouwolff, J.; Humphry, K.J.; Köster, S.; Duan, H.; et al. Droplet-based microfluidic platforms for the encapsulation and screening of mammalian cells and multicellular organisms. Chem. Biol. 2008, 15, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Theberge, A.B.; Courtois, F.; Schaerli, Y.; Fischlechner, M.; Abell, C.; Hollfelder, F.; Huck, W.T.S. Microdroplets in microfluidics: An evolving platform for discoveries in chemistry and biology. Angew. Chem. Int. Ed. 2010, 49, 5846–5868. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.T.; Rotem, A.; Heyman, J.A.; Weitz, D.A. Droplet microfluidics for high-throughput biological assays. Lab Chip 2012, 12, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Peixinho, J.; Mullin, T. Decay of turbulence in pipe flow. Phys. Rev. Lett. 2006, 96, 094501. [Google Scholar] [CrossRef] [PubMed]
- Au, A.K.; Huynh, W.; Horowitz, L.F.; Folch, A. 3D-Printed Microfluidics. Angew. Chem. Int. Ed. 2016, 55, 3862–3881. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Urrios, A.; Kang, S.; Folch, A. The upcoming 3D-printing revolution in microfluidics. Lab Chip 2016, 16, 1720–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziaie, B.; Baldi, A.; Lei, M.; Gu, Y.; Siegel, R.A. Hard and soft micromachining for BioMEMS: Review of techniques and examples of applications in microfluidics and drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 145–172. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Ostuni, E.; Jiang, X.; Ingber, D.E. Soft lithography in Biology and Biochemistry. Annu. Rev. Biomed. Eng. 2001, 3, 335–373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-M.; Xia, Y.; Whitesides, G.M. Soft lithographic methods for nano-fabrication. J. Mater. Chem. 1997, 7, 1069–1074. [Google Scholar] [CrossRef]
- Collins, D.J.; Neild, A.; deMello, A.; Liu, A.-Q.; Ai, Y. The Poisson distribution and beyond: Methods for microfluidic droplet production and single cell encapsulation. Lab Chip 2015, 3439–3459. [Google Scholar] [CrossRef] [PubMed]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. USA 2009, 106, 14195–14200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.P.; Rees, S.S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Baedeker, M.; Ringel, M.; Panier, V.; Schulze, U. Market watch: 2017 FDA drug approvals: Number rebounds but average value slips. Nat. Rev. Drug Discov. 2018, 17, 87. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Vohora, D. Drug Discovery and Development: An Overview; Elsevier Inc.: New York, NY, USA, 2017; ISBN 9780128021033. [Google Scholar]
- Janzen, W.P. Screening technologies for small molecule discovery: The state of the art. Chem. Biol. 2014, 21, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Q.; Liu, W.; He, Z.; Lin, J.M. Recent advances in microfluidic 3D cellular scaffolds for drug assays. TrAC Trends Anal. Chem. 2017, 87, 19–31. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, Q.; Nie, J.; Fu, J.; He, Y. From Microfluidic Paper-Based Analytical Devices to Paper-Based Biofluidics with Integrated Continuous Perfusion. ACS Biomater. Sci. Eng. 2017, 3, 601–607. [Google Scholar] [CrossRef]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramjee, M.K.; Patel, S. Continuous-flow injection microfluidic thrombin assays: The effect of binding kinetics on observed enzyme inhibition. Anal. Biochem. 2017, 528, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Hajba, L.; Guttman, A. Continuous-flow-based microfluidic systems for therapeutic monoclonal antibody production and organ-on-a-chip drug testing. J. Flow Chem. 2017, 7, 118–123. [Google Scholar] [CrossRef]
- Caviglia, C.; Zór, K.; Montini, L.; Tilli, V.; Canepa, S.; Melander, F.; Muhammad, H.B.; Carminati, M.; Ferrari, G.; Raiteri, R.; et al. Impedimetric toxicity assay in microfluidics using free and liposome-encapsulated anticancer drugs. Anal. Chem. 2015, 87, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, L.; Burbaum, J.; Dunn, D. Miniaturisation of HTS assays. Innov. Pharm. Technol. 2000, 5, 12–17. [Google Scholar]
