Abstract
Microglia, the unique and motile immune cells of the central nervous system (CNS), function as a security guard in maintaining CNS homeostasis, primarily through calcium signaling. The calcium dynamics in microglia control important functions such as phagocytosis, cytokine release, and migration. Calcium dysregulation in microglia has been linked to several CNS disorders, like Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), and ischemic stroke (IS). Calcium entering through channels such as voltage-gated calcium channels (VGCCs), store-operated calcium entry (SOCE), and transient receptor potential (TRP) channels is essential for microglial activation and pro-inflammatory responses. Under pathological conditions, like the formation of amyloid-β plaques in AD, aggregation of α-synuclein in PD, and oxidative stress in MS, calcium dysregulation exacerbates neuroinflammation, mitochondrial dysfunction, and neurodegeneration. Therapeutic strategies targeting calcium signaling pathways, using calcium channel blockers and antioxidant interventions, show promise for alleviating microglial activation and slowing down disease progression. This review summarizes the underlying mechanisms of microglial calcium dysregulation and potential therapeutic benefits for restoring microglial calcium balance in CNS disorders.
1. Introduction
Microglia and monocytes are essential immune cells with distinct yet overlapping roles in the central nervous system (CNS) and peripheral immune responses, respectively. Microglia, the resident immune cells of the CNS, originate from erythromyeloid precursors in the yolk sac during embryonic development, migrating into the CNS before birth and remaining throughout life. Unlike monocytes, which are continuously renewed from hematopoietic stem cells (HSCs) in the bone marrow, microglia are self-renewing and do not rely on post-natal hematopoiesis. They are distributed across all regions of the CNS, with the highest density in the gray matter [1]. These highly specialized, long-lived cells play critical roles in immune surveillance, tissue remodeling, and the regulation of neuronal activity.
Microglial cells in neighboring areas scan partly overlapping areas of the brain parenchyma, suggesting that they monitor its microenvironment as a surveillance mechanism. In spite of this, microglial cells maintain a certain spatial separation by avoiding direct contact with each other. It may be important to avoid contact between microglial cells in order to protect their individual surveillance territories and prevent excessive activation or interference with neighboring cells. Microglial cells, on the other hand, have a high affinity for other types of cortical cells, such as neurons and astrocytes [2,3,4].
Ramified microglial cells, a subset of microglia, are highly branched with small cell bodies and fine processes. In their homeostatic state, they continuously survey the brain parenchyma and communicate with neurons and astrocytes, performing vital functions such as synaptic pruning, monitoring and eliminating synapses, and promoting adult neurogenesis. These cells help reorganize neuronal networks through plasticity-driven reorganization, a process involving the modification of synapses in response to developmental or environmental changes [5]. Furthermore, microglia perform neuroprotective roles by clearing apoptotic cells and releasing neurotrophic factors, which support neuronal health. Microglia, specifically ramified microglial cells, phagocytose apoptotic neurons and help to integrate adult-born neurons into preexisting neuronal networks in areas of the brain where adult neurogenesis occurs. Importantly, under certain conditions, phagocytic microglia show no global morphological signs of activation while performing this phagocytosis [6].
The survival and activity of microglia are heavily dependent on the colony-stimulating factor 1 (CSF-1) and its receptor CSF1R, which is also a receptor for interleukin 34 (IL-34), another key player in microglial maintenance. Depleting microglia through genetic manipulation or pharmacological inhibition of CSF1R results in significant microglial loss, though this approach may also affect peripheral immune cells that express CSF1R. Microglia maintain homeostasis by constantly surveying their environment, relying on pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), to detect damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs). Upon activation, microglia transition from a surveillant to an amoeboid shape, engaging in phagocytosis and the production of pro-inflammatory cytokines. Despite their role in neuroinflammation, microglia also possess neuroprotective functions, such as clearing apoptotic cells, releasing neurotrophic factors, and modulating immune responses. The balance between their protective and harmful effects depends on the signals received from the surrounding environment. Activated microglia can express both pro-inflammatory cytokines (e.g., IL-1β, TNF-α) and anti-inflammatory mediators (e.g., IL-10, TGF-β), depending on the context. Microglia require TGF-β signaling to remain in their surveillant state but not for survival [7,8].
Monocytes, on the other hand, are peripheral myeloid cells originating from the fetal liver during embryogenesis and are continuously replenished in adults from bone marrow-derived HSCs. In mice, monocytes are categorized into two main types: inflammatory monocytes (Ly6Chi) and patrolling monocytes (Ly6Clo), while in humans, the equivalent cells are classical, intermediate, and non-classical monocytes. Under normal conditions, the blood–brain barrier (BBB) restricts monocytes from entering the CNS, but during inflammation, monocytes can infiltrate the CNS and differentiate into monocyte-derived macrophages (MDMs) or monocyte-derived dendritic cells (moDCs). These monocyte-derived cells can adopt microglia-like phenotypes, complicating the distinction between resident microglia and infiltrating cells, particularly in neuroinflammatory conditions, like MS and Alzheimer’s disease (AD). These engrafted monocytes exhibit calcium transients in response to ATP, Bz-ATP, and UDP, similar to surveillant resident microglia. However, there may be differences in the amplitudes of calcium transients between these two cell types [9]. Animal models using techniques such as microglia-depletion drugs, gene silencing, and in vivo imaging have been instrumental in advancing our understanding of microglial and monocyte functions in CNS diseases. These models, though not perfectly replicating human pathology, provide valuable insights into the distinct roles of these immune cells and offer potential therapeutic targets for neurodegenerative and neuroinflammatory disorders [10,11,12,13].
One of the most critical pathways regulating microglial activity is calcium (Ca2+) signaling, a tightly controlled process that governs functions such as migration, phagocytosis, cytokine release, and cellular metabolism. Calcium signals in microglia, triggered by various extracellular stimuli and mediated by specific calcium channels, pumps, and exchangers, play pivotal roles in both physiological and pathological conditions. However, in pathological states, such as neurodegenerative diseases and ischemia, dysregulation of calcium signaling in microglia leads to chronic activation, resulting in excessive neuroinflammation and neuronal damage. Microglial dysregulation in calcium signaling is a hallmark of several neurodegenerative diseases, including AD, Parkinson’s disease (PD), multiple sclerosis (MS), and ischemic stroke. For instance, in AD, amyloid-β (Aβ) plaques disrupt microglial calcium homeostasis, leading to abnormal calcium influx and the overproduction of pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6. These cytokines, along with reactive oxygen species (ROS) and excitotoxins, such as glutamate, exacerbate neuroinflammation and contribute to the progression of neurodegeneration. Similarly, in PD, misfolded α-synuclein aggregates stimulate microglial activation, leading to calcium dysregulation, mitochondrial dysfunction, and further neuronal loss in regions such as the substantia nigra. In MS, the chronic activation of microglia and associated calcium imbalances contribute to demyelination and the degeneration of neurons. These disease processes underscore the complex interplay between microglial activation, calcium signaling, and neuroinflammation [14,15,16].
This review aims to explore the molecular mechanisms of calcium signaling in microglia. We will examine how dysregulated calcium signaling contributes to neuroinflammation, mitochondrial dysfunction, and synaptic pruning in neurodegenerative diseases like AD, PD, MS, and ischemic stroke. Additionally, we will discuss therapeutic strategies to restore calcium homeostasis in microglia, focusing on interventions, such as calcium channel blockers and antioxidants, which hold promise in reducing neuroinflammation and slowing disease progression.
2. Calcium Signaling in Microglia
Microglia, as sole immune cells of the CNS, play a vital role in the brain’s response to injury, inflammation, and neurodegenerative diseases. In vitro, evidence suggests that microglia’s executive functions depend on iCa2+ signaling. However, little is known about microglial calcium signaling in the healthy and diseased brain in vivo. According to in vivo/in situ findings, surveillant microglia in the brain rarely exhibit spontaneous calcium transients and instead respond to cell or tissue damage in their microenvironment. These calcium transients are believed to be mediated by releasing calcium from intracellular stores and activating specific receptors on the microglial cell surface. The occurrence of spontaneous calcium transients in microglia is distinct from the calcium signaling observed in neighboring astrocytes, which have different characteristics and response patterns [17].
- Calcium channels, pumps, and exchangers in microglia
Microglia orchestrate calcium ion balance, signaling, and function via calcium channels, pumps, and exchangers that serve as portals for Ca2+ to enter or exit the cells and preserve intracellular Ca2+ (iCa2+) homeostasis [18]. The microglial Ca2+ channels include store-operated Ca2+ entry (SOCE), transient receptor potentials (TRP), and voltage-gated Ca2+ channels (VGCCs), allowing Ca2+ to enter the cell. Ca2+ extrusion from the cell occurs mainly through the Ca2+-ATPase and the Na+/Ca2+ exchanger (NCX) proteins.
- i.
- SOCE (store-operated Ca2+ Entry) channels
SOCE maintains calcium homeostasis by releasing Ca2+ into the endoplasmic reticulum (ER) when the Ca2+ store is depleted due to the functional and physical connection. Ca2+ release from ER in microglia is known to be regulated by Ca2+ itself or messengers like inositol-1,4,5-triphosphate (IP3). Stimulation of various receptors activates phospholipase C (PLC), leading to the hydrolysis of phosphoinositide-4,5-bisphosphate (PIP2) into IP3 and 1,2-diacylglycerol (DAG). IP3 induces Ca2+ release from intracellular stores, while DAG (diacylglycerol) contributes to Ca2+ entry from the extracellular compartment. IP3 binds to its receptor (IP3R) on ER walls, releasing stored Ca2+ into the cytosol [19]. Two primary elements of the SOCE channel, stromal interaction molecule (STIM) proteins and Orai channels, form the structure of calcium release-activated calcium (ICRAC) channels in the plasma membrane. STIM proteins, situated within the ER, function as calcium sensors and undergo structural alterations in response to the depletion of iCa2+ stores. Located in the plasma membrane, Orai channels engage with activated STIM proteins, facilitating the entry of Ca2+ into microglial cells [20]. The reuptake of calcium by the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) signifies the end of SOCE, prompting the return of STIM and Orai proteins to their original locations. STIM1, STIM2, and Orai1 play key roles in controlling SOCE and purinergic activation in microglia [21,22].
- ii.
- TRP (Transient receptor potential) channels
Most of them are weakly voltage-sensitive, non-selective ion channels that are involved in cytokine production, migration, and proliferation—all crucial cellular functions in microglia [23]. Because these channels typically have low conductance, they can function for longer periods of time without over-saturating the cell with Ca2+ [24]. Several TRP family members are expressed in microglia, including TRPV (transient receptor potential vanilloid), TRPM (transient receptor potential melastatin), and TRPC (transient receptor potential canonical)
- TRPV
Among the six members of TRPV subfamilies (TRPV1-V6), TRPV1, TRPV4, and TRPV2 have been reported to be involved in calcium homeostasis expressed in microglia. TRPV1 activity is associated with altered Ca2+ concentration, resulting in mitochondrial disruption and microglial cell death. Moreover, the release of cytochrome C from mitochondria upon TRPV1 channel activation caused microglia to degenerate [25]. TRPV1 also plays an important role in ATP-induced NLRP3 inflammasome activation through mediating Ca2+ influx and phosphorylation of phosphatase PP2A in microglia [26]. TRPV4 channels are non-selective cation channels with a high permeability to calcium and magnesium ions compared to sodium ions, and the activation of these channels results in calcium influx [27]. TRPV2 channels are also expressed in microglia, acting as calcium-permeable cation channels activated by heat, ligands, and mechanical stresses [28]. A study found that inducible nitric oxide synthase/nitric oxide signaling inhibits microglial proliferation through TRPV2-mediated calcium influx and p21 expression [29].
- TRPM
TRPM2 regulates iCa2+ and enhances H2O2-induced Ca2+ signaling, influencing immune responses and neuroinflammation [30]. Evidence also shows that TRPM2 is present in human microglial cells and contributes to calcium influx [31]. A study identified a TRPM7-like current in rat microglia, which is constitutively active and depends on tyrosine phosphorylation. TRPM7 channels are inhibited by elevated intracellular Mg2+ but are not affected by a wide range of iCa2+ concentrations. So, this suggests the importance of TRPM7 in microglia functions that rely on iCa2+ elevations [32]. Another study reveals that ProBDNF (precursor forms of mature Brain-Derived Neurotrophic Factor) induces a sustained elevation of iCa2+ through binding to the p75 neurotrophin receptor, possibly mediated by TRPM7 channels and Rac1 activation. It also increases the relative surface expression of TRPM7 [33].
- TRPC
The TRPC subfamily consists of Ca2+-permeable non-selective cation channels (TRPC1–C7) highly expressed in various brain regions. As far as we know, only TRPC1 and TRPC3 are active in microglia. TRPC1 is a cation non-selective calcium-permeable channel and is thought to be involved in SOCE [34]. TRPC channels, particularly TRPC1, can combine with other TRPC subunits, like TRPC3, TRPC6, and TRPC7, responding to signals from G protein-coupled receptors (GPCRs). These channels play key roles in brain function, such as maintaining the blood–brain barrier, facilitating communication between neurons and glia, and participating in brain injury processes.
TRPC channels, specifically TRPC3, TRPC6, and TRPC7, are critically involved in Ca2+ dysregulation following brain injuries, like ischemic stroke, traumatic brain injury (TBI), and epilepsy, leading to excitotoxicity and secondary brain injury. Activation of Gαq-type GPCRs leads to excessive Ca2+ entry via these TRPC channels, particularly in cerebellar Purkinje neurons, resulting in dendritic damage and neuronal dysfunction. Knockout models, such as Trpc3KO and Trpc1/3/6/7QKO, demonstrated significant neuroprotection by reducing Ca2+ entry and excitotoxicity, decreasing secondary injury in both cerebral and cerebellar regions. These findings position TRPC channels as promising therapeutic targets for neuroprotection in brain injuries characterized by excitotoxicity and secondary damage [35].
In addition, TRPC5 has been linked to neuroinflammation and memory impairment. A study demonstrated that upregulating TRPC5 in hippocampal excitatory synapses improved memory deficits associated with neuroinflammation in Cx3cr1CreERIL-10−/− mice. These mice, with IL-10 (an anti-inflammatory cytokine), knocked out in microglia, showed spatial memory deficits, elevated pro-inflammatory cytokines, such as IL-1β and IL-18, and reduced synaptic proteins, indicating impaired synaptic transmission. Upregulating TRPC5 in these neurons alleviated memory impairment by inhibiting the NLRP3 inflammasome and reducing neuroinflammation, highlighting the potential of TRPC5 as a therapeutic target for neuroinflammation-related cognitive decline [36].
