The Cytoscan HD Array in the Diagnosis of Neurodevelopmental Disorders

 ,
, 


Abstract
:1. Introduction
2. Chromosomal Microarray Platforms
3. Cytoscan HD Platform: An Overview
4. Clinical Applications of Cytoscan HD Array in Neurodevelopmental Disorders
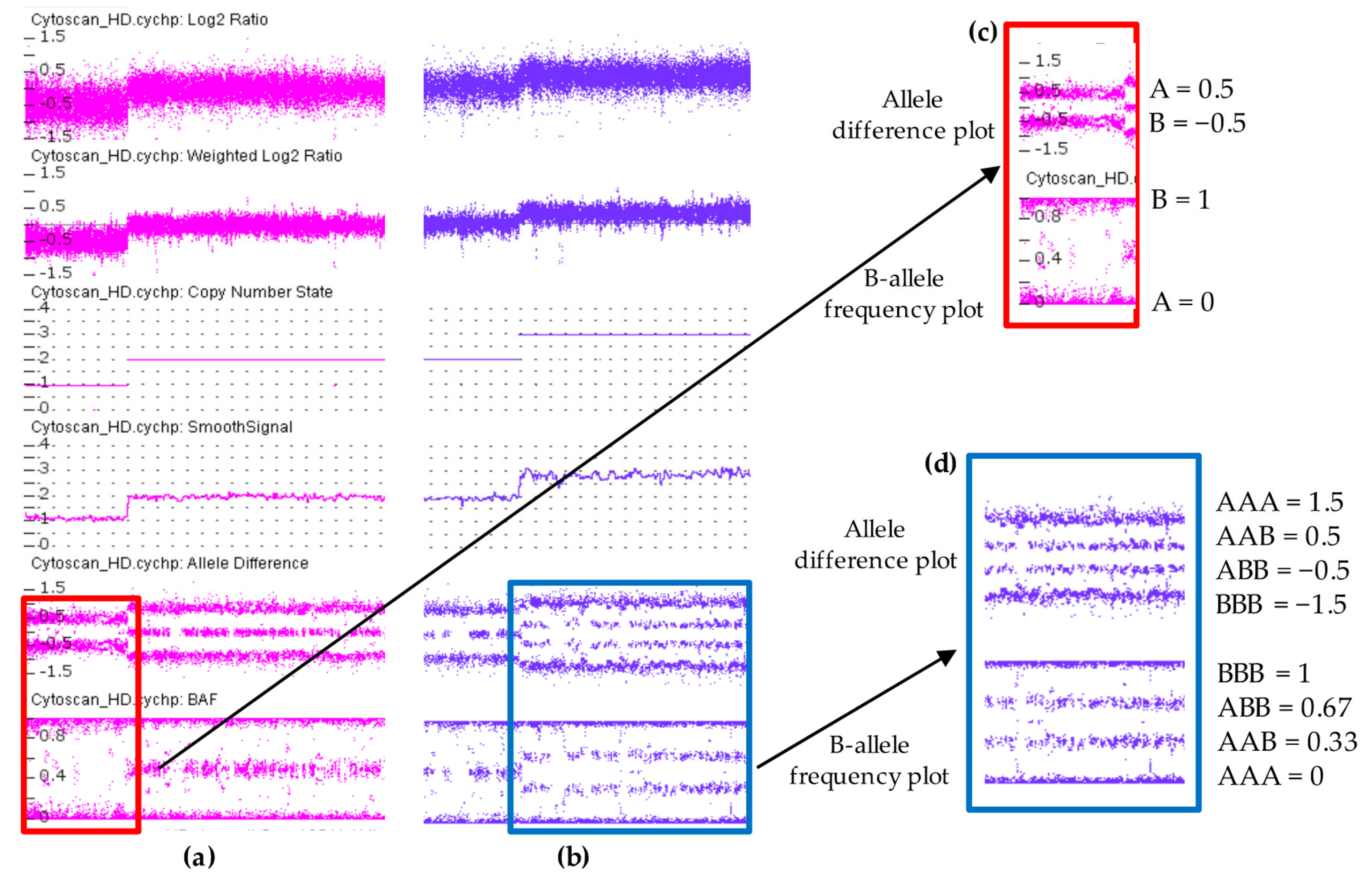
5. Clinical Interpretation of Copy Number Variations
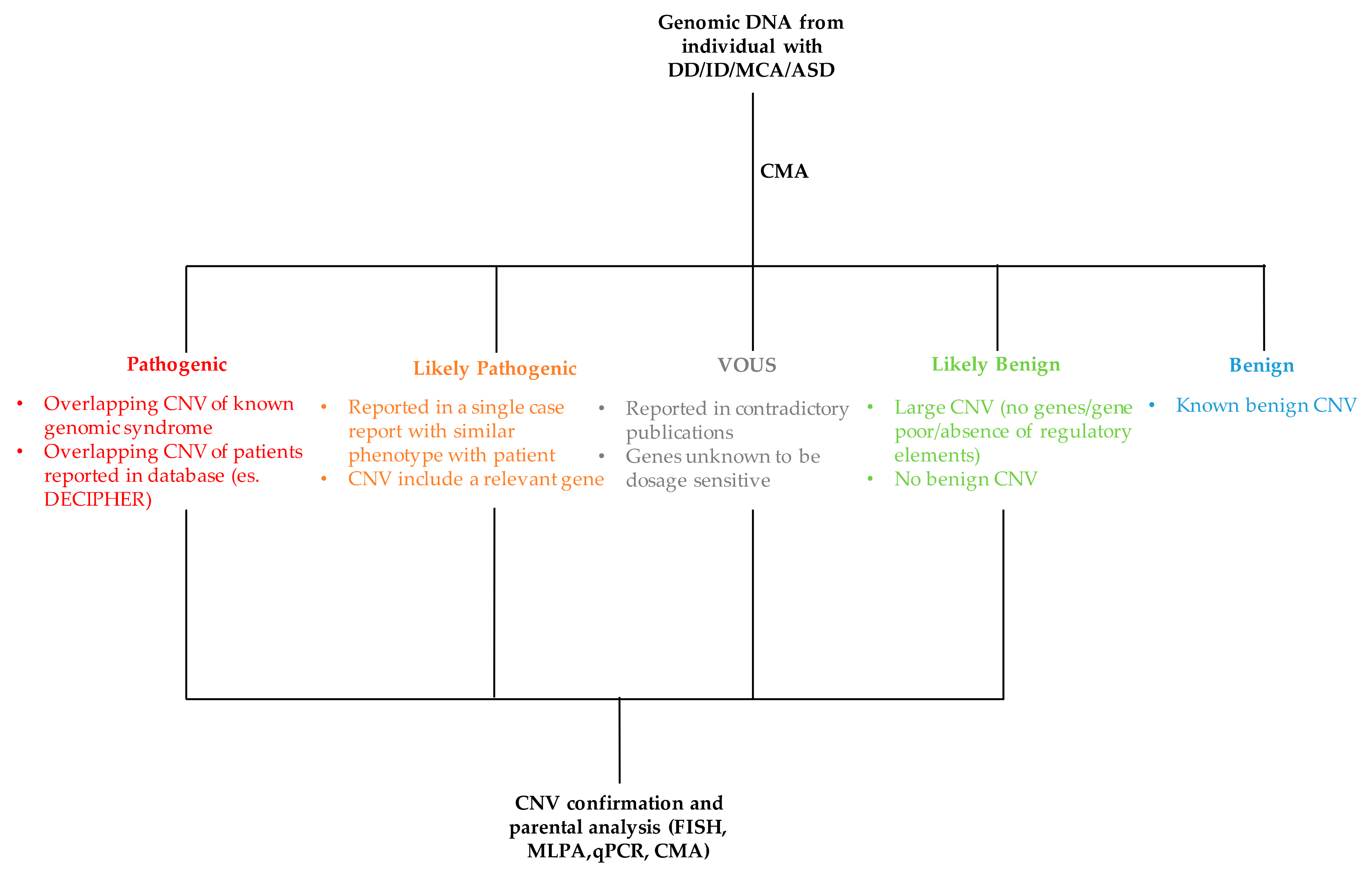
- Copy number variations size. Although there is a positive correlation between the increase of CNV size and its clinical relevance, this is not to be taken as a general rule. Large CNVs have been described as polymorphisms as otherwise small CNVs involving a single gene can be pathogenic.
- Gene content. The gene content of a CNV should be carefully evaluated for clinical association with the phenotype of proband. One should be verified if a gene or a group of genes, included in a duplication or deletion, are dosage-sensitive and associated with diseases. In this process, some considerations are important. First, if a gene is reported to be associated with a clinical phenotype when deleted or mutated, the duplication of the same gene may have no clinical relevance. Also, intragenic duplications may be pathogenic altering coding sequence, in contrast intronic deletions may have no clinical effect. If no mutation is reported in clinical literature for a gene, then it is recommended to avoid any conclusion of pathogenicity only on the basis of in silico analysis or in vitro and/or animal studies. A deletion of a gene associated with an autosomal recessive disorder may suggest the presence of a mutation on the second allele. Moreover, a CNV without genes in its interval generally is not reported in clinical laboratories. Another consideration is on CNV confirmation. Small deletions and duplications can be confirmed using quantitative-PCR (qPCR) and MLPA, while large CN (deletions >150 kb and duplications >400 kb) can be validated by other technique such as FISH and microarray. Despite the majority of duplications are in tandem, in a subset of cases the duplicated material resides on a different chromosome or in an atypical location on the chromosome of origin due to an unbalanced translocation or an inversion. In this context, FISH analysis is useful for a better characterization of the underline mechanism and for appropriate recurrence risk calculation.