- Lariosa-Willingham, K.D.; Rosler, E.S.; Tung, J.S.; Dugas, J.C.; Collins, T.L.; Leonoudakis, D. A high throughput drug screening assay to identify compounds that promote oligodendrocyte differentiation using acutely dissociated and purified oligodendrocyte precursor cells. BMC Res. Notes 2016, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.D.; Land, K.M.; O’Connor, R.M.; Fritz, H.M. High-throughput screen of drug repurposing library identifies inhibitors of Sarcocystis neurona growth. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Walters, M.A. The essential roles of chemistry in high-throughput screening triage. Future Med. Chem. 2014, 6, 1265–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, L.M.; Fuerst, P. The future of high-throughput screening. J. Biomol. Screen. 2008, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Baumann, H.; Matthews, H.; Li, M.; Hu, J.J.; Willison, K.R. A high-throughput in vitro translation screen towards discovery of novel antimalarial protein translation inhibitors. bioRxiv 2018, 44. [Google Scholar] [CrossRef]
- Brennan, J.C.; Tillitt, D.E. Development of a dual luciferase activity and fluorescamine protein assay adapted to a 384 micro-well plate format: Reducing variability in human luciferase transactivation cell lines aimed at endocrine active substances. Toxicol. In Vitro 2018, 47, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, M.; Long, Y.; Fargue, S.; Southall, N.; Hu, X.; McKew, J.C.; Danpure, C.J.; Zheng, W. High throughput cell-based assay for identification of glycolate oxidase inhibitors as a potential treatment for Primary Hyperoxaluria Type 1. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Narahari, J.; Ho, P.; Casalena, D.; Nguyen, V.; Cirbaite, E.; Hughes, D.; Daly, J.; Webb, B. Characterization and Use of TurboLuc Luciferase as a Reporter for High-Throughput Assays. Biochemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Norcliffe, J.L.; Mina, J.G.; Alvarez, E.; Cantizani, J.; De Dios-Anton, F.; Colmenarejo, G.; Silva Gonzalez-Del Valle, S.; Marco, M.; Fiandor, J.M.; Martin, J.J.; et al. Identifying inhibitors of the Leishmania inositol phosphorylceramide synthase with antiprotozoal activity using a yeast-based assay and ultra-high throughput screening platform. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wölcke, J.; Ullmann, D. Miniaturized HTS technologies-uHTS. Drug Discov. Today 2001, 6, 637–646. [Google Scholar] [CrossRef]
- Smith, A. How small should you go? Nature 2002, 418, 457. [Google Scholar] [CrossRef]
- Beske, O.E.; Goldbard, S. High-throughput cell analysis using multiplexed array technologies. Drug Discov. Today 2002, 7, S131–S135. [Google Scholar] [CrossRef]
- Gosalia, D.N.; Diamond, S.L. Printing chemical libraries on microarrays for fluid phase nanoliter reactions. Proc. Natl. Acad. Sci. USA 2003, 100, 8721–8726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitre, E.; Schulze, M.; Cumme, G.A.; Rößler, F.; Rausch, T.; Rhode, H. Turbo-mixing in microplates. J. Biomol. Screen. 2007, 12, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Perez, R.; Fan, Z.H.; Garcia-Cordero, J.L. Evaporation-Driven Bioassays in Suspended Droplets. Anal. Chem. 2016, 88, 7312–7317. [Google Scholar] [CrossRef] [PubMed]
- Küster, S.K.; Pabst, M.; Zenobi, R.; Dittrich, P.S. Screening for protein phosphorylation using nanoscale reactions on microdroplet arrays. Angew. Chem. Int. Ed. 2015, 54, 1671–1675. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cordero, J.L.; Fan, Z.H. Sessile droplets for chemical and biological assays. Lab Chip 2017, 17, 2150–2166. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.; Brien, R.; Njus, Z.; Kalwa, U.; Pandey, S. Motorized actuation system to perform droplet operations on printed plastic sheets. Lab Chip 2016, 16, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Casavant, B.P.; Berthier, E.; Theberge, A.B.; Berthier, J.; Montanez-Sauri, S.I.; Bischel, L.L.; Brakke, K.; Hedman, C.J.; Bushman, W.; Keller, N.P.; et al. Suspended microfluidics. Proc. Natl. Acad. Sci. USA 2013, 110, 10111–10116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, S.B.; Zhang, T.; Day, J.H.; Su, X.; Wilson, I.Z.; Berthier, E.; Theberge, A.B. Upgrading well plates using open microfluidic patterning. Lab Chip 2017, 17, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wang, L. Passive and active droplet generation with microfluidics: A review. Lab Chip 2017, 17, 34–75. [Google Scholar] [CrossRef] [PubMed]