Furthermore, TRPC3 channels play an important role in sustaining BDNF (Brain-Derived Neurotrophic Factor)-induced intracellular calcium (iCa2+) elevation in microglial cells. BDNF rapidly increases the surface expression of TRPC3 channels, suppressing nitric oxide (NO) production induced by TNFα. This suggests that TRPC3 channels are essential for the BDNF-mediated regulation of iCa2+ and NO levels in activated microglia, which may have implications for the inflammatory responses and pathophysiology of neuropsychiatric disorders. Together, these findings underscore the diverse and critical roles TRPC channels play in brain function, injury response, and disease mechanisms, making them promising targets for therapeutic intervention [14].
- iii.
- VGCC (voltage-gated Ca2+ channels)
Microglia express L-type voltage-gated calcium channels (VGCCs) that facilitate the influx of Ca2+. These channels regulate iCa2+ levels and are involved in various microglial functions [14]. VGCCs are classified into multiple families, namely, Cav1, Cav2, and Cav3, each comprising numerous subtypes. The Cav1.2, 1.3, and 2.2 subtypes of specific channels have been identified in microglia. These channels regulate iCa2+ levels in response to membrane depolarization. These channels are involved in microglial activation, superoxide production, and modulation of neuroinflammatory responses. Despite ongoing debate regarding their presence in microglia, several studies have confirmed the expression of VGCC genes and proteins, highlighting their role in neurodegenerative conditions [37,38].
- iv.
- Sodium/Calcium Exchangers (NCXs)
Sodium/calcium exchangers (NCXs) are membrane transporters that play a critical role in regulating iCa2+ homeostasis by exchanging three sodium ions (Na+) for one calcium ion (Ca2+) across the plasma membrane [20,39]. In microglia, these exchangers are key players in balancing Ca2+ levels, which in turn affect various cellular functions, including inflammation, migration, and neuroprotection [40]. The NCX operates bidirectionally, either expelling Ca2+ from the cell while importing Na+ (forward mode) or bringing Ca2+ into the cell while exporting Na+ (reverse mode). The direction depends on the electrochemical gradients of Na+ and Ca2+ [41]. Under physiological conditions, the NCX usually functions in forward mode, lowering iCa2+ levels. However, in pathophysiological conditions, like inflammation, the exchanger can switch to reverse mode, increasing iCa2+. Inflammatory stimuli, such as lipopolysaccharide (LPS) or pro-inflammatory cytokines, can modulate NCX activity, influencing the shift between forward and reverse modes [42]. This shift in NCX function can exacerbate or mitigate Ca2+-dependent inflammatory responses, impacting processes like NO production and ROS generation. In neurodegenerative diseases, like AD and PD, dysregulation of the NCX can lead to abnormal Ca2+ homeostasis, contributing to chronic microglial activation. The reverse mode of the NCX, leading to increased iCa2+, can enhance the production of pro-inflammatory mediators, worsening neuronal damage. Under conditions of oxidative stress or during the resolution of inflammation, NCX activity helps to restore Ca2+ balance, promoting a shift from a pro-inflammatory to an anti-inflammatory microglial phenotype. Proper NCX function is crucial for preventing excessive iCa2+ accumulation, which can lead to mitochondrial dysfunction and cell death [43].
- v.
- Sarcoplasmic/Endoplasmic Reticulum Ca2+ ATPase (SERCA)
SERCA plays a major part in maintaining the iCa2+ concentration by pumping Ca2+ from the cytoplasm to the luminal space of the sarco/ER against a concentration gradient. The cytoplasmic portion of SERCA hydrolyzes adenosine triphosphate (ATP), producing the energy required for active transport [43,44]. Three types of SERCA genes have been recognized in mammals, each having multiple splice variants. Among these, SERCA2b stands out as the isoform most abundantly expressed in the brain. In activated microglia, there is an upregulation of SERCA2b expression, likely as a response to the increased cytosolic calcium concentration. The upregulation of SERCA2b suggests its involvement in restoring calcium homeostasis in these cells. The inhibition of SERCA activity using the inhibitor, Thapsigargin, affects calcium dynamics in microglia and has effects on migration and phagocytosis [45].
- vi.
- Plasma membrane Ca2+ATPase pump (PMCAs)
The PMCA pump is responsible for maintaining the concentration gradient between extracellular and cytosolic Ca2+ levels. It actively pumps Ca2+ out of the cell, ensuring that cytosolic Ca2+ levels remain low. PMCA pumps, along with the NCX, play a crucial role in maintaining the concentration gradient of Ca2+ across the plasma membrane. They help in sequestering cytosolic Ca2+ and preventing its accumulation, which is essential for the induction of numerous Ca2+-dependent signaling cascades [38]. Dysfunction of PMCA, a key regulator of calcium homeostasis, can result in persistent elevation of cytosolic calcium levels. This disturbance may contribute to neurotoxicity and accelerate the onset of neurodegenerative diseases and cognitive impairments with aging. Regions of the brain expressing both TRP and PMCA may exhibit heightened sensitivity in calcium concentration regulation. Aberrant expression of TRP or PMCA subtypes in these specific brain areas may pose a greater risk for neurodegenerative disorders compared to other regions [46].
- vii.
- Piezo1 Channel
Mechanotransduction, the process by which cells convert mechanical forces into biochemical signals, is essential for many biological processes and is mediated by mechanosensitive ion channels, such as Piezo1 and Piezo2, first identified by Coste et al. in 2010 [47]. While Piezo2 primarily functions in detecting touch and proprioception, Piezo1 is widely expressed across both sensory and non-sensory tissues, playing a key role in cellular responses. In the CNS, cells like microglia are exposed to various mechanical forces, and Piezo1 enables them to detect and respond to environmental changes, such as tissue stiffness, through a process called durotaxis [48].
Piezo1 is crucial for regulating microglial activation and the inflammatory response in different conditions, such as lipopolysaccharide (LPS) stimulation and high-glucose environments. When activated, Piezo1 allows Ca2+ to flow into cells, triggering downstream signaling pathways, notably JNK1 and mTOR, which are vital for processes like cell survival, proliferation, migration, and differentiation. Acting as a second messenger, Ca2+ activates these pathways, leading to the production of inflammatory cytokines such as IL-1, IL-6, and TNF-α. In high-glucose conditions, Piezo1 upregulation disrupts microglial proliferation and migration, weakening their immune response. However, inhibiting Piezo1 in these conditions has been shown to improve cell viability and restore migration, indicating that Piezo1 overactivation contributes to microglial dysfunction. Excessive Piezo1 activation in high-glucose environments results in large Ca2+ peaks, disrupting the communication between the ER and mitochondria and further impairing microglial function. Inhibiting Piezo1 under these conditions has been found to restore normal inflammatory responses through JNK1 signaling, mitigating the harmful effects of excessive Ca2+ influx. Additionally, LPS activates microglia via toll-like receptor 4 (TLR4), triggering the NF-κB signaling pathway through ERK1/2 and p38 MAPK and leading to the production of pro-inflammatory cytokines, like TNF-α and IL-6. However, activating Piezo1 with Yoda1 has been found to inhibit LPS-induced NF-κB activation, suggesting that Piezo1-Ca2+ signaling can reduce microglial activation and pro-inflammatory cytokine production, with NF-κB acting as a key mediator. Piezo1 also interacts with other ion channels, such as TRPM7, which plays a role in Ca2+ signaling during inflammatory responses, suggesting a potential link between the two, although further research is needed. Overall, Piezo1 is central to regulating microglial mechanosensitivity and inflammation, making it a promising therapeutic target for neurodegenerative and neuroinflammatory diseases where microglial dysfunction is a critical factor [49,50,51].
- viii.
- Receptor-Operated Calcium Channels
- Purinergic Receptors
Changes in iCa2+ levels induced by ATP play crucial roles in cellular signal transduction, connecting external stimuli with cellular responses. Microglial cells respond to ATP in a concentration-dependent manner, inducing Ca2+ transients via activation of purinergic receptors (P2Rs). Microglia expresses both types of P2Rs: P2X receptors (specifically P2X4 and P2X7) and P2Y receptors (including P2Y2, P2Y6, and P2Y12). The increase in iCa2+ concentration mediated by P2Rs plays a pivotal role in modulating microglial cell activation and regulating associated functions. These functions encompass chemotaxis (via P2Y12 receptors), pro-inflammatory substance release (through P2X4 receptors), phagocytosis (via P2Y6 receptors), and proliferation (mediated by P2X7 receptors) [52,53]. ATP triggers current and induces iCa2+ elevations in mouse and human microglia. In mice, ATP prompts the release of IL-1β and IL-6 from microglia, while in rats, it induces chemotaxis and the secretion of plasminogen and TNF-α. ATP also activates the nuclear factor of activated T-cells (NFAT), which regulates early inflammatory gene expression, as well as NF-κB, controlling cytokine expression and apoptosis. Additionally, ATP stimulates the phosphorylation of MAPK [54,55,56]. Activation of P2Y receptors (P2YR) leads to increased iCa2+ levels via depletion of SOCE. Alternatively, activation of P2X receptors (P2XR) is associated with the influx of Na+ and Ca2+ ions and the efflux of K+ ions through non-selective cationic channels, resulting in cellular depolarization. An interesting aspect of purinergic stimulation in microglia is the dependency of cellular responses on the concentration of the agonist. For example, increased ATP levels (in the millimolar range) that activate the P2X7 receptor subtype markedly increase the release of inflammatory mediators by microglia. Nevertheless, additional ionotropic P2X receptor family members that are sensitive to reduced ATP levels also have significant roles in mediating brain microglial inflammatory responses. Activation of SOC in human microglia is influenced by ATP binding to a P2X receptor (not P2X7) at lower ATP concentrations, in addition to the P2Y receptor-dependent depletion of internal Ca stores. This modulation is consistent with depolarization-induced SOC inhibition and Na+ influx mediated by P2X receptors [57,58].
- Adrenergic receptors
Adrenergic receptors (ARs) also known as adreno-receptors, are classified as α or β receptors. Those two classes are further subdivided into α-1, α-2, β-1, β-2, and β-3. α-1 and α-2 have three subtypes. Microglial cells were found to express mRNAs encoding α1A, α2A, β1, and β2 ARs. These receptors respond to neurotransmitters, such as norepinephrine (NE) and epinephrine, which are released by neurons and other cells, including the adrenal glands [59,60]. Activation of α1-ARs triggers an elevation in the turnover of IP3, leading to a subsequent increase in iCa2+ [61].
- Glutamate receptors
Glutamate, the primary excitatory neurotransmitter in the brain, has been shown to interact with receptors expressed on microglia, allowing these immune cells to respond to glutamate in both the developing and mature brain. Microglia express both metabotropic glutamate receptors (mGluRs), primarily group I, and ionotropic glutamate receptors (iGluRs), allowing them to respond to glutamate. The primary ionotropic glutamate receptors found in microglia are AMPA receptors (AMPARs) and NMDA receptors (NMDARs). While AMPARs in astrocytes are typically calcium permeable, those in microglia are often calcium impermeable due to the presence of the GluA2 subunit [62,63,64].
The expression of AMPARs in microglia varies depending on the conditions. In neurodegenerative diseases, where GluA2 expression is reduced, microglia may become more responsive to glutamate or kainate (KA), leading to increased calcium influx and enhanced neuroinflammation. Studies with GluA2-deficient mice (GluA2−/−) revealed that the absence of GluA2 leads to increased calcium permeability and higher release of pro-inflammatory cytokines, like TNF-α. A study investigating the role of the GluA2 subunit in microglial AMPA receptor function found that in normal microglia (GluA2+/+), glutamate-induced currents gradually declined over time following inflammation, but this decline was absent in GluA2-deficient microglia (GluA2−/−), indicating that GluA2 helps regulate AMPAR activity under inflammatory conditions; the absence of GluA2 leads to sustained AMPAR activation, potentially increasing calcium influx and pro-inflammatory responses, which could exacerbate neuroinflammation and contribute to neurodegenerative disease progression [65,66]. Another iGluR is NMDA (N-methyl-D-aspartate) receptors, which are the prominent Ca2+ influx channels. The activation of NMDA receptors involves the binding of glutamate to the receptor, which induces a conformational change in the receptor structure. This conformational change allows the influx of calcium ions to enter the cell [67,68].
mGluRs can increase iCa2+ concentration by mobilizing calcium from IP3-sensitive (IP3) and ryanodine-sensitive calcium stores. Both types of stores are functionally linked to calcium-permeable channels present in the plasma membrane. The increase in iCa2+ levels mediated by mGluRs can activate calcium-sensitive potassium channels and calcium-dependent non-selective cationic channels. These effects of mGluRs are often attributed to the release of calcium from ryanodine-sensitive stores rather than Ins(1,4,5)P3-sensitive stores, indicating the existence of close functional interactions between mGluRs, iCa2+ stores, and calcium-sensitive ion channels in the membrane [69,70]. PARP-1 (poly(ADP-ribose) polymerase-1) is a nuclear enzyme involved in microglial pro-inflammatory responses. It is activated through pathways that involve Ca2+ influx-dependent ERK1/2 (extracellular signal-regulated protein kinases 1 and 2)-mediated phosphorylation. The activation of PARP-1 by calcium influx, particularly through NMDARs, can lead to enhanced microglial activation and proliferation, linking glutamate signaling to chronic neuroinflammation [71].
- Calcium-Sensing Receptor (CaSR)
The calcium-sensing receptor (CaSR) is a GPCR primarily responsible for detecting and regulating extracellular calcium concentrations. It plays a crucial role in modulating iCa2+ signaling by detecting changes in extracellular calcium levels and translating these signals into various intracellular responses. CaSR couples with phospholipase Cβ, leading to the production of IP3 and DAG, which subsequently elevate iCa2+ levels [72]. CaSR enhances calcium influx through various channels, including CRAC channels, influencing key calcium signaling pathways. This modulation of calcium influx allows CaSR to regulate the release of pro-inflammatory cytokines and other mediators in microglia [73]. Dysregulation of CaSR and calcium signaling in microglia has been implicated in several neurodegenerative diseases. Dysregulation of this receptor can trigger programmed cell death pathways, such as apoptosis and necroptosis, commonly observed in AD and PD [74]. Dysregulation in CaSR causes impaired NCX function that contributes to dopaminergic neuron loss and promotes neuroinflammatory pathways in PD [75]. According to a study, the expression of CaSR increases in microglia as well as in neurons and astrocytes during post-subarachnoid hemorrhage. CaSR activation promotes early brain injury, possibly through the CaMKII (calmodulin-dependent protein kinase II)/NLRP3 signaling pathway [76].