- Databases. The are many public catalogs available for CNV interpretation. Among these the most used are the Database of Genomic Variants (DGV; http://dgv.tcag.ca/dvg/app/home), the Database of Chromosomal Imbalance and Phenotype in Human using Ensembl Resource (DECIPHER; https://decipher.sanger.ac.uk) and the Clinical Genome Resource (ClinGen; https://www.clinicalgenome.org). The DGV include human genomic structural variations found in healthy individuals and collected from worldwide studies. Although not present in ACMG recommendations, some authors suggest considering a CNV benign if present in at least three control individuals with the same orientation (deletion/duplication) [37]. The DECIPHER contains data from patients including both clinical phenotypes and genomic rearrangements. The ClinGen is a National Institutes of Health (NHI)-funded resource of clinically annotated genes and variants for use in precision medicine and research. ClinGen has a curated genome-wide dosage sensitivity map which can be used for the clinical interpretation of CNV. This resource provides evidence-based correlations between haploinsufficiency (loss) or triplosensitivity (gain) of a gene or genomic regions and clinical phenotypes. In addition, ClinGen provides CNV data from contributing laboratories and their classification, displayed in the NCBI ClinVar database. Finally, in-house or national reference database could be useful to construct a CNV map characterizing regional populations.
- Parental analysis. The inheritance of a CNV by an affected parent may support its pathogenicity. However, this event may be coincidental. When available, the evaluation of additional familial members may be useful to verify if the CNV continues to segregate with the phenotype. The inheritance of a CNV by an unaffected parent may not exclude its pathogenicity due to incomplete penetrance, variable expression, parent of origin imprinting effects or mosaic CNV in parent. Also, as reported above, the occurrence of an autosomal recessive disorder should be taken into consideration.
6. Conclusions
Funding
Conflicts of Interest
References
- Zarrei, M.; MacDoanld, J.R.; Merico, D. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; Barbosa, M.; Maciel, P. Recurrent copy number variations as risk factors for neurodevelopmental disorders: Critical overview and analysis of clinical implications. J. Med. Genet. 2016, 53, 73–90. [Google Scholar] [CrossRef] [PubMed]
