Venomics: A Mini-Review


{kind=link}
{kind=link}
Abstract
:1. Introduction
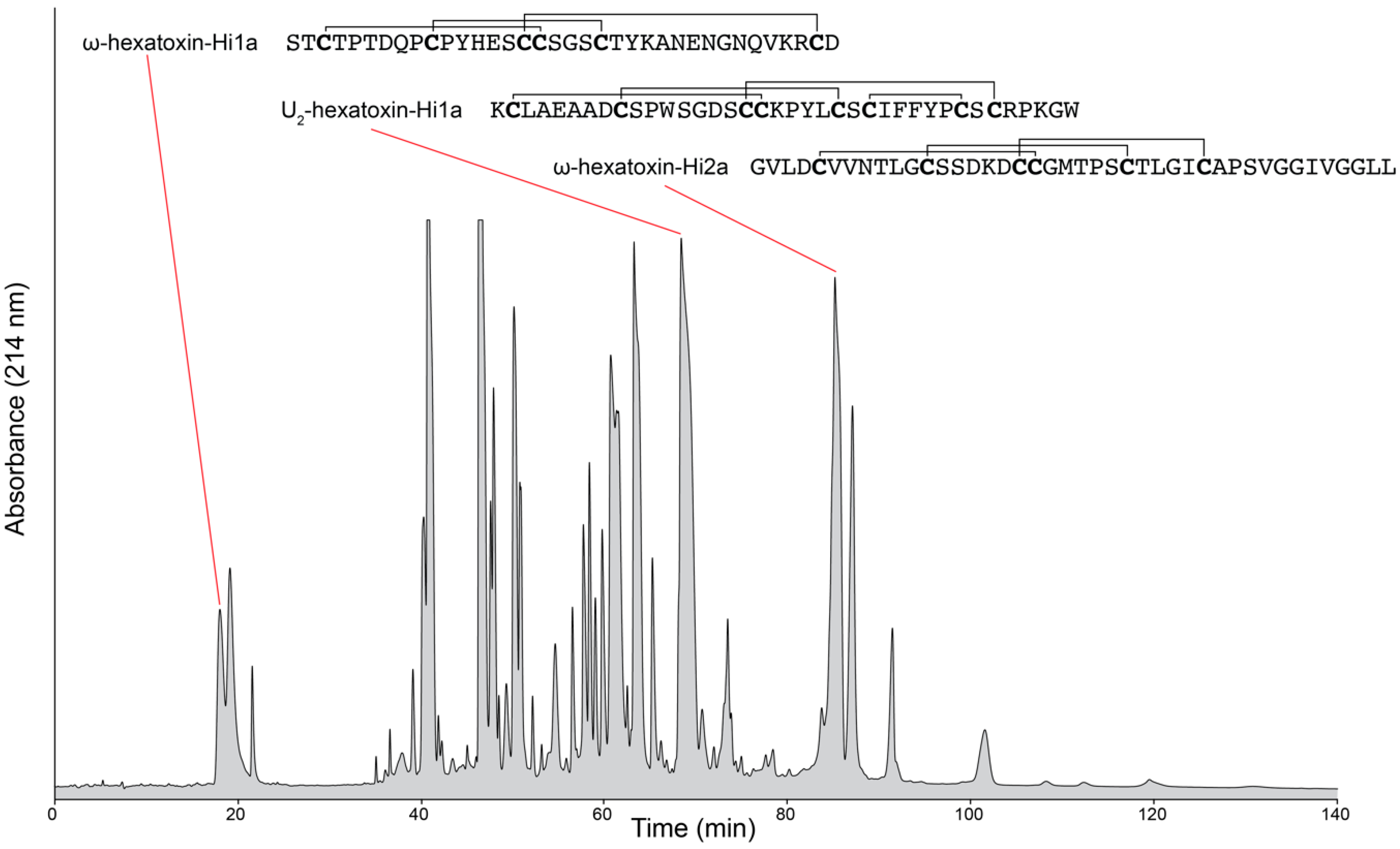
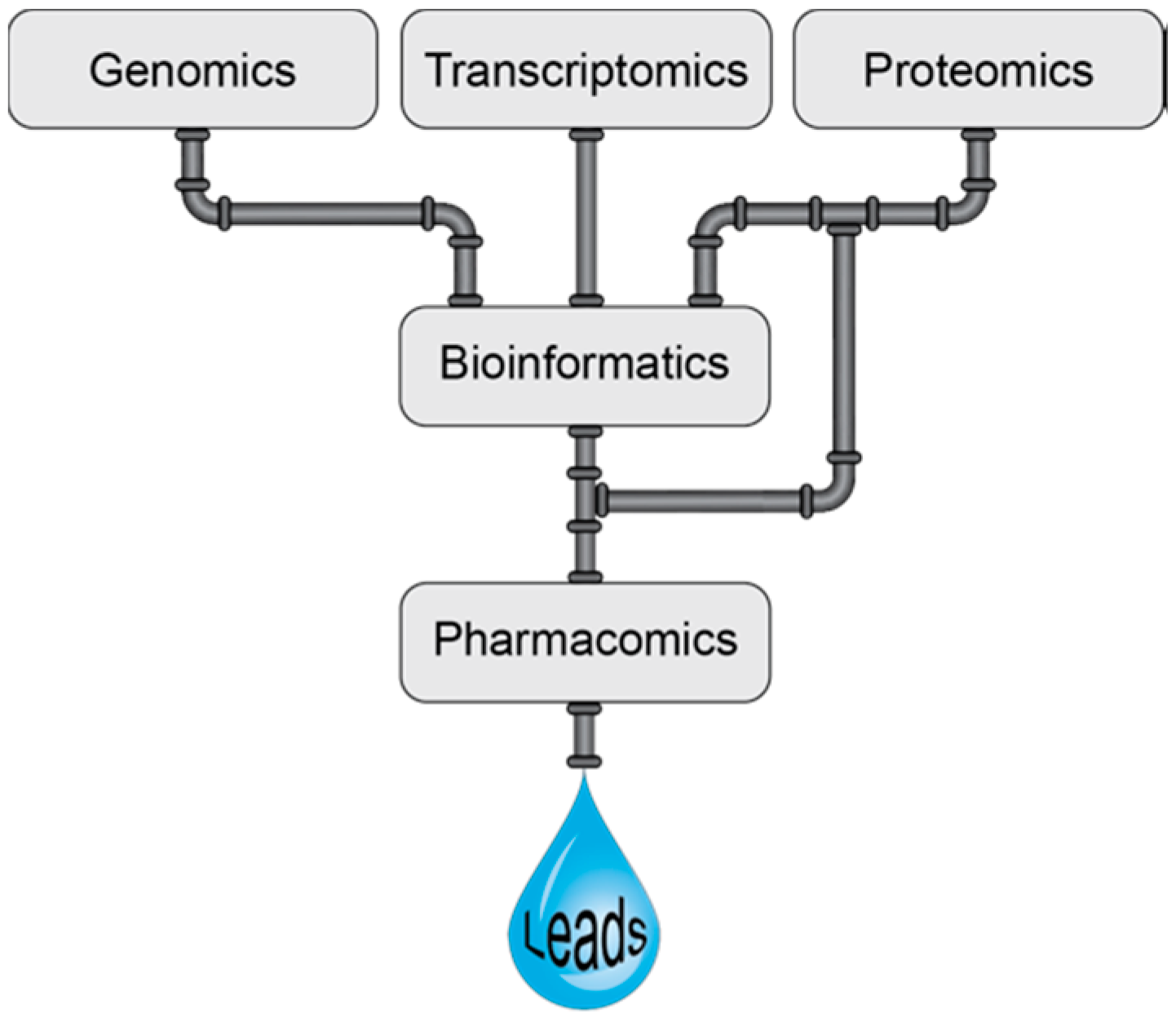
2. Venomics
2.1. Genomics
2.2. Transcriptomics
2.3. Proteomics
2.4. Bioinformatics
2.5. High-Throughtput Assay Screening
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wilson, D.; Boyle, G.M.; McIntyre, L.; Nolan, M.J.; Parsons, P.G.; Smith, J.J.; Tribolet, L.; Loukas, A.; Liddell, M.J.; Rash, L.D.; et al. The aromatic head group of spider toxin polyamines influences toxicity to cancer cells. Toxins 2017, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Himaya, S.W.A.; Lewis, R.J. Venomics-accelerated cone snail venom peptide discovery. Int. J. Mol. Sci. 2018, 19, 788. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Herzig, V.; King, G.F.; Alewood, P.F. The insecticidal potential of venom peptides. Cell. Mol. Life Sci. 2013, 70, 3665–3693. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Lewis, R.J.; Dutertre, S. Towards an integrated venomics approach for accelerated conopeptide discovery. Toxicon 2012, 60, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; King, G.F. Venomics as a drug discovery platform. Expert Rev. Proteom. 2009, 6, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, S. Venomics in medicinal chemistry. Future Med. Chem. 2014, 6, 1609–1610. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Vetter, I.; Himaya, S.W.; Alewood, P.F.; Lewis, R.J.; Dutertre, S. Transcriptome and proteome of Conus planorbis identify the nicotinic receptors as primary target for the defensive venom. Proteomics 2015, 15, 4030–4040. [Google Scholar] [CrossRef] [PubMed]
- Garb, J.E. Extraction of venom and venom gland microdissections from spiders for proteomic and transcriptomic analyses. J. Vis. Exp. 2014, e51618. [Google Scholar] [CrossRef] [PubMed]
- Fuzita, F.J.; Pinkse, M.W.; Patane, J.S.; Verhaert, P.D.; Lopes, A.R. High throughput techniques to reveal the molecular physiology and evolution of digestion in spiders. BMC Genom. 2016, 17, 716. [Google Scholar] [CrossRef] [PubMed]
- Juarez, P.; Sanz, L.; Calvete, J.J. Snake venomics: Characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 2004, 4, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Oldrati, V.; Arrell, M.; Violette, A.; Perret, F.; Sprungli, X.; Wolfender, J.L.; Stocklin, R. Advances in venomics. Mol. Biosyst. 2016, 12, 3530–3543. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.S.; Chaumeil, P.A.; Kunert, A.; Kaas, Q.; Thang, M.W.C.; Le, L.; Nuhn, M.; Herzig, V.; Saez, N.J.; Cristofori-Armstrong, B.; et al. ArachnoServer 3.0: An online resource for automated discovery, analysis and annotation of spider toxins. Bioinformatics 2018, 34, 1074–1076. [Google Scholar] [CrossRef] [PubMed]