- b.
- Intracellular calcium dynamics and regulation
Microglial Ca2+ signaling is a crucial pathway for signal transduction in these immune cells, with fluctuations in intracellular free calcium levels (iCa2+) being vital for the release of trophic factors, NO, and both pro- and anti-inflammatory cytokines. The dynamics of iCa2+ in microglia play a fundamental role in regulating essential cellular functions, including cytokine production, phagocytosis, cell motility, and their responses to CNS injury [77,78,79]. Calcium influx into microglial cells primarily occurs via VGCCs, receptor-operated channels (ROCs), and purinergic receptors (P2X and P2Y), with ionotropic and metabotropic glutamate receptors (iGluRs and mGluRs) also contributing to calcium entry. Additional pathways, such as transient receptor potential (TRP) channels and calcium release-activated channels (CRACs), including Orai1, are crucial for maintaining calcium homeostasis, particularly when iCa2+ stores within the endoplasmic reticulum (ER) are depleted. Internally, calcium is released from the ER through IP3R and ryanodine receptors (RyRs) in response to signaling molecules like IP3, generated by PLC. SERCA helps restore calcium levels in the ER by pumping calcium back from the cytosol, while PMCA and sodium–calcium exchangers (NCXs) extrude excess calcium from the cell. Under inflammatory conditions, the NCX can reverse its function, increasing iCa2+ and worsening neurodegenerative processes [16,17]. Mitochondria also regulate calcium by taking it up through the mitochondrial calcium uniporter (MCU) and voltage-dependent anion channels (VDACs), playing a key role in ATP production. However, excessive mitochondrial calcium can generate ROS and initiate apoptosis via cytochrome c release through the permeability transition pore (PTP), contributing to neurodegeneration. Calcium-binding proteins (CBPs) buffer cytosolic calcium to prevent toxicity, and disruptions in calcium homeostasis can activate the NLRP3 inflammasome, leading to the release of pro-inflammatory cytokines, such as IL-1β and IL-18, driving neuroinflammation—a hallmark of neurodegenerative diseases, like AD and PD [80,81,82]. TREM2 (Triggering Receptor Expressed on Myeloid Cells 2) is a crucial regulator of microglial calcium signaling, particularly in the context of neurodegenerative diseases. TREM2, interacting with the DAP12 adaptor protein, influences calcium dynamics by activating downstream pathways, such as PLC, which leads to calcium release from ER stores. Mutations in TREM2, as seen in AD, can dysregulate this calcium signaling, resulting in hyperactive microglial responses, excessive inflammation, and impaired clearance of amyloid-beta plaques. In TREM2 knockout models, microglia show altered calcium transients, particularly through purinergic signaling involving P2Y receptors, leading to impaired motility and responses to injury [9,83]. Microglial calcium signaling is modulated by various external stimuli, including ATP, cytokines, and neurotransmitters. For instance, ATP can bind to P2X receptors to facilitate direct calcium influx or to P2Y receptors to release calcium from intracellular stores. Inflammatory stimuli, like lipopolysaccharides (LPS) and cytokines, such as TNF-α, can further elevate calcium levels, activating pathways like nuclear factor-kappa B (NF-κB), which promotes pro-inflammatory cytokine production. Amyloid-beta plaques in AD exacerbate this calcium dysregulation, increasing ROS production and promoting neuronal damage [84]. In young adult brains, microglial calcium transients are infrequent but play a role in synaptic pruning, critical for neuronal circuitry maintenance. Dysregulated calcium signaling impairs synaptic pruning, which has been linked to neurodevelopmental disorders like autism and schizophrenia. Additionally, microglia express receptors for neurotransmitters, such as serotonin, histamine, acetylcholine, and substance P, which trigger calcium transients essential for immune functions. However, microglia’s resting membrane potential of approximately −40 mV results in a weaker driving force for calcium influx compared to neurons or astrocytes. Despite this, microglia still exhibit significant calcium dynamics, crucial for immune surveillance and CNS homeostasis. Microglial Ca2+ transients can also occur in response to local tissue damage, known as damage-induced Ca2+ transients (DICTs), mediated by ATP binding to P2Y receptors and requiring calcium release from intracellular stores. These transients are specific to microglia and contribute to their morphological response to tissue injury. In contrast, nearby astrocytes either do not exhibit these transients or show delayed responses. The prevention of these calcium signals by agents like BAPTA AM highlights their role in microglial migration toward injury sites. Overall, microglial calcium signaling is crucial not only for immediate immune responses but also for long-term neuroinflammatory processes and neurodegeneration. Disruptions in calcium homeostasis, whether through TREM2 dysregulation or other pathways, have significant implications for neurodegenerative diseases, such as AD and PD, presenting therapeutic opportunities targeting these calcium regulatory mechanisms [9,17,85]. Figure 1 illustrates the regulation of iCa2+ in microglia, highlighting the pathways involved in calcium influx, release, buffering, and extrusion.
Figure 1.
This figure illustrates the complex regulation of calcium (Ca2+) signaling in microglia, highlighting the pathways of calcium influx, its release from intracellular stores, and its involvement in cellular responses, such as inflammation and gene expression. Calcium enters the cell through various channels, including transient receptor potential (TRP) channels, voltage-gated calcium channels (VGCCs), receptor-operated channels (ROCs), P2X receptors (P2XRs), P2Y receptors (P2YRs), ionotropic glutamate receptors (iGluRs), and the Piezo1 channel, while Inositol 1,4,5-Trisphosphate Receptors (IP3R) and ryanodine receptors (RyR) mediate its release from the endoplasmic reticulum (ER). Calcium is also taken up by mitochondria through the mitochondrial calcium uniporter (MCU), influencing ATP production and the generation of reactive oxygen species (ROS). The figure shows how G protein-coupled receptors (GPCRs) (e.g., Muscarinic Acetylcholine Receptors (mAChRs), metabotropic glutamate receptors (mGluRs), calcium-sensing receptors (CaSRs), and Adenosine Receptors, like A1, A2A, A2B, and A3) activate second messenger pathways, such as phospholipase C (PLC) and cyclic nucleotides, which regulate calcium levels and activate downstream effectors like protein kinase C (PKC), protein kinase A (PKA), and protein kinase G (PKG). Calcium is buffered by proteins such as calreticulin, and it can also influence nuclear gene expression. Additionally, adrenergic receptors, endothelin receptors (ETa, ETb), Brain-Derived Neurotrophic Factor (BDNF) receptors, Gamma-Aminobutyric Acid (GABA) receptors, platelet-activating factor (PAF) receptors, and Thrombin Protease-Activated Receptors (PARs) affect calcium (Ca2+) signaling, while Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) modulates inflammatory processes. Agonists are indicated by green dashed lines, and antagonistic pathways are shown by red dashed lines.
Microglial calcium signaling plays a pivotal role in neuroinflammation and neurodegeneration, with multiple pathways regulating iCa2+ dynamics. Future studies are necessary to explore the intricate regulation of these calcium pathways to better understand their therapeutic potential in combating neurodegenerative diseases.
3. Functional Consequences of Aberrant Calcium Signaling in Microglia
Calcium transients, also known as calcium elevations, have been implicated in regulating various stages of neuronal development, including migration, differentiation, and survival [86]. The diagnosis and treatment of neurodegenerative diseases, such as AD, PD, ALS, etc., are important both in basic science and medicine. Even though extensive research has been conducted into the causes of these diseases, no cure has been found yet [87]. Aberrant calcium signaling in microglia has significant effects on several processes essential for brain health and function, contributing to neuroinflammation, synaptic dysfunction, and other pathological outcomes in various neurodegenerative and neurodevelopmental disorders [10].
- a.
- Microglial activation and pro-inflammatory responses
There are many different signals present in the brain parenchyma that control microglial function. Certain signals, known as “Off” signals, can prevent the activation of microglial cells in the normal, healthy brain. These “Off” signals include cell surface molecules, like CD200 and CD47, that belong to the immunoglobulin superfamily. Furthermore, as “Off” signals to prevent microglial activation, the chemokine fractalkine (CX3CL1) and the anti-inflammatory cytokine transforming growth factor-beta (TNF-β) function. Neurotransmitters, which are chemical messengers in the brain, can have a calming effect on microglia. This is because the presence of neurotransmitters signals normal neuronal activity, which indicates that there is no need for microglial activation. When the “Off” signals are no longer present or when certain “On” signals appear, microglia are activated. For example, ATP or glutamate, which are released by endangered or impaired neurons, can act as “On” signals and trigger microglial activation [88,89]. When microglia are activated, they undergo certain changes in their appearance. They have enlarged cell bodies (somata) and their processes become retracted and thickened. These changes are indicative of an activated phenotype of microglia [90]. TREM2 knockout microglia show a greater sensitivity to changes in cytosolic calcium levels, with higher speeds at lower calcium levels and a more dramatic reduction in cell speed at high calcium levels. Higher expression of P2Y12 and P2Y13 receptors in TREM2 knockout cells may make them more susceptible to the transient STOP signal caused by high cytosolic calcium in microglia when ADP is present. In TREM2 knockout microglia, decreasing cytosolic calcium increases mean speed, displacement, and straighter paths, indicating a larger dynamic range in controlling motility in response to prolonged calcium elevations. For patients with AD who have mutations that cause TREM2 to lose its function, new therapeutic approaches may involve reducing purinergic signaling or directly modifying calcium signaling. Potential strategies to regulate microglial activation and motility include pharmacologically targeting P2Y12 receptors and employing CRAC (Orai1) channel blockers to selectively inhibit sustained calcium signals. Furthermore, in TREM2 knockout microglia, decreasing purinergic receptor activity may restore chemotactic migration. These findings have implications for understanding the consequences of TREM2 signaling and suggest potential therapeutic targets beyond TREM2 in microglial activation and motility [91].
These insights into microglial signaling highlight the complex regulatory mechanisms governing microglial activation and motility. Targeting these pathways presents promising avenues for developing therapeutic strategies to modulate microglial function in neurodegenerative diseases.
- b.
- Cytokine release and neuroinflammation
Microglia are described as the “immune sentinels” of the brain, meaning they constantly survey their surrounding brain parenchyma for any signs of danger or damage. The activation of microglia is triggered by specific molecules called pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), which are released during infection or injury. In the case of danger or injury, microglia undergo various changes in their behavior, such as changes in their movement, shape, and gene expression. They also release different molecules that can either promote inflammation (pro-inflammatory) or reduce inflammation (anti-inflammatory) [10,92]. Initially, microglia were thought to have two distinct states: a surveillant state where they have a ramified shape and an activated state where they have a roundish, macrophage-like shape. However, it is now known that microglia can have a wide range of functional and morphological states that do not necessarily overlap. Different functional states, such as pro-inflammatory or anti-inflammatory, can be associated with similar or even identical morphological states. Microglia can also switch their functional state without any obvious changes in their morphology [89]. Microglia are the first responders to inflammation in the CNS, and their response is characterized by various changes in morphology and function. Following inflammation, microglia extend their processes towards the site of injury or insult and modify their morphology [90]. They release cytokines and other mediators, contributing to the inflammatory response. Aberrant calcium signaling in microglia can lead to the release of cytokines, which are inflammatory mediators involved in neuroinflammation. Dysregulated calcium signaling can result in an exaggerated or prolonged activation of microglia, leading to an excessive production of pro-inflammatory cytokines. Neuroinflammation is characterized by the activation of microglia and astrocytes, resulting in changes in gene expression, cellular structure, and function. Peripheral cytokines can access the brain and promote cytokine production by astrocytes and other cells within the CNS, further exacerbating neuroinflammation [93]. Sustained exposure to cytokines and other inflammatory mediators can impair microglial function, leading to a decrease in directed process motility and phagocytic activity. Elevated iCa2+ levels can trigger the activation of calcium-dependent signaling pathways, such as the NF-κB pathway, which plays a crucial role in cytokine production. Calcium signaling can also modulate the expression and release of specific cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), which are key mediators of neuroinflammation. Abnormal calcium signaling in microglia may disrupt the delicate balance of cytokine production and regulation, contributing to the progression and severity of neuroinflammatory diseases. Abnormal calcium signaling in microglia can also lead to the activation of NADPH oxidase, resulting in the production of ROS and oxidative stress, which can contribute to neuronal damage and cell death [94].
Research reveals microglia’s dynamic role as immune sentinels in the brain, with dysregulated calcium signaling contributing to neuroinflammation and damage. The understanding of microglia’s various functional and morphological states and their impact on their responses to inflammation and injury in neurodegenerative diseases remains limited.
- c.
- Phagocytosis and clearance of cellular debris
Dysregulated calcium signaling in microglia can impair phagocytic activity and clearance of cellular debris, leading to the accumulation of toxic protein aggregates and contributing to the progression of neurodegenerative diseases [10]. During early epilepsy development, aberrant calcium signaling in microglia occurs following status epilepticus (SE), leading to prolonged calcium transients. This signaling, distinct from normal activity, is triggered by UDP-P2Y6 signaling in response to seizures. It is crucial for lysosome upregulation and the production of pro-inflammatory cytokines TNF-α and IL-1β. P2Y6 knockout mice exhibit failures in lysosome upregulation, similar to effects seen when microglial calcium signaling is reduced. Microglia in the hippocampus with P2Y6 expression can engulf neurons, leading to decreased CA3 neuron survival and impaired cognition. This aberrant calcium signaling is associated with phagocytic and pro-inflammatory functions, contributing to disease progression in epilepsy. Lysosome upregulation in microglia aids in phagocytic clearance of cellular debris and pathogens, contributing to inflammation resolution and tissue repair. However, excessive lysosome activity can harm neurons, as increased production of pro-inflammatory cytokines, like TNF- α and IL-1 β, leads to neurodegeneration and immunopathic damage. This combination of phagocytic activity and cytokine production may drive ongoing neuronal loss and neurodegeneration in epilepsy [95].
Understanding immune cells’ role in neurological conditions is limited, with gaps in understanding dysregulated signaling, impaired cellular clearance, inflammation, and long-term effects. Comprehensive in vivo studies are required to understand disease progression and targeted strategies.
- d.
- Synaptic pruning and neuronal circuitry modulation
Synaptic pruning is a critical process in brain development, involving the elimination of unnecessary or weak synapses to refine and optimize neuronal circuitry for efficient communication. Microglia, the resident immune cells of the CNS, play a key role in this process by engulfing and removing excess synapses. The complement system, involving proteins like C1q and C3, is central to tagging synapses for removal. These complement proteins bind to weak or inactive synapses, marking them for elimination through opsonization. Microglia, expressing receptors such as complement receptor 3 (CR3), recognize these tagged synapses and phagocytose them, ensuring that only functional synapses are preserved [96,97,98].