- Weinmeister, R.; Freeman, E.; Eperon, I.C.; Stuart, A.M.; Hudson, A.J. Single-Fluorophore Detection in Femtoliter Droplets Generated by Flow Focusing. ACS Nano 2015, 9, 9718–9730. [Google Scholar] [CrossRef] [PubMed]
- Bringer, M.R.; Gerts, C.J.; Song, H.; Tice, J.D.; Ismagilov, R.F. Microfluidic systems for chemical kinetics that rely on chaotic mixing in droplets. Phil. Trans. R. Soc. Lond. A 2004, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Duits, M.H.G.; Mugele, F. Droplets formation and merging in two-phase flow microfluidics. Int. J. Mol. Sci. 2011, 12, 2572–2597. [Google Scholar] [CrossRef] [PubMed]
- Kunstmann-Olsen, C.; Hanczyc, M.M.; Hoyland, J.; Rasmussen, S.; Rubahn, H.G. Uniform droplet splitting and detection using Lab-on-Chip flow cytometry on a microfluidic PDMS device. Sens. Actuators B Chem. 2016, 229, 7–13. [Google Scholar] [CrossRef]
- Huebner, A.; Bratton, D.; Whyte, G.; Yang, M.; deMello, A.J.; Abell, C.; Hollfelder, F. Static microdroplet arrays: A microfluidic device for droplet trapping, incubation and release for enzymatic and cell-based assays. Lab Chip 2009, 9, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Frenz, L.; Blank, K.; Brouzes, E.; Griffiths, A.D. Reliable microfluidic on-chip incubation of droplets in delay-lines. Lab Chip 2009, 9, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, H.-D.; Zheng, H.; Guo, W.; Gañán-Calvo, A.M.; Ai, Y.; Tsao, C.-W.; Zhou, J.; Li, W.; Huang, Y.; Nguyen, N.-T.; et al. Active droplet sorting in microfluidics: A review. Lab Chip 2017, 17, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Bithi, S.S.; Vanapalli, S.A. Microfluidic cell isolation technology for drug testing of single tumor cells and their clusters. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.H.-H.; Li, H.; Jia, Y.; Mak, P.-I.; Martins, P.-I.; da Silva Martins, R.P.; Liu, Y.; Vong, C.M.; Wong, H.C.; Wong, P.K.; et al. Drug screening of cancer cell lines and human primary tumors using droplet microfluidics. Sci. Rep. 2017, 7, 9109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabhachandani, P.; Motwani, V.; Cohen, N.; Sarkar, S.; Torchilin, V.; Konry, T. Generation and functional assessment of 3D multicellular spheroids in droplet based microfluidics platform. Lab Chip 2016, 16, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Shembekar, N.; Hu, H.; Eustace, D.; Merten, C.A. Single-Cell Droplet Microfluidic Screening for Antibodies Specifically Binding to Target Cells. Cell Rep. 2018, 22, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Eustace, D.; Merten, C. A. Efficient cell pairing in droplets using dual-color sorting. Lab Chip 2015, 15, 3989–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Guo, F.; Xie, Y.; Zhao, Y.; Lapsley, M.I.; Wang, L.; Mai, J.D.; Costanzo, F.; Huang, T.J. Label-Free Measurements of Reaction Kinetics Using a Droplet-Based Optofluidic Device. J. Lab. Autom. 2015, 20, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, M.; Chen, X.; Chan, S.; Mohamed, T.; Rao, P.P.N.; Ren, C.L. Droplet Microfluidic System with On-Demand Trapping and Releasing of Droplet for Drug Screening Applications. Anal. Chem. 2017, 89, 910–915. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Kim, K.; Kim, J. Microfluidic System Based High Throughput Drug Screening System for Curcumin/TRAIL Combinational Chemotherapy in Human Prostate Cancer PC3 Cells. Biomol. Ther. 2014, 22, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochstetter, A.; Stellamanns, E.; Deshpande, S.; Uppaluri, S.; Engstler, M.; Pfohl, T. Microfluidics-based single cell analysis reveals drug-dependent motility changes in trypanosomes. Lab Chip 2015, 15, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Hochstetter, A. Motility, Manipulation and Controlling of Unicellular Organisms. Ph.D. Thesis, Universität Basel, Basel, Switzerland, 2014. [Google Scholar]