- South, S.T.; Lee, C.; Lamb, A.N.; Higging, A.W.; Kearney, H.M. ACMG standards and guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet. Med. 2013, 15, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Neurology. Evidence Repot: Genetic and Metabolic Testing in Children with Global Developmental Delay. Available online: https://www.aan.com/Guidelines/home/GuidelineDetail/487 (accessed on 10 July 2018).
- Nevado, J.; Mergener, R.; Palomares-Bralo, M.; Souza, K.R.; Vallespín, E.; Mena, R.; Martínez-Glez, V.; Mori, M.Á.; Santos, F.; García-Miñaur, S.; et al. New microdeletion and microduplication syndromes: A comprehensive review. Genet. Mol. Biol. 2014, 37, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Berns, A. Cancer: Gene expression in diagnosis. Nature 2000, 403, 491–492. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Cocciardi, S.; Waddell, N.; Johnstone, C.N.; Marsh, A.; Henderson, S.; Simpson, P.; da Silva, L.; kConFab Investigators; et al. DNA methylome of familial breast cancer identifies distinct profiles defined by mutation status. Am. J. Hum. Genet. 2010, 86, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Di Martino, M.T.; Scionti, F.; Agapito, G.; Guzzi, P.H.; Cannataro, M.; Tassone, P.; Tagliaferri, P. DMET (Drug Metabolism Enzymes and Transporters): A pharmacogenomic platform for precision medicine. Oncotarget 2016, 7, 54028–54050. [Google Scholar] [CrossRef] [PubMed]
- Borlot, F.; Regan, B.M.; Bassett, A.S.; Stavropoulos, D.J.; Andrade, D.M. Prevalence of Pathogenic Copy Number Variation in Adults with Pediatric-Onset Epilepsy and Intellectual Disability. JAMA Neurol. 2017, 74, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Chen, C.H.; Lee, T.T.; Peck, K. Optimization of probe length and the number of probes per gene for optimal microarray analysis of gene expression. Nucleic Acids Res. 2004, 32, e99. [Google Scholar] [CrossRef] [PubMed]
- Relógio, A.; Schwager, C.; Richter, A.; Ansorge, W.; Valcárcel, J. Optimization of oligonucleotide-based DNA microarrays. Nucleic Acids Res. 2002, 30, e51. [Google Scholar] [CrossRef] [PubMed]
- Mason-Suares, H.; Kim, W.; Grimmett, L.; Williams, E.S.; Horner, V.L.; Kunig, D.; Goldlust, I.S.; Wu, B.L.; Shen, Y.; Miller, D.T.; et al. Density matters: Comparison of array platforms for detection of copy-number variation and copy-neutral abnormalities. Genet. Med. 2013, 15, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, L.; Alesi, V.; Loddo, S.; Novelli, A.; Bottillo, I.; Battaglia, A.; Digilio, M.C.; Zampino, G.; Ertel, A.; Fortina, P.; et al. High-resolution SNP arrays in mental retardation diagnostics: How much do we gain? Eur. J. Hum. Genet. 2010, 18, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Fridman, C.; Koiffmann, C.P. Origin of uniparental disomy 15 in patients with Prader-Willi or Angelman syndrome. Am. J. Med. Genet. 2000, 94, 249–253. [Google Scholar] [CrossRef]