Microglia-mediated synaptic pruning is highly regulated by a balance between “Eat Me” and “Don’t Eat Me” signals. Proteins such as C1q and C3, along with phosphatidylserine (PS) exposed on damaged dendrites, serve as “Eat Me” signals, guiding microglia to engulf synapses. On the other hand, molecules like CD47 act as “Don’t Eat Me” signals, inhibiting phagocytosis. Disruption in this balance can lead to either excessive or insufficient pruning, contributing to neurodevelopmental and neuropsychiatric disorders, such as autism and schizophrenia. For example, in schizophrenia, overactive pruning results in the removal of too many synapses, while in autism, insufficient pruning leads to increased synaptic density and dysfunctional connectivity [99].
Key receptors, such as CX3CR1 and TREM2, on microglia further modulate synapse elimination. CX3CR1 signaling, mediated by fractalkine (CX3CL1), directs microglia to specific synapses for pruning. Mice lacking CX3CR1 exhibit impaired synaptic pruning, leading to weak synaptic transmission, reduced brain connectivity, and behavioral deficits. TREM2 recognizes PS on apoptotic or damaged synapses, marking them for removal, and its deletion has been linked to disrupted synapse elimination [100,101,102,103].
Astrocytes, another type of glial cell, also play a role in synaptic pruning by releasing complement proteins, like C1q, and responding to synaptic glutamate release. Astrocytes can modulate neuronal activity and synaptic plasticity through the release of gliotransmitters, such as glutamate, D-serine, and ATP, which in turn influence microglial activity [104,105,106].
The dysregulation of microglia-mediated synaptic pruning has been implicated in various neurodevelopmental disorders. For example, in mice with reduced microglial numbers due to the deletion of CX3CR1, both inhibitory and excitatory synapses are differentially affected, suggesting that inhibitory synapses may not be typical substrates for microglial pruning. This dysregulation can disrupt the fine tuning of neuronal circuits and impact learning, memory, and behavior [107,108,109].
Additionally, microglial activity shifts with age, leading to chronic low-level activation in older brains. These “primed” microglia are more sensitive to stimuli, potentially contributing to age-related neurodegenerative diseases, like AD. This heightened state can result in excessive inflammation and neuronal damage, further disrupting brain homeostasis and synaptic connectivity [110,111].
Studies on synaptic pruning often focus on specific synaptic populations, potentially overlooking the broader diversity of synaptic types and their varying susceptibilities to pruning. Longitudinal studies and comprehensive datasets are needed to better understand the relationship between microglial activity and neurodevelopmental disorders, aiding in targeted interventions and therapeutic strategies.
4. Microglial Calcium Dysregulation in CNS Disorder
- a.
- Alzheimer’s disease and microglial calcium dysregulation
AD is a severe neurodegenerative illness causing dementia in the elderly, characterized by tau accumulation, Aβ protein deposits, and senile plaques [112]. Dysregulation of iCa2+ homeostasis is an early hallmark of AD, leading to cellular dysfunction and degeneration.
Indeed, it has been documented that Aβ peptide 25–35 (Aβ25–35)-stimulated microglia produce a brief rise in iCa2+ [113]. The release of pro-inflammatory mediators from the microglia reaction, such as cytokines, chemokines, and NO, depends on iCa2+ for microglia executive functions. Although Aβ monomers are neurotrophic, Aβ oligomers and fibrils disrupt calcium signaling in microglia more than in normal aging, leading to abnormal calcium influx and altered pathways [114]. This disruption alters calcium homeostasis through calcium channel and transporter expression and protein modifications. A study on microglial calcium concentration changes found that spontaneous Ca2+ transients are more frequent in individual microglial processes than in their somata. The study also found that proximity to Aβ plaques strongly affected microglial Ca2+ activity, with somatic Ca2+ responses being less intense in microglia near Aβ plaques [78]. Mitochondrial dysfunction is another key aspect of AD. Excessive calcium within mitochondria triggers increased ROS production and apoptosis, exacerbating AD pathology. Both Aβ and hyperphosphorylated tau disrupt calcium homeostasis in mitochondria. Dysregulation in microglia can elevate mitochondrial calcium levels due to increased influx via the MCU and reduced efflux through the mitochondrial NCLX. These disruptions impair calcium-buffering mechanisms, worsening AD-associated calcium dysregulation [115]. Additionally, cultured fetal human microglial cells exposed to Aβ1–42 showed an increased Ca2+ response to a purinergic P2X7 receptor (P2X7R) agonist, and AD microglia consistently showed enhanced P2X7R expression [116]. The NOD (Nucleotide-binding Oligomerization Domain), LRR (Leucine-rich Repeat), and pyrin domain-containing 3 (NLRP3) inflammasomes are crucial in neurodegenerative diseases, promoting Aβ-plaque formation and tau-induced pathology. Aβ elevates iCa2+ levels, which in turn help trigger the NLRP3 inflammasome in microglia [117]. The NLRP3 inflammasome mediates the maturation of IL-1β by activating caspase-1. According to Lee et al., the murine calcium-sensing receptor (CASR) activates the NLRP3 inflammasome through increased iCa2+ and decreased cellular cyclic AMP (cAMP). CASR agonists activate the NLRP3 inflammasome without exogenous ATP, while knockdown reduces activation. Ca2+ promotes inflammasome assembly, while cAMP inhibits it [118]. Therefore, it can be said that the Ca2+-dependent activation of the NLRP3 inflammasome would then result in an increase in IL-1/-18 production, which could lead to neuronal hyperactivity, particularly near plaques. SOCE in microglia, the calcium signaling mechanism, plays a crucial role in cellular functions. Research suggests that in sporadic AD, there are alterations in the SOCE pathway in microglia [119]. When stimulated by PAF in normal conditions, microglia exhibit a quick transient depletion of calcium from ER stores, followed by sustained calcium entry through SOCE. Nevertheless, in microglia obtained from AD brain tissue or those treated with Aβ peptide (Aβ42), there are significant reductions in the amplitude of SOCE. Additionally, AD microglia confirmed decreased levels of calcium release from ER stores compared to controls. The alteration in SOCE properties within microglia may impact immune cell responses and potentially lead to neurovascular unit dysfunction in the inflamed AD brain [119]. TREM2 variants amplify the risk of developing late-onset AD (LOAD) by 24-fold [120]. TREM2, the membrane protein, regulates microglial functions, like phagocytosis and chemotaxis. Loss-of-function variants of TREM2 increase the risk of AD. Researchers found that human-induced pluripotent stem cell (iPSC) microglia lacking TREM2 show exaggerated Ca2+ signals in response to purinergic agonists, affecting cell motility [91]. In the field of AD research, the role of Ca2+ homeostasis modulator family proteins has recently drawn more attention. The CALHM family consists of six members, CALHM1-6, with CALHM1 being the most extensively studied. CALHM1 forms a channel that allows the permeation of ions and substrates, including ATP and calcium, in a voltage-dependent manner. A single-nucleotide polymorphism in the CALHM1 gene has been associated with an increased risk of early-onset AD [121]. CALHM1 regulates cytosolic Ca2+ concentrations and Aβ levels. In a recent study, it was shown that the expression of CALHM2 is increased in the microglia of an AD mouse model, leading to reduced Aβ deposition, neuroinflammation, and cognitive impairments. Knockout of CALHM2 inhibited microglial pro-inflammatory activity but increased phagocytic activity, restoring the balance between inflammation and phagocytosis. Moreover, the knockout of CALHM2 reduced acute LPS-induced neuroinflammation [116,122]. Table 1 summarizes the major aspects of calcium homeostasis disruptions in AD, focusing on how these disruptions impact inflammatory mediator release, mitochondrial dysfunction, and the altered signaling pathways caused by amyloid plaques.

Table 1.
Summary of the key findings of microglial calcium dysregulation in Alzheimer’s disease. This table highlights the major aspects of calcium homeostasis disruptions in microglia, including their impact on inflammatory mediator release, mitochondrial dysfunction, SOCE pathway alterations, and the role of amyloid plaques in exacerbating calcium-related abnormalities in AD pathology.
Although numerous research studies have demonstrated that elevated Ca2+ concentration can increase the formation of Aβ plaque and neurofibrillary tangles and impair learning and memory in AD, the underlying mechanisms by which Ca2+ influences the development of progression of AD remain largely unexplored. Furthermore, it is still debated whether Ca2+ dysregulation is an upstream factor in the disease process or a downstream effect of AD pathology. Despite uncertainties about the causes and molecular basis, the finding of microglial Ca2+ dyshomeostasis or altered calcium signaling in AD supports the possibility that novel brain-penetrant, microglial-specific calcium channel antagonists or related drugs targeting microglial calcium signaling could hold therapeutic potential.
- b.
- Parkinson’s disease and altered calcium signaling in microglia
PD is a neurodegenerative disorder characterized by motor symptoms associated with the accumulation of misfolded α-synuclein into Lewy bodies and the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) [123]. PD belongs to a group of diseases called synucleinopathies [124]. In PD, chronic microglial activation, known as microgliosis, results from persistent inflammation, misfolded α-synuclein, and neuronal death, leading to excessive release of pro-inflammatory cytokines, like TNF-α, IL-1β, and interferon-γ, along with ROS, contributing to neurodegeneration. This heightened sensitivity of microglia to misfolded proteins and cellular damage causes persistent calcium influx via VGCCs and receptor-operated calcium channels (ROCs), fueling ongoing inflammation. Excessive calcium influx disrupts homeostasis, causing mitochondrial dysfunction, energy deficits, and increased ROS production, which collectively accelerate neuronal death in the SNpc, a hallmark of PD’s motor symptoms [38]. Emerging evidence highlights the crucial role of iCa2+dysregulation in PD, where a shift from Cav1.2 to Cav1.3 channels in dopaminergic neurons elevates cytosolic calcium, leading to further mitochondrial dysfunction and neuronal loss [125].
Additionally, irregular calcium release from the ER via RyRs exacerbates calcium imbalance, contributing to the progression of PD. In PD, P2X7 receptor signaling plays a significant role in promoting the production of ROS and initiating apoptotic processes, contributing to neuronal death. In animal models of hemi-parkinsonism created by 6-OHDA lesioning of the basal ganglia, blocking P2X7 receptors and P2Y6 receptors has been shown to increase dopamine production, reduce abnormal rotational behavior, and prevent cell death [126]. Additionally, the P2X1 receptor has been implicated in PD, where ATP-mediated activation induces lysosomal dysfunction and α-synuclein aggregation in vitro. Inhibiting P2X1 or genetically knocking it out has been found to improve lysosomal function and reduce α-synuclein aggregation. These findings highlight purinergic receptors as key mediators that link neurodegeneration with harmful inflammatory processes in PD [127]. Table 2 summarizes the major aspects of calcium homeostasis disruptions in PD and how these disruptions impact Cav1.2 and Cav1.3 channels and are particularly implicated in the dysfunction of dopaminergic neurons. The table also highlights the role of RyRs and mitochondrial disturbances in exacerbating PD pathology.
Table 2.
Summary of the key findings of altered calcium signaling in microglia in Parkinson’s disease. This table highlights the main aspects of microglial dysfunction in Parkinson’s disease, including chronic microglial activation, disrupted calcium homeostasis, and associated neurodegenerative processes. The table also emphasizes specific calcium channels and purinergic receptors involved in exacerbating neuroinflammation and neuronal loss, along with key molecular pathways implicated in the progression of the disease.
Given that microglia are motile cells and Ca2+ is a crucial metal ion used by cells to respond and adapt to constantly changing surrounding environments, understanding the role of microglial Ca2+ signaling in microglial motility and its implication for neuronal behavior is critical for gaining insight into synucleinopathy diseases, including PD. PD-associated proteins, like alpha-synuclein, can form calcium-permeable pores in the plasma membrane, and their binding to Ca2+ promotes the aggregation of cytoplasmic alpha-synuclein. While blocking microglia-specific calcium channels leads to dopaminergic neuron degeneration and impairs behavioral deficit, questions remain about how synuclein-induced calcium dyshomeostasis or calcium-induced alpha-synuclein aggregation affects microglial behavior, particularly the movement of microglial processes and the cell body, their connections with neuronal synapses, and changes in neuronal synaptic plasticity.
- c.
- Multiple sclerosis (MS) and calcium-mediated neuroinflammation
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the CNS that leads to focal lesions in both gray and white matter along with widespread neurodegeneration [129]. Experimental autoimmune encephalomyelitis (EAE), an established animal model for studying MS, attributes the disease’s pathogenesis to T-cell auto-reactivity against CNS antigens. A key feature of MS is the activation of microglia, which is closely linked to calcium dysregulation within the CNS. In both MS and EAE lesions, microglia produce pro-inflammatory cytokines, ROS, and reactive nitrogen species (RNS), like NO, proteases, and glutamate, all contributing to inflammation [130,131]. Excitotoxicity, a process where excessive neuronal activity leads to cell death, is commonly observed in MS and EAE [132]. It is driven by the overactivation of AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and kainic acid (KA) receptors, and blocking these receptors has been shown to reduce EAE severity [132]. Elevated glutamate receptor activity in EAE may result from T-cell interactions or the release of TNF-α, which, in turn, stimulates microglia to release more glutamate, further intensifying excitotoxicity [133,134]. High glutamate levels lead to increased iCa2+ concentrations, triggering excitotoxicity through ROS production and mitochondrial calcium overload [132,135]. Oxidative stress, a hallmark of MS, activates TRPM2 channels, which are highly sensitive to ROS. Activation of TRPM2 channels in microglia results in the production of pro-inflammatory mediators, exacerbating neuroinflammation [136]. Chronic CNS inflammation in MS is marked by energy imbalances and demyelination, which lead to dysfunction and improper distribution of various ion channels. These disturbances converge on Ca2+ overload, which is the main driver of neurotoxicity. Excess calcium activates degradative enzymes, impairs mitochondrial function, and disrupts axonal transport, creating a feedback loop that further elevates calcium levels [130]. This neuroaxonal calcium overload is fueled by multiple sources, including calcium release from intracellular stores (like the ER), entry through VGCCs, and other cation channels. Additionally, TRPV1 channels are upregulated in the microglia of MS and EAE models. TRPV1 channels regulate ATP-induced NLRP3 inflammasome activation by mediating calcium influx and modulating phosphatase PP2A activity. Studies suggest that deleting TRPV1 can reduce neuroinflammation by inhibiting NLRP3 inflammasome activation in EAE. The TRPV1-Ca2+-PP2A pathway may be a critical regulator of NLRP3 inflammasome activation, identifying TRPV1 as a potential therapeutic target for CNS inflammatory diseases [26]. Table 3 provides an overview of the pathological mechanisms in MS, including the effects of excitotoxicity, oxidative stress, and TRPV1-mediated calcium influx that activate inflammasomes and drive neuroinflammation.