- Babb, P.L.; Lahens, N.F.; Correa-Garhwal, S.M.; Nicholson, D.N.; Kim, E.J.; Hogenesch, J.B.; Kuntner, M.; Higgins, L.; Hayashi, C.Y.; Agnarsson, I.; et al. The Nephila clavipes genome highlights the diversity of spider silk genes and their complex expression. Nat. Genet. 2017, 49, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Wilson, R.K.; Eichler, E.E. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015, 16, 627–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Zhang, Y.; Hu, W.; Xu, D.; Tao, H.; Yang, X.; Li, Y.; Jiang, L.; Liang, S. Molecular diversification of peptide toxins from the tarantula Haplopelma hainanum (Ornithoctonus hainana) venom based on transcriptomic, peptidomic, and genomic analyses. J. Proteome Res. 2010, 9, 2550–2564. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Zuo, X.P.; Chai, Z.F.; Ji, Y.H. The cDNA and genomic DNA organization of a novel toxin SHT-I from spider Ornithoctonus huwena. Acta Biochim. Biophys. Sin. 2004, 36, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, J.; Peng, L.; Zhang, Y.; Xiong, X.; Liang, S. Genomic organization and cloning of novel genes encoding toxin-like peptides of three superfamilies from the spider Orinithoctonus huwena. Peptides 2008, 29, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.S.; Wilson, D.; Mattick, J.S.; King, G.F. The lethal toxin from Australian funnel-web spiders is encoded by an intronless gene. PLoS ONE 2012, 7, e43699. [Google Scholar] [CrossRef] [PubMed]
- Danilevich, V.; Grishin, E. The genes encoding black widow spider neurotoxins are intronless. Russ. J. Bioorg. Chem. 2000, 26, 838–843. [Google Scholar] [CrossRef]
- Krapcho, K.J.; Kral, R.M., Jr.; Vanwagenen, B.C.; Eppler, K.G.; Morgan, T.K. Characterization and cloning of insecticidal peptides from the primitive weaving spider Diguetia canities. Insect Biochem. Mol. Biol. 1995, 25, 991–1000. [Google Scholar] [CrossRef]
- Olivera, B.M.; Walker, C.; Cartier, G.E.; Hooper, D.; Santos, A.D.; Schoenfeld, R.; Shetty, R.; Watkins, M.; Bandyopadhyay, P.; Hillyard, D.R. Speciation of cone snails and interspecific hyperdivergence of their venom peptides. Potential evolutionary significance of introns. Ann. N. Y. Acad. Sci. 1999, 870, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Schwager, E.E.; Sharma, P.P.; Clarke, T.; Leite, D.J.; Wierschin, T.; Pechmann, M.; Akiyama-Oda, Y.; Esposito, L.; Bechsgaard, J.; Bilde, T. The house spider genome reveals an ancient whole-genome duplication during arachnid evolution. BMC Biol. 2017, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Sanggaard, K.W.; Bechsgaard, J.S.; Fang, X.; Duan, J.; Dyrlund, T.F.; Gupta, V.; Jiang, X.; Cheng, L.; Fan, D.; Feng, Y.; et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 