- Schwartz, S. Clinical utility of single nucleotide polymorphism arrays. Clin. Lab. Med. 2011, 31, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Hoppman, N.; Rumilla, K.; Lauer, E.; Kearney, H.; Thorland, E. Patterns of homozygosity in patients with uniparental disomy: Detection rate and suggested reporting thresholds for SNP microarrays. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Kearney, J.B.; Conlin, L.K. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: Consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 2011, 31, 595–613. [Google Scholar] [CrossRef] [PubMed]
- Rehder, C.W.; David, K.L.; Hirsch, B.; Toriello, H.V.; Wilson, C.M.; Kearney, H.M. American College of Medical Genetics and Genomics: Standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genet. Med. 2013, 15, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Wiszniewska, J.; Bi, W.; Shaw, C.; Stankiewicz, P.; Kang, S.H.; Pursley, A.N.; Lalani, S.; Hixson, P.; Gambin, T.; Tsai, C.H.; et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur. J. Hum. Genet. 2014, 22, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Ballif, B.C.; Rorem, E.A.; Sundin, K.; Lincicum, M.; Gaskin, S.; Coppinger, J.; Kashork, C.D.; Shaffer, L.G.; Bejjani, B.A. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am. J. Med. Genet. A 2006, 140, 2757–2767. [Google Scholar] [CrossRef] [PubMed]
- Conlin, L.K.; Thiel, B.D.; Bonnemann, C.G.; Medne, L.; Ernst, L.M.; Zackai, E.H.; Deardorff, M.A.; Krantz, I.D.; Hakonarson, H.; Spinner, N.B. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum. Mol. Genet. 2010, 19, 19–1263. [Google Scholar] [CrossRef] [PubMed]
- Feenstra, I.; Hanemaaijer, N.; Sikkema-Raddatz, B.; Yntema, H.; Dijkhuizen, T.; Lugtenberg, D.; Verheij, J.; Green, A.; Hordijk, R.; Reardon, W.; et al. Balanced into array: Genome-wide array analysis in 54 patients with an apparently balanced de novo chromosome rearrangement and a meta-analysis. Eur. J. Hum. Genet. 2011, 19, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- De Gregori, M.; Ciccone, R.; Magini, P.; Pramparo, T.; Gimelli, S.; Messa, J.; Novara, F.; Vetro, A.; Rossi, E.; Maraschio, P.; et al. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: A study of 59 patients. J. Med. Genet. 2007, 44, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lin, S.; Huang, L.; He, Z.; Huang, X.; Zhou, Y.; Fang, Q.; Luo, Y. Application of chromosomal microarray analysis in prenatal diagnosis of fetal growth restriction. Prenat. Diagn. 