Table 3.
This table summarizes the role of calcium in the neuroinflammatory processes of multiple sclerosis, detailing mechanisms like excitotoxicity, oxidative stress responses via TRPM2, and calcium influx through TRPV1, which activate inflammasomes in microglia. These processes are critical in driving the neurodegenerative and demyelinating effects observed in MS.
Microglia are involved in all stages of MS, and microglial calcium signaling plays a crucial role in coordinating the pathological response in MS. Microglia detect increased extracellular Ca2+ concentrations through microglia-specific channels, which triggers their migration to the site of an MS lesion. Although microglia are strongly implicated in MS pathology, their role, whether beneficial or detrimental, remains debated, as they contribute to both inflammation and remyelination. It would be valuable to further investigate the role of microglia-specific calcium channels and purinergic receptor signaling in regulating microglial calcium activity, particularly by attenuating purinergic signaling and blocking calcium channels, to determine whether the modified phagocytic microglia alter the pathophysiology and phenotypes of MS. A recent study documented that clusters of activated microglia and macrophages, known as microglia nodules, contribute to the increased severity of MS lesions [137]. So, it would be interesting to study the impact of extracellular and intracellular Ca2+ concentration on the movement of microglia toward the lesions and formation of microglia nodules and whether blocking calcium signaling could reverse microglia nodules associated with MS lesions.
- d.
- Microglial calcium response to Ischemic Stroke
Ischemic stroke (IS) occurs when a blood vessel in the brain or neck is blocked, often due to thrombosis, embolism, or stenosis, leading to reduced blood flow and oxygen deprivation. The resulting ischemia causes necrosis in the core tissue and impaired function in the surrounding penumbra. Disruption of ATP production due to ischemia impairs the Na+/K+ transporter, leading to cellular depolarization and Ca2+ influx, while glutamate accumulation in the extracellular space triggers excitotoxicity and apoptosis [138].
IS activates microglia into the pro-inflammatory M1 phenotype, releasing factors like TNF-α and IL-1β, and enzymes such as iNOS, which drive a cytokine storm and exacerbate neuronal damage [139,140]. This M1 activation disrupts mitochondrial dynamics by promoting fission and inhibiting fusion, leading to fragmented and dysfunctional mitochondria with reduced ATP and increased ROS levels. Damaged microglial mitochondria can even enter neurons, worsening mitochondrial dysfunction and neuronal injury [139]. Hypoxia and hypoglycemia conditions further elevate iCa2+ in microglia via NCX1 and TRPM2 channels, while TRPM2 and TRPM7 channels play key roles in delayed calcium deregulation, promoting membrane depolarization, mitochondrial damage, and ultimately contributing to neuronal injury in IS [141]. Microglia have been identified as key players in the propagation of CSD. A recent investigation conducted by Liu et al. utilized high-speed 2-photon microscopy and genetically encoded calcium indicators to explore iCa2+ dynamics in cortical microglia during IS. The researchers postulated that acute IS disrupts the regulation of intracellular microglial calcium activity, with these calcium transients aligning with and contributing to the incidence of CSD. The study provided convincing evidence supporting the notion that ischemic injury triggers recurrent and progressive CSD, accompanied by frequent calcium transients in microglia. In vivo imaging unveiled periodic calcium activity in microglia during the hyperacute phase of IS, with increased activity within the initial 6 h post-occlusion and larger amplitudes observed at later time points. Furthermore, the study demonstrated that peripheral inflammation induced by LPS produced markedly greater microglial calcium transients during CSD, effectively mimicking the sterile inflammation characteristic of IS. In vivo investigations additionally indicated that calcium influx through CRAC channels contributes, at least in part, to CSD-associated microglial calcium activity. Focal cerebral ischemia induced by middle cerebral artery occlusion (MCAo) in mice was shown to induce recurring and progressive CSD, leading to frequent calcium transients in microglia. Elevated levels of microglial calcium observed in vitro have been associated with microglial activation and the subsequent release of inflammatory molecules, ultimately contributing to neuronal death or injury [142,143].
As outlined in Table 4, disruptions in ATP synthesis and TRPM2/TRPM7 channel activation led to widespread calcium dysregulation, contributing to excitotoxicity and neuronal injury. Additionally, the role of cortical spreading depolarization (CSD) and peripheral inflammation in amplifying these effects is highlighted.
Table 4.
This table reviews the findings on ischemic stroke, explaining how microglial calcium responses are disrupted during ischemia, leading to excessive calcium influx, excitotoxicity, and neuronal injury. The table also discusses the role of CSD and how peripheral inflammation exacerbates calcium signaling abnormalities in microglia during stroke.
- e.
- Psychiatric disorders and microglial calcium abnormalities
The concept of a “psychiatric or mental disorder” is broad and lacks a precise boundary, with various definitions proposed, including the one in the DSM (Diagnostic and Statistical Manual of Mental Disorders). According to the DSM-V, a psychiatric disorder is characterized by a noticeable behavioral or psychological pattern that causes significant distress or impairment. It is not a typical reaction to common stressors or losses, nor a culturally sanctioned response to specific events [144]. Major Depressive Disorder (MDD) is a complex psychiatric condition marked by abnormal synaptic plasticity. Recent evidence suggests that microglia play a crucial role in regulating synaptic plasticity [145]. Calcium is involved in the regulation of neurotransmitter release, synaptic plasticity, and neuronal signaling, which are all implicated in depression. Dysregulation of calcium homeostasis has been observed in depression, with alterations in calcium channels and calcium-dependent signaling pathways [146]. Patients with depression show increased peripheral blood inflammatory biomarkers, including cytokines, which interact with various pathophysiologic domains involved in depression. Increased cytokine levels in MDD may lead to neuroinflammation, potentially impacting neurotrophic support, glutamate release, oxidative stress, excitotoxicity, and glial element loss [147]. Excessive glutamate release activates microglial glutamate receptors, resulting in calcium influx into microglia, further contributing to calcium dysregulation [132]. This calcium influx triggers signaling pathways that promote the release of pro-inflammatory cytokines. Meta-analyses have shown that pro-inflammatory cytokines, like TNF-α and IL-6, are consistently elevated in depressed patients [148,149,150]. Given the involvement of microglia in neuroinflammation, several studies have already suggested that MDD could be considered a microglia disease. Therefore, it would be of interest to investigate the direct involvement of microglial calcium signaling in the pathophysiology of MDD. Although some studies have reported an increased risk of depression associated with the use of calcium channel blockers, the clinical significance of brain-penetrant calcium channel blockers in reducing MDD has yet to be determined.
Schizophrenia is a chronic mental disorder characterized by symptoms such as paranoia and hallucinations [151]. While the exact etiology remains unclear, neuroinflammation and elevated peripheral benzodiazepine receptor expression have been implicated in its pathophysiology. PET imaging reveals neuroinflammation in schizophrenic patients, particularly during psychosis [152]. iCa2+ signaling, regulated by the ER, is crucial for microglial function and is linked to schizophrenia [153]. Activated microglia contribute to neurotoxicity, leading to neuroanatomical changes and cognitive symptoms. Microglial iCa2+ signaling is considered a therapeutic target for the treatment of schizophrenia and antipsychotic drugs, like Aripiprazole, reduce inflammation by inhibiting NO and TNF-α production in IFN-γ-activated microglia, primarily by lowering iCa2+ concentration. Additionally, typical (e.g., Risperidone) and atypical (e.g., Haloperidol) antipsychotics have been found to suppress pro-inflammatory cytokine and NO release from activated microglia by reducing iCa2+ elevations [153,154]. Despite experimental findings on the association of microglial iCa2+ signaling with the pathophysiology of schizophrenia, an open-label pilot study suggested that calcium channel blockers should only be used as an add-on therapy to antipsychotics. However, further studies are needed to explore novel calcium channel blockers targeting microglia-specific calcium channels or signaling pathways.
Bipolar disorder (BD) encompasses serious conditions, including bipolar I (characterized by full-blown manic episodes) and bipolar II (featuring hypomanic episodes and major depression). These disorders significantly impair psychosocial functioning and are linked to a 10–20-year reduction in life expectancy. Microglia are implicated in the abnormal immune and inflammatory signaling that contribute to all stages of BD. Neuropathological studies have reported reduced glial cells, including microglia, in the prefrontal cortex and limbic system of BD patients. Changes in microglial calcium signaling may drive the neuroinflammatory processes seen in BD [155]. Mitochondria and the ER, connected by mitochondria-associated membranes (MAMs), regulate iCa2+ levels and facilitate the assembly of NLRP3 inflammasomes. These complexes detect ROS from damaged mitochondria, leading to pro-inflammatory cytokine release. The sigma-1 receptor (SIGMAR-1) acts on iCa2+ concentration in microglia and is associated with MAMs and plays a key role in modulating inflammatory signaling in microglia and other neuropsychiatric models [156,157]. SIGMAR-1 also mitigates apoptotic and inflammatory signals, regulates calcium channels (IP3R), and enhances neuroplasticity by promoting BDNF expression. In conditions like BD, where the unfolded protein response (UPR) is overwhelmed, excess calcium, ROS, and other stress signals push the cell towards apoptosis or autophagy [156,158]. The accumulation of unfolded proteins further activates NLRP3 inflammasomes, contributing to inflammation [159,160]. Moreover, the influx of calcium ions into microglia is closely linked to the activation of NLRP3 inflammasomes. Although NLRP3 inflammasome serves as a key mediator of inflammatory signaling in BD, the contribution of calcium-dependent microglial NLRP3 inflammasome in the pathophysiology and phenotype of BD is yet to be determined. Similarly, the significance of microglial sigma-1 receptor-mediated calcium homeostasis in the pathophysiology of BD is crucial to explore in order to improve BD therapy. Table 5 explores how these interactions contribute to synaptic dysfunction, neuroinflammation, and the therapeutic potential of targeting calcium signaling in these conditions.
Table 5.
The key findings of microglial calcium dysregulation in psychiatric disorders. This table summarizes the major mechanisms and pathophysiological effects of calcium dysregulation in microglia within various psychiatric disorders, including Major Depressive Disorder (MDD), schizophrenia, and bipolar disorder (BD). It highlights how altered calcium signaling contributes to neuroinflammation, synaptic dysfunction, and neurodegeneration, alongside specific findings that demonstrate the involvement of calcium channels, signaling pathways, and mitochondrial-ER interactions in these processes.
5. Therapeutic Strategies Targeting Microglial Calcium Signaling
Calcium signaling is essential for microglial function, and its dysregulation is implicated in various neurodegenerative diseases and inflammatory conditions. Targeting microglial calcium signaling pathways presents a promising therapeutic strategy to modulate neuroinflammation and promote neuroprotection.
- a.
- Calcium channel blockers
Calcium channel blockers (CCBs) obstruct the passage of extracellular calcium by blocking ion-specific channels that extend across the cellular membrane. Microglia have various types of calcium channels, as discussed earlier. Ca2+ channels offer a potential target for altering microglia phenotype in several neurological disorders. CCBs offer neuroprotection and exhibit anti-amyloid, anti-tau, anti-phospholipase, anti-platelet, antioxidant, and anti-inflammatory activity, making them potential drugs for AD prevention or treatment [161]. VGCC blockers are primarily composed of three main components: dihydropyridines, benzothiazepines, and phenylalkylamine.
Nimodipine is a dihydropyridine L-type CCB that has been proposed as a potential treatment for AD and PD. It is a powerful blocker of cell damage caused by monomeric and oligomeric Aβ [162]. A study showed that Nimodipine reduced neuroinflammation by inhibiting microglial activation, suggesting potential protection against degeneration [163].
Nimodipine demonstrated effectiveness in PD by attenuating neurotoxic effects in both cellular and animal models of the disease. It significantly reduced 1-methyl-4-phenyl pyridinium ion-induced mitochondrial damage, iCa2+ levels, and loss of mitochondrial membrane potential [164]. Nicardipine is a second-generation dihydropyridine-type CCB that significantly inhibits microglia-related neuroinflammatory responses. It inhibits microglial cell migration and the release of NO, as well as the expression of iNOS and cyclooxygenase-2 (COX-2) [165]. Nicardipine’s beneficial use in acute ischemic stroke has been studied in few humans or animals [166]. Isradipine, an FDA-approved CCB, attenuates Aβ oligomer toxicity by suppressing calcium influx and Cav1.2 expression. It is well tolerated in AD models, reduces tau burden, and improves autophagy function [117]. A pilot study demonstrated the potential of isradipine for the improvement of cognitive functioning in patients with schizophrenia [167]. When compared to other L-type VGCCs, nifedipine is known to preferentially block Cav1.2 channels. Nifedipine exhibits an additional potential mechanism of action in AD by modulating Aβ-peptide production. According to a study, nifedipine reduces the generation of Aβ-peptide in cell culture, and this effect is unrelated to its impact on calcium flux. The reduction in Aβ production is attributed to nifedipine’s modulation of components within the gamma-secretase complex, such as presenilin-1 and Pen-2. Furthermore, nifedipine lowers levels of orphan-G-coupled protein receptor 3, which is suggested to contribute to the stability of the gamma-secretase complex [168]. Verapamil, a phenylalkylamine, has gained approval for treating various cardiac conditions due to its ability to block VGCCs, particularly the L-type channels. In a human microglia cell culture treated with Aβ25–35, verapamil was shown to inhibit the rise of calcium concentration. These results imply that while verapamil may not directly protect neurons from Aβ toxicity in certain experimental conditions, it could still modulate calcium levels in microglia, which are involved in the neuroinflammatory response associated with AD [169]. A study investigated the neuroprotective effect of verapamil against LPS-induced neuroinflammation in mice and assessed whether the timing of verapamil administration influenced its effectiveness. Neuroinflammation was evidenced by elevated microglia markers, inflammatory cytokines, iCa2+, and compromised mitochondria. Pretreatment with verapamil significantly improved brain architecture and behavioral performance, with the timing of administration impacting its efficacy [170]. Another study showed that verapamil improved short-term memory, enhancing exploratory behavior in a murine model of AD. This drug reduced the surveillance speed of microglia processes and altered microglia morphology in the cortex [171]. The use of two CCBs, bepridil and nitrendipine, demonstrated a notable improvement in EAE, the animal model for MS. Despite distinct structural and functional characteristics, both bepridil and nitrendipine share the common feature of inhibiting calcium influx through L-type VGCCs [172]. Amlodipine, another CCB, has been shown to reduce microglial inflammatory phenotypes in hypertensive mice, suggesting its potential to mitigate cognitive dysfunction linked to hypertension-induced neuroinflammation in aging [173]. Another study found that in dementia patients with hypertension, amlodipine reduced mortality risk, while nifedipine increased it, especially in AD and PD patients. Amlodipine also lowered IS risk in AD patients compared to other CCBs [174]. The neuroprotective anti-dementia effects of diltiazem, a non-dihydropyridine benzothiazepine CCB, was reported to significantly improve cognitive impairments in a rat model of sporadic AD. Diltiazem reduced pro-inflammatory markers, such as TNF-α and IL-1β. Furthermore, it restored the increased Aβ expression in the brain tissues [175]. In another study by Lintunen et al., diltiazem was found to be associated with a decreased risk of psychiatric hospitalization in bipolar disorder [175]. A systematic review of randomized controlled trials (RCTs) found that for nimodipine, flunarizine, isradipine, nicardipine, fasudil, and lifarizine, none of the trials demonstrated a convincing beneficial effect of CCBs in reducing mortality, dependency, adverse events, or other adverse clinical outcomes in people with acute ischemic stroke [176]. On the other hand, nifedipine, nicardipine, nimodipine, diltiazem, flunarizine, and other CCBs have shown promising potential in the treatment of certain psychiatric disorders [177]. Table 6 summarizes the key findings regarding the neuroprotective effects of various CCBs across different neurological conditions.