2014, 5, 3765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ari, S.; Arikan, M. Next-generation sequencing: Advantages, disadvantages and future. In Plant Omics-Trends and Applications; Hakeem, K.R., Tombuloglu, H., Tombuloglu, G., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 109–136. [Google Scholar]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; Gurtowski, J.; Ethe-Sayers, S.; Deshpande, P.; Schatz, M.C.; McCombie, W.R. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015, 25, 1750–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.D.; Nguyen, S.H.; Ganesamoorthy, D.; Elliott, A.G.; Cooper, M.A.; Coin, L.J. Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nat. Commun. 2017, 8, 14515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, T.J.; Abbenante, G.; Tyndall, J.D.; Halliday, J.; Lewis, R.J. Isolation and characterization of a cone snail protease with homology to CRISP proteins of the pathogenesis-related protein superfamily. J. Biol. Chem. 2003, 278, 31105–31110. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Johnson, W.E.; O’Brien, S.J.; Vasconcelos, V.; Antunes, A. Evolution of CRISPs associated with toxicoferan-reptilian venom and mammalian reproduction. Mol. Biol. Evol. 2012, 29, 1807–1822. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Turner, D.J. Low-bias, strand-specific transcriptome Illumina sequencing by on-flowcell reverse transcription (FRT-seq). Nat. Protoc. 2011, 6, 1736–1747. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D.; Raz, T.; Kieu, A.; Jones, D.R.; Giladi, E.; Thayer, E.; Thompson, J.F.; Letovsky, S.; Milos, P.; Causey, M. Quantification of the yeast transcriptome by single-molecule sequencing. Nat. Biotechnol. 2009, 27, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Kozarewa, I.; Ning, Z.; Quail, M.A.; Sanders, M.J.; Berriman, M.; Turner, D.J. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Methods 2009, 6, 291–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamanova, L.; Andrews, R.M.; James, K.D.; Sheridan, E.M.; Ellis, P.D.; Langford, C.F.; Ost, T.W.; Collins, J.E.; Turner, D.J. FRT-seq: Amplification-free, strand-specific transcriptome sequencing. Nat. Methods 2010, 7, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Platt, A.R.; Jones, D.R.; Reifenberger, J.G.; Sass, L.E.; McInerney, P.; Thompson, J.F.; Bowers, J.; Jarosz, M.; Milos, P.M. Direct RNA sequencing. Nature 2009, 461, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Thomas, S.; Underwood, J.G.; Tseng, E.; Holloway, A.K.; on behalf of the Bench to Basinet CvDC Informatics Subcommittee. Long-read sequencing of chicken transcripts and identification of new transcript isoforms. PLoS ONE 2014, 9, e94650. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, M.T.; Rajadinakaran, G.; Graveley, B.R. Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol. 2015, 16, 204. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Undheim, E.A.; Jauss, R.-T.; Jenner, R.A. Venomics of remipede crustaceans reveals novel peptide diversity and illuminates the venom’s biological role. Toxins 2017, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Dekan, Z.; Smout, M.J.; Wilson, D.; Dutertre, S.; Vetter, I.; Lewis, R.J.; Loukas, A.; Daly, N.L.; Alewood, P.F. Conotoxin Φ-MiXXVIIA from the superfamily G2 employs a novel cysteine framework that mimics granulin and displays anti-apoptotic activity. Angew. Chem. 2017, 129, 15169–15172. [Google Scholar] [CrossRef]
- Graves, P.R.; Haystead, T.A. Molecular biologist’s guide to proteomics. Microbiol. Mol. Biol. Rev. 2002, 66, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Melani, R.D.; Nogueira, F.C.S.; Domont, G.B. It is time for top-down venomics. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Petras, D.; Calderon-Celis, F.; Lomonte, B.; Encinar, J.R.; Sanz-Medel, A. Protein-species quantitative venomics: Looking through a crystal ball. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 27. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J. Venomics: Integrative venom proteomics and beyond. Biochem. J. 2017, 474, 611–634. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Quinton, L.; Nicholson, G.M. Venomics: Unravelling the complexity of animal venoms with mass spectrometry. J. Mass Spectrom. 2008, 43, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.; Sunagar, K.; Herzig, V.; Kely, L.; Low, D.H.; Jackson, T.N.; Jones, A.; Kurniawan, N.; King, G.F.; Ali, S.A.; et al. A proteomics and transcriptomics investigation of the venom from the barychelid spider Trittame loki (brush-foot trapdoor). Toxins 2013, 5, 2488–2503. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, D.C.; Aerts, M.; Danneels, E.; Devreese, B. Bee, wasp and ant venomics pave the way for a component-resolved diagnosis of sting allergy. J. Proteom. 2009, 72, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Sandra, K.; Devreese, B.; Van Beeumen, J.; Stals, I.; Claeyssens, M. The Q-Trap mass spectrometer, a novel tool in the study of protein glycosylation. J. Am. Soc. Mass Spectrom. 2004, 15, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Harvey, D.J. Identification of protein-bound carbohydrates by mass spectrometry. Proteomics 2001, 1, 311–328. [Google Scholar] [CrossRef]