2016, 36, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, F.B.; Machado-Neto, J.A.; Bertini, V.H.L.L.; Velloso, E.D.R.P.; Ratis, C.A.; Calado, R.T.; Simões, B.P.; Rego, E.M.; Traina, F. Single-nucleotide polymorphism array (SNP-A) improves the identification of chromosomal abnormalities by metaphase cytogenetics in myelodysplastic syndrome. J. Clin. Pathol. 2017, 70, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.R.; Pinto, I.P.; Minasi, L.B.; de Melo, A.V.; da Cruz e Cunha, D.M.; Cruz, A.S.; Ribeiro, C.L.; da Silva, C.C.; de Melo e Silva, D.; da Cruz, A.D. Screening for intellectual disability using high-resolution CMA technology in a retrospective cohort from Central Brazil. PLoS ONE 2014, 9, e103117. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lei, T.; Fu, F.; Li, R.; Jing, X.; Yang, X.; Liu, J.; Li, D.; Liao, C. Application of chromosome microarray analysis in patients with unexplained developmental delay/intellectual disability in South China. Pediatr. Neonatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bruno, D.L.; Ganesamoorthy, D.; Schoumans, J.; Bankier, A.; Coman, D.; Delatycki, M.; Gardner, R.J.; Hunter, M.; James, P.A.; Kannu, P.; et al. Detection of cryptic pathogenic copy number variations and constitutional loss of heterozygosity using high resolution SNP microarray analysis in 117 patients referred for cytogenetic analysis and impact on clinical practice. J. Med. Genet. 2009, 46, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Adam, S.; Arbour, L.; Armstrong, L.; Baross, A.; Birch, P.; Boerkoel, C.; Chan, S.; Chai, D.; Delaney, A.D.; et al. Detection of pathogenic copy number variants in children with idiopathic intellectual disability using 500 K SNP array genomic hybridization. BMC Genom. 2009, 16, 526. [Google Scholar] [CrossRef] [PubMed]
- Zarrei, M.; Fehlings, D.L.; Mawjee, K.; Switzer, L.; Thiruvahindrapuram, B.; Walker, S.; Merico, D.; Casallo, G.; Uddin, M.; MacDonald, J.R.; et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet. Med. 2018, 20, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Al-Qattan, S.M.; Wakil, S.M.; Anazi, S.; Alazami, A.M.; Patel, N.; Shaheen, R.; Shamseldin, H.E.; Hagos, S.T.; AlDossari, H.M.; Salih, M.A.; et al. The clinical utility of molecular karyotyping for neurocognitive phenotypes in a consanguineous population. Genet. Med. 2015, 17, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Asadollahi, R.; Oneda, B.; Joset, P.; Azzarello-Burri, S.; Bartholdi, D.; Steindl, K.; Vincent, M.; Cobilanschi, J.; Sticht, H.; Baldinger, R.; et al. The clinical significance of small copy number variants in neurodevelopmental disorders. J. Med. Genet. 