Table 6.
This table outlines the therapeutic potential of various calcium channel blockers (CCBs) in neurodegenerative and psychiatric disorders. The key findings include their effects on neuroinflammation, calcium dysregulation, and cognitive outcomes in diseases like Alzheimer’s, Parkinson’s, and multiple sclerosis. Each CCB is noted for its specific mechanisms, such as reducing Aβ toxicity, modulating calcium influx, and improving neuronal health.
CCBs have shown potential for neuroprotection by inhibiting calcium influx in microglia, offering therapeutic benefits for various neurological conditions. By blocking specific ion channels, CCBs have been shown to reduce inflammation, oxidative stress, and excitotoxicity, which are common in neurodegenerative diseases. While some CCBs, like nimodipine and nicardipine, demonstrate promise in conditions, such as AD and PD, the clinical outcomes in acute IS remains inconsistent. Overall, CCBs could offer targeted intervention strategies for neuroinflammation and calcium dysregulation-related disorders.
- b.
- Immunomodulatory agents and anti-inflammatory approaches
Immunomodulation involves regulating immune system responses to achieve therapeutic outcomes. Neurodegenerative diseases are often associated with neuroinflammation, marked by microglial activation, pro-inflammatory cytokine release, and neuronal damage. Modulating microglial activation from the pro-inflammatory phenotype to the neuroprotective phenotype is a potential therapeutic strategy. Targeting calcium signaling pathways in microglia is central to immunomodulation since these pathways are key to microglial activation. Ethyl pyruvate (EP) has shown neuroprotective effects in AD and PD models by inhibiting high mobility group box 1 (HMGB1) secretion, reducing pro-inflammatory molecule production, and suppressing NLRP3 inflammasome activation [180,181,182]. EP also directly chelates Ca2+, contributing to its anti-inflammatory effects [181]. In PD, HMGB1 released from microglia activates the NF-κB pathway, leading to the production of neurotoxic factors, making HMGB1 a target for immunomodulation [183]. AMPK activation is another promising strategy, as it inhibits pro-inflammatory cytokine expression and NF-κB activation in glial cells. Compounds like AICAR, berberine, resveratrol, and metformin reduce microglial hyperactivation by activating AMPK, offering therapeutic potential for neurodegenerative diseases [184]. Non-steroidal anti-inflammatory drugs (NSAIDs) have been explored for neuroprotection, although the results have been mixed. While some studies failed to show efficacy, recent findings suggest protective effects of diclofenac and fenamates in AD [185]. These drugs reduce pro-inflammatory mediator release from microglia, mitigating AD pathology. Ibuprofen and non-aspirin NSAIDs showed a reduced risk of PD, suggesting a potential protective effect [186]. Table 7 summarizes the key findings on immunomodulatory and anti-inflammatory approaches.
Table 7.
This table highlights the role of immunomodulatory agents and anti-inflammatory strategies in managing neurodegenerative diseases. The agents listed, including ethyl pyruvate, AMPK activators, and NSAIDs, are described for their ability to modulate microglial activation, suppress pro-inflammatory signaling pathways, and reduce neuroinflammation. These strategies offer potential therapeutic benefits by targeting the molecular processes driving neuroinflammation.
Targeting calcium signaling pathways and utilizing agents like ethyl pyruvate, AMPK activators, and NSAIDs for immunomodulatory and anti-inflammatory approaches have shown neuroprotective potential. Continued exploration of these pathways may pave the way for novel therapeutic approaches, offering hope for mitigating disease progression and improving patient outcomes in the future.
- c.
- Neuroprotective strategies by antioxidant interventions
Neuroinflammation and oxidative stress are hallmarks of neurodegeneration, contributing to disease progression. The crosstalk between Ca2+ and redox signals is crucial for brain physiological functions, but excessive Ca2+ release can trigger pathological responses and neuronal death under oxidative stress conditions [188,189,190]. Recently, it was reported that microglia contain NADPH oxidases, which are highly regulated enzymes that produce ROS, and mitochondrial respiration is the major source [191]. Excessive stimulation of NADPH oxidases and disrupts the electron transport chain or tricarbonic acid cycle, leading to oxidative stress damaging cell structures [188]. Targeting microglia pharmacologically offers a promising approach to regulating ROS production. Natural antioxidant products like curcumin, resveratrol, cannabidiol, flavonoids, and sulforaphane have shown the potential to suppress microglial activation and mitigate neuroinflammation [192]. By modulating microglia, these compounds could help control redox imbalance and neuroinflammation in neurodegenerative diseases, offering potential therapeutic benefits. Curcumin, a popular anti-inflammatory drug, amends Ca2+ dysregulation in microglia by inhibiting P2X7 receptor activation [193]. Curcumin is also observed to inhibit TNF activity, reduce the formation of β-amyloid plaques, and protect brain cells from harmful substances. So, it suggests that this naturally occurring antioxidant compound could be a promising approach for treating AD [194]. The findings of a study pointed out that resveratrol inhibited the release of cytokines from activated microglia and attenuated changes in the NF-κB signaling pathway induced by LPS. It suggests that resveratrol can mitigate microglial overactivation and protect against neuronal injury induced by microglial activation [195,196]. Cannabidiol was found to enhance microglial phagocytosis via TRP channel activation, causing an influx of calcium into the cell [197,198]. A study conducted by Madhi et al. showed that Ginsenoside Re (G-Re) significantly inhibited the production of IL-6, TNF-α, NO, and ROS in microglial cells without affecting cell viability [199]. Furthermore, G-Re suppressed the phosphorylation of Ca2+/calmodulin-dependent protein kinase (CAMK)2, CAMK4, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinases (JNKs), indicating its inhibitory effect on the CAMK/ERK/JNK/NF-κB signaling pathway in microglial cells [199]. Calcium signaling plays a crucial role in activating the CAMK/ERK/JNK/NF-κB pathway in microglia, which in turn regulates microglial function and contributes to neuroinflammation in various neurological disorders. Table 8 summarizes the key findings on antioxidant interventions.
Table 8.
This table summarizes the effects of various antioxidant compounds in reducing oxidative stress and microglial activation in neurodegenerative diseases. Agents like curcumin, resveratrol, cannabidiol, and flavonoids are presented for their ability to modulate redox balance, inhibit pro-inflammatory signaling pathways, and protect against neuronal damage. The table emphasizes the crosstalk between calcium signaling and redox pathways in neuroprotection.
Natural antioxidants can target microglial activity and modulate calcium and redox signaling pathways. Although the complexity of neurodegenerative diseases and variability in individual responses to antioxidants pose significant challenges, these compounds hold the potential to mitigate neurodegenerative disease progression and enhance neuroprotection.
- d.
- Modulation of microglial calcium signaling for synaptic plasticity
Targeting the pathways via which calcium dynamics in microglia affect neuronal function and synaptic transmission is necessary to modify microglial calcium signaling for synaptic plasticity. The term “synaptic plasticity” describes how synapses, or the connections between neurons, can alter in strength and effectiveness in response to experience and activity [5]. Microglia play multifaceted roles in synaptic plasticity, ranging from synaptic pruning and maturation to modulation of synaptic transmission and immune surveillance, including processes like long-term potentiation (LTP) and long-term depression (LTD) [201]. Microglia have a complex impact on synaptic plasticity, which depends on the balance between the pro-inflammatory and anti-inflammatory cytokines they release. Various molecules, such as PI3K, BDNF, CREB, CD200, TNFα, and IL-1β, from microglia, contribute to either enhancing or inhibiting synaptic plasticity [201]. In a study on acute neuroinflammation induced by microbial neuraminidase, microglial activation was observed in the amygdala, leading to anxiety-like behavior. The results have shown that modulating mitochondrial calcium signaling may influence microglial activation and the production of inflammatory mediators, thus mitigating neuro-inflammatory responses and enhancing synaptic plasticity [202]. Synaptic plasticity is disrupted in diseases like AD due to microglial activation into the pro-inflammatory state. Therapeutic targets in a study modulated microglial activity by modifying TREM2 activity to shift microglia towards a neuroprotective phenotype and inhibiting CSF1R signaling to prevent synaptic loss and cognitive decline [203]. The endocannabinoid (eCB) system comprises cannabinoid receptors CB1R and CB2R. The eCB modulation of microglial phenotypes involves shifting microglia from the M1 state to the M2 state. This shift supports synaptic plasticity by reducing inflammation, promoting tissue repair, and protecting neurons from damage. The mechanisms include the activation of CB2R, the modulation of gene expression, and the reduction in neurotoxicity [204]. Table 9 summarizes the key findings on microglial calcium signaling for synaptic plasticity.
Table 9.
Summary of the key findings on microglial calcium signaling for synaptic plasticity. This table provides insights into how microglial calcium signaling influences synaptic plasticity, highlighting pathways and targets like TREM2, CSF1R, mitochondrial calcium modulation, and the endocannabinoid system. The findings focus on how these pathways affect synaptic transmission, long-term potentiation (LTP), and cognitive function, emphasizing the potential for therapeutic modulation in conditions like Alzheimer’s and schizophrenia.
The complexities of microglial roles in synaptic plasticity and the variability of molecular pathways involved limit the understanding of how to precisely target microglial calcium signaling for therapeutic purposes. Additionally, translating these findings into effective treatments for neurodegenerative diseases remains challenging.
6. Conclusions
Research on microglial calcium signaling has highlighted its pivotal role in CNS disorders, ranging from neurodegenerative diseases like Alzheimer’s and Parkinson’s to neurodevelopmental and psychiatric disorders such as schizophrenia and autism. Key findings demonstrate that microglial calcium dysregulation contributes to aberrant immune responses, neuroinflammation, disrupted synaptic pruning, and impaired neuroprotection, all of which drive the progression of CNS disorders. Microglial calcium signaling is mediated by a complex interplay of channels, receptors, and intracellular dynamics. Store-operated calcium entry (SOCE), transient receptor potential (TRP) channels, and voltage-gated calcium channels (VGCCs) are among the primary pathways that maintain calcium homeostasis and influence microglial functions. Dysregulation in these pathways leads to sustained calcium elevations that trigger pro-inflammatory responses, enhance cytokine production, and contribute to oxidative stress and neurotoxicity. Recent studies highlight that targeting calcium signaling holds promise for therapeutic intervention. Strategies involving calcium channel blockers (CCBs), modulation of purinergic and glutamate receptors, and targeting iCa2+ dynamics have shown potential in mitigating neuroinflammatory responses and improving outcomes in CNS disorders. For instance, drugs like nimodipine and isradipine have demonstrated neuroprotective effects by attenuating calcium influx and reducing pro-inflammatory signaling. Future research should aim to further elucidate the precise mechanisms by which microglial calcium signaling interacts with various pathological processes in the CNS. Recent advancements in two-photon calcium imaging using fluorescent or luminescent Ca2+ indicators that enable the simultaneous detection of microglial calcium signals and neuronal activity would further validate the significance of calcium signals in CNS disease pathology. By applying microglia-specific calcium channel blockers or genetically knocked out calcium channels in microglia, it may become possible to record the movement of microglial processes and their process interaction intensities with neuronal synapses in freely moving, awake mice of various neurodegenerative diseases models. This approach would help clarify the relative contribution of microglial calcium signaling to disease pathology. In addition, designing and developing pharmacological agents targeting microglia-specific calcium channels and receptors is critical for assessing the exact function in healthy conditions. Furthermore, exploring personalized medicine approaches that target microglial calcium dynamics based on patient-specific genetic profiles and disease stages could lead to more effective treatments for neurodegenerative and neuropsychiatric disorders. Understanding the intricate role of microglial calcium signaling will not only enhance our knowledge of CNS pathophysiology but also open new avenues for therapeutic strategies aimed at preserving cognitive function and improving patient outcomes across a broad spectrum of CNS disorders.
7. Limitations of This Study
Current knowledge in this field is largely derived from animal models, which may not fully recapitulate human pathology due to species-specific differences in immune responses and calcium signaling. The distinction between resident microglia and infiltrating monocyte-derived macrophages in neuroinflammatory contexts poses a significant challenge, often resulting in experimental ambiguities. Furthermore, the heterogeneity of microglia across various brain regions, stages of disease progression, and among individual patients complicates the generalization of findings. Temporal and spatial dynamics of calcium signaling are not consistently captured, as many studies focus on discrete time points or regions.
In vitro evidence suggests that microglial functions are closely associated with intracellular calcium signaling. However, knowledge of microglial calcium dynamics in healthy and diseased brains in vivo remains limited. In vivo and in situ findings indicate that surveillant microglia rarely exhibit spontaneous calcium transients, typically responding only to cell or tissue damage in their microenvironment. These transients are thought to be mediated by the release of calcium from intracellular stores and the activation of specific surface receptors.