- Kuzmenkov, A.I.; Krylov, N.A.; Chugunov, A.O.; Grishin, E.V.; Vassilevski, A.A. Kalium: A database of potassium channel toxins from scorpion venom. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Roly, Z.Y.; Hakim, M.A.; Zahan, A.S.; Hossain, M.M.; Reza, M.A. ISOB: A Database of Indigenous Snake Species of Bangladesh with respective known venom composition. Bioinformation 2015, 11, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.; Nolan, M.J.; Field, M.; Daly, N.L. The Venom Gland Transcriptome of a Species of Spider, Phlogius sp.; Australian Institute for Tropical Health and Medicine, James Cook University: Smithfield, Australia, 2016. [Google Scholar]
- Lavergne, V.; Dutertre, S.; Jin, A.-H.; Lewis, R.J.; Taft, R.J.; Alewood, P.F. Systematic interrogation of the Conus marmoreus venom duct transcriptome with ConoSorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genom. 2013, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Davis, J.L.; Rash, L.D.; Anangi, R.; Mobli, M.; Alewood, P.F.; Lewis, R.J.; King, G.F. Venomics: A new paradigm for natural products-based drug discovery. Amino Acids 2011, 40, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Hasaballah, N.; Vetter, I. Pharmacological screening technologies for venom peptide discovery. Neuropharmacology 2017, 127, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Milward, E.A.; Daneshi, N.; Johnstone, D.M. Emerging real-time technologies in molecular medicine and the evolution of integrated ‘pharmacomics’ approaches to personalized medicine and drug discovery. Pharmacol. Ther. 2012, 136, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Kuyucak, S.; Norton, R.S. Computational approaches for designing potent and selective analogs of peptide toxins as novel therapeutics. Future Med. Chem. 2014, 6, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Danneels, E.L.; Formesyn, E.M.; de Graaf, D.C. Exploring the potential of venom from Nasonia vitripennis as therapeutic agent with high-throughput screening tools. Toxins 2015, 7, 2051–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalbantsoy, A.; Hempel, B.-F.; Petras, D.; Heiss, P.; Göçmen, B.; Iğci, N.; Yildiz, M.Z.; Süssmuth, R.D. Combined venom profiling and cytotoxicity screening of the Radde’s mountain viper (Montivipera raddei) and Mount Bulgar Viper (Montivipera bulgardaghica) with potent cytotoxicity against human A549 lung carcinoma cells. Toxicon 2017, 135, 71–83. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, D.; Daly, N.L. Venomics: A Mini-Review. High-Throughput 2018, 7, 19. https://doi.org/10.3390/ht7030019
Wilson D, Daly NL. Venomics: A Mini-Review. High-Throughput. 2018; 7(3):19. https://doi.org/10.3390/ht7030019
Chicago/Turabian StyleWilson, David, and Norelle L. Daly. 2018. "Venomics: A Mini-Review" High-Throughput 7, no. 3: 19. https://doi.org/10.3390/ht7030019
APA StyleWilson, D., & Daly, N. L. (2018). Venomics: A Mini-Review. High-Throughput, 7(3), 19. https://doi.org/10.3390/ht7030019