2014, 51, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenbeck, D.; Williams, C.L.; Drazba, K.; Descartes, M.; Korf, B.R.; Rutledge, S.L.; Lose, E.J.; Robin, N.H.; Carroll, A.J.; Mikhail, F.M. Clinical relevance of small copy-number variants in chromosomal microarray clinical testing. Genet. Med. 2017, 19, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Qiu, W.; Wang, L.; Gu, X.; Yu, Y. Exonic deletions of AUTS2 in Chinese patients with developmental delay and intellectual disability. Am. J. Med. Genet. A 2016, 170, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Thorald, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. A Working Group of the American College of Medical Genetics (ACMG) Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variations. Genome Med. 2011, 13, 680–685. [Google Scholar] [CrossRef]
- Koolen, D.A.; Pfundt, R.; de Leeuw, N.; Hehir-Kwa, J.Y.; Nillesen, W.M.; Neefs, I.; Scheltinga, I.; Sistermans, E.; Smeets, D.; Brunner, H.G.; et al. Genomic Microarrays in Mental Retardation: A Practical Workflow for Diagnostic Applications. Hum. Mutat. 2009, 30, 283–292. [Google Scholar] [CrossRef] [PubMed]
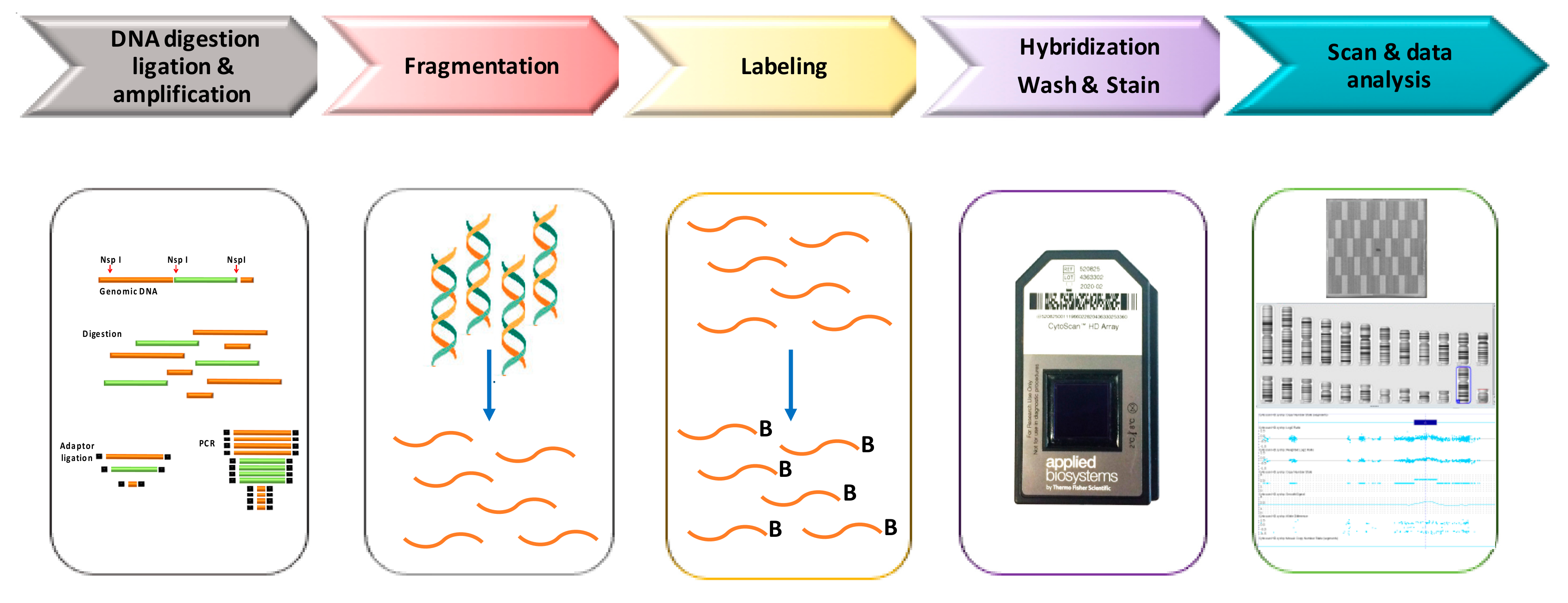



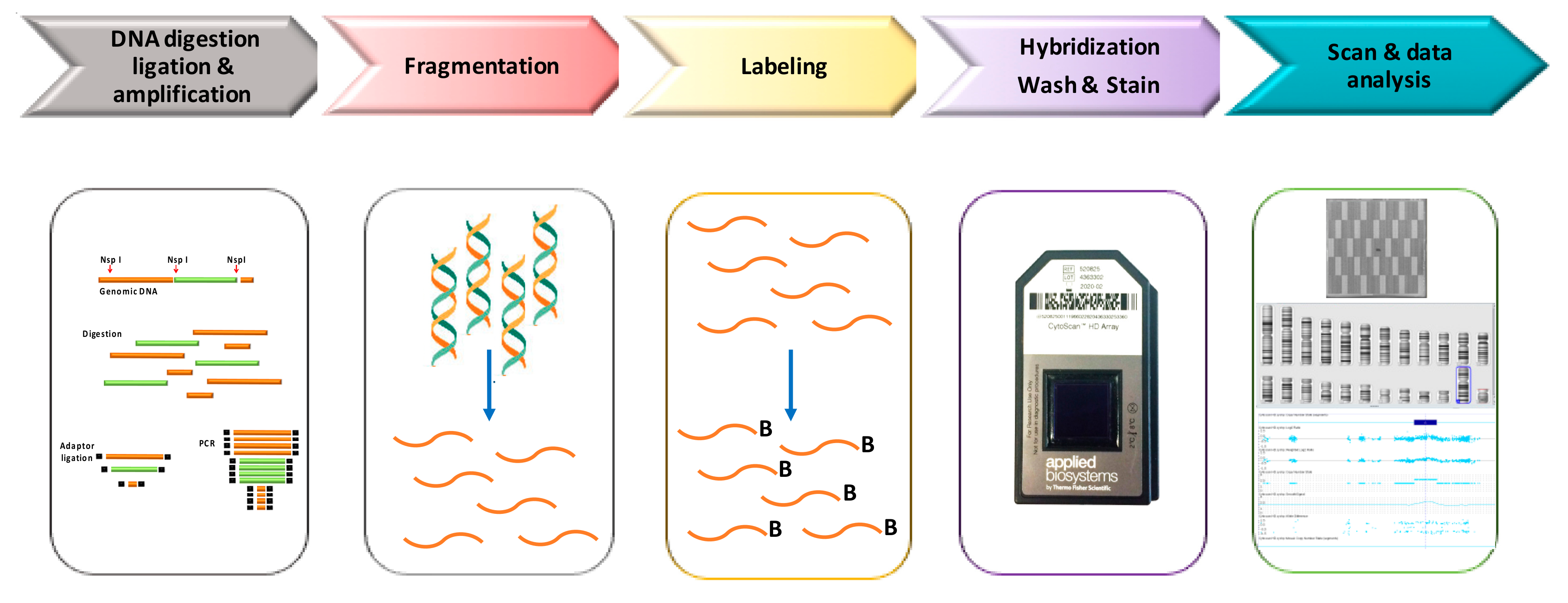{kind=link}
{kind=link}
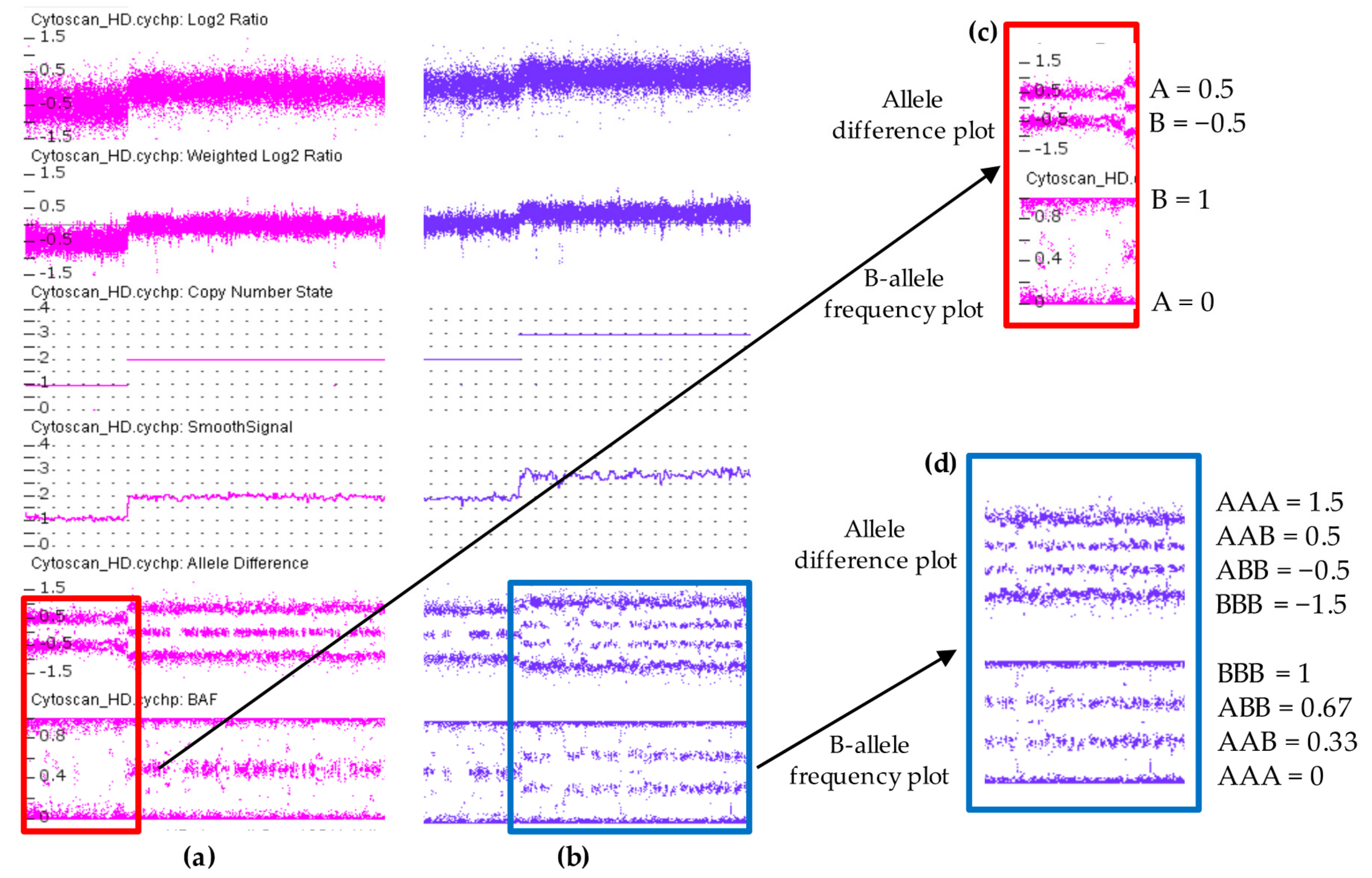{kind=link}
{kind=link}
| SNP-array | a-CGH | a-CGH CN + SNP |
|---|---|---|
| Oligonucleotide probe length: ~25 bp | Oligonucleotide probe length: 60–70 bp | Oligonucleotide probe length: 60–70 bp |
| Copy number probe + SNP probe (high density) | Copy number probe only | Copy number probe + SNP probe (low or mid density) |
| Hybridization of DNA test only | Hybridization of DNA test and DNA reference | Hybridization of DNA test and DNA reference |
| Detection of UPD and consanguinity | No detection of UPD and consanguinity | Detection of UPD and consanguinity |
| Reference | Patients (n) | Disorder | CMA Platform | CNV Size (kb) | Origin | CNV Interpretation | |||
|---|---|---|---|---|---|---|---|---|---|
| De Novo n (%) | Inherited n (%) | Pathogenic n (%) | Lakely Pathogenic n (%) | VOUS n (%) | |||||
| Pereira et al. [27] | 15 | ID | Cytoscan HD | ≥100 | 9 (50) | 9 (50) | 4 (22) | 4 (22) | 10 (56) |
| Wang et al. [28] | 489 | ID | Cytoscan HD | ≥100 | 141 (70%) | 60 (30%) | 122 (61%) | 4 (2) | 75 (37) |
| Zarrei et al. [31] | 97 | CP | Cytoscan HD | ≥10 | 9 (30) | 21 (70) | 4 (13.3) | 1 (3.3) | 25 (83.4) |
| Al-Qattan et al. [32] | 183 | DD/ID | Cytoscan HD | ≥200 | 40 (90) * | 4 (10) * | 40 (81.6) | 5 (10.2) | 4 (8.2) |
| Affymetrix SNP Array 6.0 | |||||||||