Although targeting calcium signaling pathways holds therapeutic promise, clinical evidence supporting the efficacy of such interventions in humans is scarce, and concerns regarding off-target effects on other CNS cells remain unresolved. Additionally, this review predominantly addresses well-known neurodegenerative diseases, leaving significant gaps in the understanding of microglial calcium dysregulation in conditions such as traumatic brain injury and advanced psychiatric disorders. A major challenge for therapeutic development lies in balancing the neuroprotective functions of microglia with the necessity of mitigating detrimental neuroinflammatory responses.
Author Contributions
M.O.R., J.E.P. and J.H. conceptualized and supervised the study. A.R.H., F.T., M.A., M.T.I., R.R. and M.O.R. wrote the manuscript. A.B., J.E.P., J.H. and M.O.R. reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no competing interests in this work.
References
- Hattori, Y. The behavior and functions of embryonic microglia. Anat. Sci. Int. 2022, 97, 1. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System–Associated Macrophages—From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Vazquez, A.L.; Cui, X.T. Revealing in vivo cellular mechanisms of cerebral microbleeds on neurons and microglia across cortical layers. iScience 2024, 27, 109371. [Google Scholar] [CrossRef]
- Petry, P.; Oschwald, A.; Kierdorf, K. Microglial tissue surveillance: The never-resting gardener in the developing and adult CNS. Eur. J. Immunol. 2023, 53, 2250232. [Google Scholar] [CrossRef]
- Valizadeh, A.; Asl, M.M. Synaptic plasticity during brain development: Implications for therapeutic reorganization of neural circuits. In Encyclopedia of Child and Adolescent Health, 1st ed.; Academic Press: Cambridge, MA, USA, 2023; Volume 1, pp. 14–24. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, D.R.; Blanco-Luquin, I.; Mendioroz, M. The Participation of Microglia in Neurogenesis: A Review. Brain Sci. 2021, 11, 658. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef]
- Casali, B.T.; Reed-Geaghan, E.G. Microglial Function and Regulation during Development, Homeostasis and Alzheimer’s Disease. Cells 2021, 10, 957. [Google Scholar] [CrossRef] [PubMed]
- Brawek, B.; Garaschuk, O. Microglial Calcium Signaling in the Adult, Aged and Diseased Brain; Elsevier Ltd.: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets; Springer Nature: Berlin/Heidelberg, Germany, 2023. [Google Scholar] [CrossRef]
- Parajuli, B.; Koizumi, S. Strategies for Manipulating Microglia to Determine Their Role in the Healthy and Diseased Brain. Neurochem. Res. 2022, 48, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yong, V.W. Microglia: Custodians of the Central Nervous System. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2024. [Google Scholar] [CrossRef]
- Spiteri, A.G.; Wishart, C.L.; Pamphlett, R.; Locatelli, G.; King, N.J.C. Microglia and monocytes in inflammatory CNS disease: Integrating phenotype and function. Acta Neuropathol. 2021, 143, 179–224. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Zhang, J.; Li, A.; Yuan, Z.; Cheng, J. Roles of microglial calcium channels in neurodegenerative diseases. Hum. Brain 2024, 3. [Google Scholar] [CrossRef]
- Marzan, D.E.; Brügger-Verdon, V.; West, B.L.; Liddelow, S.; Samanta, J.; Salzer, J.L. Activated microglia drive demyelination via CSF1R signaling. Glia 2021, 69, 1583–1604. [Google Scholar] [CrossRef] [PubMed]
- Berdnikov, A.K.; Atyakshin, D.A.; Nikolaeva, L.V.; Salmina, A.B. Regulation of calcium homeostasis in microglia: Current view on the pathogenesis and correction of neuroinflammation. Opera Med. Physiol. 2023, 10, 58–82. [Google Scholar] [CrossRef]
- Pan, K.; Garaschuk, O. The role of intracellular calcium-store-mediated calcium signals in in vivo sensor and effector functions of microglia. J. Physiol. 2023, 601, 4203–4215. [Google Scholar] [CrossRef] [PubMed]
- Granzotto, A.; McQuade, A.; Chadarevian, J.P.; Davtyan, H.; Sensi, S.L.; Parker, I.; Blurton-Jones, M.; Smith, I. ER and SOCE Ca2+ signals are not required for directed cell migration in human microglia. Cell Calcium 2024, 123, 102923. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. Regulation of calcium homeostasis and flux between the endoplasmic reticulum and the cytosol. J. Biol. Chem. 2022, 298, 102061. [Google Scholar] [CrossRef]
- Luo, L.; Song, S.; Ezenwukwa, C.C.; Jalali, S.; Sun, B.; Sun, D. Ion Channels and Transporters in Microglial Function in Physiology and Brain Diseases Lanxin. Neurochem. Int. 2020, 142, 104925. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Alqinyah, M.; Vella, S.; Moreno, S.N.J.; Hooks, S. Regulator of G protein Signaling 10 (RGS10) functionally interacts with store-operated calcium entry machinery to regulate COX-2 and TNF-α in Microglia. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Alqinyah, M.; Vella, S.; Dean, P.; Almutairi, F.; Davila-Rivera, R.; Rayatpisheh, S.; Wohlschlegel, J.; Moreno, S.; Hooks, S.B.; et al. RGS10 physically and functionally interacts with STIM2 and requires store-operated calcium entry to regulate pro-inflammatory gene expression in microglia. Cell Signal 2021, 83, 109974. [Google Scholar] [CrossRef]
- Nazıroğlu, M.; Radu, B.M.; Cucu, D. Editorial: Transient receptor potential (TRP) ion channels in non-excitable cells. Front. Physiol. 2023, 14, 1213332. [Google Scholar] [CrossRef]
- Hong, C.; Jeong, B.; Park, H.J.; Chung, J.Y.; Lee, J.E.; Kim, J.; Shin, Y.C.; So, I. TRP Channels as Emerging Therapeutic Targets for Neurodegenerative Diseases. Front. Physiol. 2020, 11, 521830. [Google Scholar] [CrossRef]
- Wang, C.; Huang, W.; Lu, J.; Chen, H.; Yu, Z. TRPV1-Mediated Microglial Autophagy Attenuates Alzheimer’s Disease-Associated Pathology and Cognitive Decline. Front. Pharmacol. 2022, 12, 763866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hou, B.; Liang, P.; Lu, X.; Wu, Y.; Zhang, X.; Fan, Y.; Liu, Y.; Chen, T.; Liu, W.; et al. TRPV1 channel mediates NLRP3 inflammasome-dependent neuroinflammation in microglia. Cell Death Dis. 2021, 12, 1159. [Google Scholar] [CrossRef]
- Montaño, L.M.; Carbajal-García, A.; Casas-Hernández, M.F.; Arredondo-Zamarripa, D.; Reyes-García, J. TRPV4 Activation during Guinea Pig Airway Smooth Muscle Contraction Promotes Ca2+ and Na+ Influx. Pharmaceuticals 2024, 17, 293. [Google Scholar] [CrossRef] [PubMed]
- Nadezhdin, K.D.; Neuberger, A.; Sobolevsky, A.I. Structural snapshots of the mechanism of TRPV2 channel activation by small-molecule agonists. Cell Calcium 2022, 105, 102607. [Google Scholar] [CrossRef] [PubMed]
- Maksoud, M.J.E.; Tellios, V.; Lu, W.-Y. Nitric oxide attenuates microglia proliferation by sequentially facilitating calcium influx through TRPV2 channels, activating NFATC2, and increasing p21 transcription. Cell Cycle 2021, 20, 417–433. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhu, T.; Hu, H.; Zhao, Y.; Ying, Y.; Luo, X.; Ling, Y.; Chen, Z.; Ji, H.; Jiang, P.; et al. TRPM2 ion channel is involved in the aggravation of cognitive impairment and down regulation of epilepsy threshold in pentylenetetrazole-induced kindling mice. Brain Res. Bull. 2020, 155, 48–60. [Google Scholar] [CrossRef]
- Akyuva, Y.; Nazıroğlu, M.; Yıldızhan, K. Selenium prevents interferon-gamma induced activation of TRPM2 channel and inhibits inflammation, mitochondrial oxidative stress, and apoptosis in microglia. Metab. Brain Dis. 2021, 36, 285–298. [Google Scholar] [CrossRef]
- Liu, M.; Liu, H.; Feng, F.; Krook-Magnuson, E.; Dudley, S.C. TRPM7 kinase mediates hypomagnesemia-induced seizure-related death. Sci. Rep. 2023, 13, 1–13. [Google Scholar] [CrossRef]
- Mizoguchi, Y.; Ohgidani, M.; Haraguchi, Y.; Murakawa-Hirachi, T.; Kato, T.A.; Monji, A. ProBDNF induces sustained elevation of intracellular Ca2+ possibly mediated by TRPM7 channels in rodent microglial cells. Glia 2021, 69, 1694–1708. [Google Scholar] [CrossRef]
- Qian, H.; Zhang, H.N.; Gao, T.; Wang, X.S.; Wang, X.; Yu, M.Y.; Li, M.K.; Huang, J. Upregulation of TRPC1 in microglia promotes neutrophil infiltration after ischemic stroke. Brain Res. Bull. 2024, 208, 110894. [Google Scholar] [CrossRef]
- Parmar, J.; von Jonquieres, G.; Gorlamandala, N.; Chung, B.; Craig, A.J.; Pinyon, J.L.; Birnbaumer, L.; Klugmann, M.; Moor-house, A.J.; Power, J.M.; et al. TRPC Channels Activated by G Protein-Coupled Receptors Drive Ca2+ Dysregulation Leading to Secondary Brain Injury in the Mouse Model. Transl. Stroke Res. 2024, 15, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Ren, J.; Ma, Y.; Ozathaley, A.; Yuan, W.; Ni, H.; Li, D.; Liu, Z. Upregulation of TRPC5 in hippocampal excitatory synapses improves memory impairment associated with neuroinflammation in microglia knockout IL-10 mice. J. Neuroinflamm. 2021, 18, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Trus, M.; Atlas, D. Non-ionotropic voltage-gated calcium channel signaling. Channels 2024, 18, 2341077. [Google Scholar] [CrossRef] [PubMed]
- Hopp, S.C. Targeting microglia L-type voltage-dependent calcium channels for the treatment of central nervous system disorders. J. Neurosci. Res. 2020, 99, 141–162. [Google Scholar] [CrossRef]
- Xue, J.; Zeng, W.; Han, Y.; John, S.; Ottolia, M.; Jiang, Y. Structural mechanisms of the human cardiac sodium-calcium exchanger NCX1. Nat. Commun. 2023, 14, 1–11. [Google Scholar] [CrossRef]
- Poshtkohi, A.; Wade, J.; McDaid, L.; Liu, J.; Dallas, M.; Bithell, A. Mathematical modelling of human P2X-mediated plasma membrane electrophysiology and calcium dynamics in microglia. PLoS Comput. Biol. 2021, 17, e1009520. [Google Scholar] [CrossRef] [PubMed]
- Héja, L.; Kardos, J. NCX activity generates spontaneous Ca2+ oscillations in the astrocytic leaflet microdomain. Cell Calcium 2020, 86, 102137. [Google Scholar] [CrossRef]
- Tedeschi, V.; Sisalli, M.J.; Pannaccione, A.; Piccialli, I.; Molinaro, P.; Annunziato, L.; Secondo, A. Na+/Ca2+ exchanger isoform 1 (NCX1) and canonical transient receptor potential channel 6 (TRPC6) are recruited by STIM1 to mediate Store-Operated Calcium Entry in primary cortical neurons. Cell Calcium 2021, 101, 102525. [Google Scholar] [CrossRef]
- Orädd, F.; Magkakis, K.; Prabudiansyah, I.; Levantino, M.; Inaba, K.; Andersson, M. Tracking the structural dynamics of SERCA regulation in real time. Biophys. J. 2023, 122, 58a. [Google Scholar] [CrossRef]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepúlveda, M.R.; Vangheluwe, P. Primary Active Ca2+ Transport Systems in Health and Disease. Cold Spring Harb. Perspect. Biol. 2019, 12, a035113. [Google Scholar] [CrossRef]
- Morales-ropero, J.M.; Arroyo-Urea, S.; Neubrand, V.E.; Martín-Oliva, D.; Marín-Teva, J.L.; Cuadros, M.A.; Vangheluwe, P.; Navascués, J.; Mata, A.M.; Sepúlveda, M.R. The endoplasmic reticulum Ca2+-ATPase SERCA2b is upregulated in activated microglia and its inhibition causes opposite effects on migration and phagocytosis. Glia 2020, 69, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Lee, J.Y.; Park, C.; Kim, Y.H.; Park, C. The Role of TRP Channels and PMCA in Brain Disorders: Intracellular Calcium and pH Homeostasis. Front. Cell Dev. Biol. 2021, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell Biol. 2020, 22, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Jia, X.; Wood, I.; Jiang, L.H. Piezo1 channel-mediated Ca2+ signaling inhibits lipopolysaccharide-induced activation of the NF-κB inflammatory signaling pathway and generation of TNF-α and IL-6 in microglial cells. Glia 2023, 71, 848–865. [Google Scholar] [CrossRef]
- Zong, B.; Yu, F.; Zhang, X.; Pang, Y.; Zhao, W.; Sun, P.; Li, L. Mechanosensitive Piezo1 channel in physiology and pathophysiology of the central nervous system. Ageing Res. Rev. 2023, 90, 102026. [Google Scholar] [CrossRef]
- Liu, H.; Bian, W.; Yang, D.; Yang, M.; Luo, H. Inhibiting the Piezo1 channel protects microglia from acute hyperglycaemia damage through the JNK1 and mTOR signalling pathways. Life Sci. 2021, 264, 118667. [Google Scholar] [CrossRef]
- Imraish, A.; Abu-Thiab, T.; Hammad, H. P2X and P2Y Receptor Antagonists Reduce Inflammation in ATPinduced Microglia. Pharm. Pract. 2023, 21, 1–7. [Google Scholar] [CrossRef]
- Chisari, M.; Barraco, M.; Bucolo, C.; Ciranna, L.; Sortino, M.A. Purinergic ionotropic P2X7 and metabotropic glutamate mGlu5 receptors crosstalk influences pro-inflammatory conditions in microglia. Eur. J. Pharmacol. 2023, 938, 175389. [Google Scholar] [CrossRef]