- Ding, X.; Njus, Z.; Kong, T.; Su, W.; Ho, C.-M.; Pandey, S. Effective drug combination for Caenorhabditis elegans nematodes discovered by output-driven feedback system control technique. Sci. Adv. 2017, 3, eaao1254. [Google Scholar] [CrossRef] [PubMed]
- Derda, R.; Laromaine, A.; Mammoto, A.; Tang, S.K.Y.; Mammoto, T.; Ingber, D.E. Paper-supported 3D cell culture for tissue-based bioassays. Proc. Natl. Acad. Sci. USA 2009, 106, 18457–18462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Wu, Y.; Fu, J.-Z.; Wu, W.-B. Fabrication of paper-based microfluidic analysis devices: A review. RSC Adv. 2015, 5, 78109–78127. [Google Scholar] [CrossRef]
- Zhang, Y.; Nguyen, N.-T. Magnetic digital microfluidics-A review. Lab Chip 2017, 17, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Samiei, E.; Tabrizian, M.; Hoorfar, M. A review of digital microfluidics as portable platforms for lab-on a-chip applications. Lab Chip 2016, 16, 2376–2396. [Google Scholar] [CrossRef] [PubMed]
- Hvastkovs, E.G.; Rusling, J.F. State-of-the-Art Metabolic Toxicity Screening and Pathway Evaluation. Anal. Chem. 2016, 88, 4584–4599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jebrail, M.J.; Assem, N.; Mudrik, J.M.; Dryden, M.D.M.; Lin, K.; Yudin, A.K.; Wheeler, A.R. Combinatorial Synthesis of Peptidomimetics Using Digital Microfluidics. J. Flow Chem. 2012, 2, 103–107. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Waqued, S.C.; Nodurft, D.T.; Devarenne, T.P.; Yakovlev, V.V.; Han, A. Raman spectroscopy compatible PDMS droplet microfluidic culture and analysis platform towards on-chip lipidomics. Analyst 2017, 142, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, J.; Deng, K.; Shih, S.C.C.; Gao, J.; Adams, P.D.; Singh, A.K.; Northen, T.R. On-chip integration of droplet microfluidics and nanostructure-initiator mass spectrometry for enzyme screening. Lab Chip 2017, 17, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Mesbah, K.; Thai, R.; Bregant, S.; Malloggi, F. DMF-MALDI: Droplet based microfluidic combined to MALDI-TOF for focused peptide detection. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Technique | Microtiter Well Plates | Droplet Microfluidics | Single Phase Microfluidics |
|---|---|---|---|
| Pros | Parallelizable | Extremely high parallel throughput | Very fast readout |
| Miniaturization | Post processing possible | Continuous process possible | |
| Many commercial options available | Commercial options are currently emerging Highly customizable | Allows motility and viability assay High control over concentration via diffusion Highly customizable | |
| Neutral | Current gold standard | Emerging technology (start-ups) | Niche technology |
| Cons | Long assay durations (days) | No industrial standard yet | Lower throughput |
| Evaporation | Needs additional handling of oil and surfactants to generate droplets | Sample volumes are not isolated | |
| Local concentration gradients | Adding of compounds over time requires additional chip architecture | Unwanted diffusion Currently no commercial availability |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regnault, C.; Dheeman, D.S.; Hochstetter, A. Microfluidic Devices for Drug Assays. High-Throughput 2018, 7, 18. https://doi.org/10.3390/ht7020018
Regnault C, Dheeman DS, Hochstetter A. Microfluidic Devices for Drug Assays. High-Throughput. 2018; 7(2):18. https://doi.org/10.3390/ht7020018
Chicago/Turabian StyleRegnault, Clément, Dharmendra S. Dheeman, and Axel Hochstetter. 2018. "Microfluidic Devices for Drug Assays" High-Throughput 7, no. 2: 18. https://doi.org/10.3390/ht7020018
APA StyleRegnault, C., Dheeman, D. S., & Hochstetter, A. (2018). Microfluidic Devices for Drug Assays. High-Throughput, 7(2), 18. https://doi.org/10.3390/ht7020018