| Cyto-V2 | |||||||||
| Asadollhai et al. [33] | 714 | NDD | Cytoscan HD | <500 | 12 (46.1) | 14 (53.4) | 12 (46.1%) | 4 (15.4) | 10 (38.5) |
| Affymetrix SNP Array 6.0 | |||||||||
| Affymetrix Cytogenetics 2.7 | |||||||||
| CNV Classification | Description |
|---|---|
| Pathogenic | The CNV is documented as clinically significant in multiple peer-reviewed publications, even if penetrance and expressivity of the CNV are known to be variable |
| Benign | The CNV has been reported in multiple peer-reviewed publications or curated databases as a benign variant, particularly if the nature of the copy number variation has been well characterized and/or the CNV represents a common polymorphism |
| Uncertain clinical significance CNV (Likely pathogenic) | The CNV is described in a single case report but with well-defined breakpoints and phenotype, both specific and relevant to the patient findings. |
| A gene within the CNV interval has a very compelling gene function that is relevant and specific to the reason for patient referral | |
| Uncertain clinical significance CNV (Likely benign) | The CNV has no genes in interval but exceeds a size criterion that may be established by the laboratory. |
| The CNV is described in a small number of cases in databases of variation in the general population but does not represent a common polymorphism | |
| Uncertain clinical significance CNV (No subclassification) | The CNV contains genes, but it is not known whether the genes in the interval are dosage sensitive. |
| The CNV is described in multiple contradictory publications and/or databases, and firm conclusions regarding clinical significance are not yet established |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scionti, F.; Di Martino, M.T.; Pensabene, L.; Bruni, V.; Concolino, D. The Cytoscan HD Array in the Diagnosis of Neurodevelopmental Disorders. High-Throughput 2018, 7, 28. https://doi.org/10.3390/ht7030028
Scionti F, Di Martino MT, Pensabene L, Bruni V, Concolino D. The Cytoscan HD Array in the Diagnosis of Neurodevelopmental Disorders. High-Throughput. 2018; 7(3):28. https://doi.org/10.3390/ht7030028
Chicago/Turabian StyleScionti, Francesca, Maria Teresa Di Martino, Licia Pensabene, Valentina Bruni, and Daniela Concolino. 2018. "The Cytoscan HD Array in the Diagnosis of Neurodevelopmental Disorders" High-Throughput 7, no. 3: 28. https://doi.org/10.3390/ht7030028
APA StyleScionti, F., Di Martino, M. T., Pensabene, L., Bruni, V., & Concolino, D. (2018). The Cytoscan HD Array in the Diagnosis of Neurodevelopmental Disorders. High-Throughput, 7(3), 28. https://doi.org/10.3390/ht7030028