- Palomba, N.P.; Martinello, K.; Cocozza, G.; Casciato, S.; Mascia, A.; Di Gennaro, G.; Morace, R.; Esposito, V.; Wulff, H.; Li-matola, C.; et al. ATP-evoked intracellular Ca2+ transients shape the ionic permeability of human microglia from epileptic temporal cortex. J. Neuroinflamm. 2021, 18, 1–12. [Google Scholar] [CrossRef]
- Schädlich, I.S.; Winzer, R.; Stabernack, J.; Tolosa, E.; Magnus, T.; Rissiek, B. The role of the ATP-adenosine axis in ischemic stroke. Semin. Immunopathol. 2023, 45, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Lee, S.J.; Kim, C.H. Roles of cytokines in the temporal changes of microglial membrane currents and neuronal excitability and synaptic efficacy in ATP-induced cortical injury model. Int. J. Mol. Sci. 2021, 22, 6853. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P.; Liu, S.-C.; Hu, S.-Q.; Lu, J.-F.; Wu, C.-L.; Hu, D.-X.; Zhang, W.-J. ATP ion channel P2X purinergic receptors in inflammation response. Biomed. Pharmacother. 2023, 158, 114205. [Google Scholar] [CrossRef] [PubMed]
- Cowley, J.; Harrison, N.; Morano, I.; Chan, H.; Li, Z.; Pegg, A.; Corsi-Zuelli, F.; Jinks, E.; Ali, H.; Stevens, A.; et al. Regulation of pro-inflammatory cytokine release by purinergic signalling in a human monocyte-derived microglial model. Alzheimer’s Dement. 2023, 19, e082639. [Google Scholar] [CrossRef]
- Kaidonis, X.; Mohl, M.C.; Graham, R.M. α1-Adrenergic receptors. In Primer on the Autonomic Nervous System, 4th ed.; Academic Press: Waltham, MA, USA; Elsevier Inc.: Amsterdam, The Netherlands, 2023; pp. 43–47. [Google Scholar] [CrossRef]
- Gurevich, V.V. β-Adrenergic receptors. In Primer on the Autonomic Nervous System, 4th ed.; Academic Press: Waltham, MA, USA; Elsevier Inc.: Amsterdam, The Netherlands, 2023; pp. 53–55. [Google Scholar] [CrossRef]
- Barodia, S.K.; Sophronea, T.; Luthra, P.M. A2A R mediated modulation in IP3 levels altering the [Ca2+]i through cAMP-dependent PKA signalling pathway. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2022, 1866, 130242. [Google Scholar] [CrossRef]
- Abd-Elrahman, K.S.; Sarasija, S.; Ferguson, S.S.G. The Role of Neuroglial Metabotropic Glutamate Receptors in Alzheimer’s Disease. Curr. Neuropharmacol. 2021, 21, 273–283. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, L.; Chen, J.; Luo, Y.; Peng, Z.; Zhang, H.; Pan, Z.; Chen, Y. Metabotropic Glutamate Receptor 8 Suppresses M1 Polarization in Microglia by Alleviating Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. J. Integr. Neurosci. 2024, 23, 26. [Google Scholar] [CrossRef]
- Chen, T.S.; Huang, T.H.; Lai, M.C.; Huang, C.W. The Role of Glutamate Receptors in Epilepsy. Biomedicines 2023, 11, 783. [Google Scholar] [CrossRef]
- da Silva, J.A.C.; Schröder, N. The Role of Ca2+ Permeable AMPA Receptors in Neurodegeneration, Neurotoxicity, and Neuroinflammation. CNS Neurol. Disord. Drug Targets 2022, 22, 624–633. [Google Scholar] [CrossRef]
- He, X.; Gao, Y.; Yin, X.; Wu, X.; Ji, M.; Shen, J. GluR2 may drive neuroinflammation and cognitive impairments following peripherally repeated lipopolysaccharide exposures. Neurochem. Res. 2022, 49, 2393–2407. [Google Scholar] [CrossRef]
- Wu, E.; Zhang, J.; Zhang, J.; Zhu, S. Structural insights into gating mechanism and allosteric regulation of NMDA receptors. Curr. Opin. Neurobiol. 2023, 83, 102806. [Google Scholar] [CrossRef] [PubMed]
- Seljeset, S.; Sintsova, O.; Wang, Y.; Harb, H.Y.; Lynagh, T. Constitutive activity of ionotropic glutamate receptors via hydrophobic substitutions in the ligand-binding domain. Structure 2024, 32, 966–978.e6. [Google Scholar] [CrossRef] [PubMed]
- Jong, Y.-J.I.; Harmon, S.K.; O’Malley, K. Intracellular metabotropic glutamate receptor 5 (mGlu5) triggers ER calcium release distinct from cell surface counterparts in striatal neurons. J. Pharmacol. Exp. Ther. 2023, 385, 333. [Google Scholar] [CrossRef]
- Guo, C.; Wang, C.; He, T.; Yu, B.; Li, M.; Zhao, C.; Yuan, Y.; Chen, H. The effect of mGlu2/3 receptors on synaptic activities to different types of GABAergic interneurons in the anterior cingulate cortex. Neuropharmacology 2020, 175, 108180. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef]
- Chakravarti, B.; Chattopadhyay, N.; Brown, E.M. Signaling Through the Extracellular Calcium-Sensing Receptor (CaSR). In Calcium Signaling; Springer: Berlin/Heidelberg, Germany, 2012; Volume 740, pp. 103–142. [Google Scholar] [CrossRef]
- Tvrdik, P.; Kalani, M. In Vivo Imaging of Microglial Calcium Signaling in Brain Inflammation and Injury. Int. J. Mol. Sci. 2017, 18, 2366. [Google Scholar] [CrossRef]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef]
- Bastioli, G.; Piccirillo, S.; Graciotti, L.; Carone, M.; Sprega, G.; Taoussi, O.; Preziuso, A.; Castaldo, P. Calcium Deregulation in Neurodegeneration and Neuroinflammation in Parkinson’s Disease: Role of Calcium-Storing Organelles and Sodium–Calcium Exchanger. Cells 2024, 13, 1301. [Google Scholar] [CrossRef]
- Wang, C.; Jia, Q.; Sun, C.; Jing, C. Calcium sensing receptor contribute to early brain injury through the CaMKII/NLRP3 pathway after subarachnoid hemorrhage in mice. Biochem. Biophys. Res. Commun. 2020, 530, 651–657. [Google Scholar] [CrossRef]
- Nevelchuk, S.; Brawek, B.; Schwarz, N.; Valiente-Gabioud, A.; Wuttke, T.V.; Kovalchuk, Y.; Koch, H.; Höllig, A.; Steiner, F.; Figarella, K.; et al. Morphotype-specific calcium signaling in human microglia. J. Neuroinflamm. 2024, 21, 1–18. [Google Scholar] [CrossRef]
- Izquierdo, P.; Jolivet, R.B.; Attwell, D.; Madry, C. Amyloid plaques and normal ageing have differential effects on mi-croglial Ca2+ activity in the mouse brain. Pflugers Arch. 2024, 476, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Del Moral, M.O.; Asavapanumas, N.; Uzcátegui, N.L.; Garaschuk, O. Healthy Brain Aging Modifies Microglial Calcium Signaling In Vivo. Int. J. Mol. Sci. 2019, 20, 589. [Google Scholar] [CrossRef] [PubMed]
- Correa, B.H.M.; Moreira, C.R.; Hildebrand, M.E.; Vieira, L.B. The Role of Voltage-Gated Calcium Channels in Basal Ganglia Neurodegenerative Disorders. Curr. Neuropharmacol. 2022, 21, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, 432. [Google Scholar] [CrossRef]
- Tanwar, J.; Singh, J.B.; Motiani, R.K. Molecular machinery regulating mitochondrial calcium levels: The nuts and bolts of mitochondrial calcium dynamics. Mitochondrion 2021, 57, 9–22. [Google Scholar] [CrossRef]
- Li, Y.; Xu, H.; Wang, H.; Yang, K.; Luan, J.; Wang, S. TREM2: Potential therapeutic targeting of microglia for Alzheimer’s disease. Biomed. Pharmacother. 2023, 165, 115218. [Google Scholar] [CrossRef]
- Tewari, M.; Michalski, S.; Egan, T.M. Modulation of Microglial Function by ATP-Gated P2X7 Receptors: Studies in Rat, Mice and Human. Cells 2024, 13, 161. [Google Scholar] [CrossRef]
- Madry, C.; Kyrargyri, V.; Arancibia-Cárcamo, I.L.; Jolivet, R.; Kohsaka, S.; Bryan, R.M.; Attwell, D. Microglial Ramification, Surveillance, and Interleukin-1β Release Are Regulated by the Two-Pore Domain K+ Channel THIK-1. Neuron 2018, 97, 299–312.e6. [Google Scholar] [CrossRef]
- Díaz-Piña, D.A.; Rivera-Ramírez, N.; García-López, G.; Díaz, N.F.; Molina-Hernández, A. Calcium and Neural Stem Cell Proliferation. Int. J. Mol. Sci. 2024, 25, 4073. [Google Scholar] [CrossRef]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Riester, K.; Brawek, B.; Savitska, D.; Fröhlich, N.; Zirdum, E.; Mojtahedi, N.; Heneka, M.T.; Garaschuk, O. In vivo characterization of functional states of cortical microglia during peripheral inflammation. Brain Behav. Immun. 2020, 87, 243–255. [Google Scholar] [CrossRef]
- Karavis, M.Y.; Siafaka, I.; Vadalouca, A.; Georgoudis, G. Role of Microglia in Neuropathic Pain. Cureus 2023, 15, e43555. [Google Scholar] [CrossRef] [PubMed]
- Jairaman, A.; McQuade, A.; Granzotto, A.; Kang, Y.J.; Chadarevian, J.P.; Gandhi, S.; Parker, I.; Smith, I.; Cho, H.; Sensi, S.L.; et al. TREM2 regulates purinergic receptor-mediated calcium signaling and motility in human iPSC-derived microglia. eLife 2022, 11, e73021. [Google Scholar] [CrossRef]
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef]
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2021, 2021, 7086512. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Li, B.; Ayasoufi, K.; Simon, W.L.; Zhao, S.; Xie, M.; Thyen, G.; Hur, B.; Zheng, J.; Liang, Y.; et al. Microglial P2Y6 calcium signaling promotes phagocytosis and shapes neuroimmune responses in epileptogenesis. Neuron 2023, 112, 1959–1977. [Google Scholar] [CrossRef]
- Faust, T.E.; Gunner, G.; Schafer, D.P. Mechanisms governing activity-dependent synaptic pruning in the mammalian CNS. Nat. Rev. Neurosci. 2021, 22, 657. [Google Scholar] [CrossRef]
- Sakai, J. Core Concept: How synaptic pruning shapes neural wiring during development and, possibly, in disease. Proc. Natl. Acad. Sci. USA 2020, 117, 16096. [Google Scholar] [CrossRef]
- Han, Q.Q.; Shen, S.Y.; Liang, L.F.; Chen, X.R.; Yu, J. Complement C1q/C3-CR3 signaling pathway mediates abnormal microglial phagocytosis of synapses in a mouse model of depression. Brain Behav. Immun. 2024, 119, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Chafee, M.V.; Averbeck, B.B. Unmasking Schizophrenia: Synaptic Pruning in Adolescence Reveals a Latent Physiological Vulnerability in Prefrontal Recurrent Networks. Biol. Psychiatry 2022, 92, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Pocock, J.; Vasilopoulou, F.; Svensson, E.; Cosker, K. Microglia and TREM2. Neuropharmacology 2024, 257, 110020. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhang, L.; Xin, W.; Pan, Y.; Tatenhorst, L.; Hao, Z.; Gerner, S.T.; Huber, S.; Juenemann, M.; Butz, M.; et al. TREM2 regulates microglial lipid droplet formation and represses post-ischemic brain injury. Biomed. Pharmacother. 2024, 170, 115962. [Google Scholar] [CrossRef]
- Shen, F.-S.; Liu, C.; Sun, H.-Z.; Chen, X.-Y.; Xue, Y.; Chen, L. Emerging evidence of context-dependent synapse elimination by phagocytes in the CNS. J. Leukoc. Biol. 2024, 116, 511–522. [Google Scholar] [CrossRef]
- Ge, Y.; Yang, J.; Chen, J.; Dai, M.; Dou, X.; Yao, S.; Yao, C.; Lin, Y. Absence in CX3CR1 receptor signaling promotes post-ischemic stroke cognitive function recovery through suppressed microglial pyroptosis in mice. CNS Neurosci. Ther. 2024, 30, e14551. [Google Scholar] [CrossRef]
- Park, J.; Chung, W.S. Astrocyte-dependent circuit remodeling by synapse phagocytosis. Curr. Opin. Neurobiol. 2023, 81, 102732. [Google Scholar] [CrossRef] [PubMed]
- Stogsdill, J.A.; Harwell, C.C.; Goldman, S.A. Astrocytes as master modulators of neural networks: Synaptic functions and disease-associated dysfunction of astrocytes. Ann. N. Y Acad. Sci. 2023, 1525, 41–60. [Google Scholar] [CrossRef]
- Kofuji, P.; Araque, A. G-Protein-Coupled Receptors in Astrocyte–Neuron Communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef]
- Zhou, M.; Salinas, S.; Cornell, J.; Bui, A. Microglia regulate cognition and stress-related cognitive disorders. In Stress: Immunology and Inflammation; Handbook of Stress Series; Academic Press: Cambridge, MA, USA, 2024; Volume 5, pp. 183–197. [Google Scholar] [CrossRef]
- Orhan, F.; Malwade, S.; Khanlarkhani, N.; Gkoga, A.; Jungholm, O.; Koskuvi, M.; Lehtonen, S.; Schwieler, L.; Jardemark, K.; Tiihonen, J.; et al. Kynurenic acid promotes activity-dependent synaptic pruning in schizophrenia. bioRxiv 2023, 2023.10.19.563090. [Google Scholar] [CrossRef]
- Surala, M.; Soso-Zdravkovic, L.; Munro, D.; Rifat, A.; Ouk, K.; Vida, I.; Priller, J.; Madry, C. Lifelong absence of microglia alters hippocampal glutamatergic networks but not synapse and spine density. EMBO Rep. 2024, 25, 2348–2374. [Google Scholar] [CrossRef] [PubMed]
- Heuer, S.E.; Bloss, E.B.; Howell, G.R. Strategies to dissect microglia-synaptic interactions during aging and in Alzheimer’s disease. Neuropharmacology 2024, 254, 109987. [Google Scholar] [CrossRef] [PubMed]
- Malvaso, A.; Gatti, A.; Negro, G.; Calatozzolo, C.; Medici, V.; Poloni, T.E. Microglial Senescence and Activation in Healthy Aging and Alzheimer’s Disease: Systematic Review and Neuropathological Scoring. Cells 2023, 12, 2824. [Google Scholar] [CrossRef]
- Rathi, K.; Bhole, R.; Bansode, P.; Motghare, N. Navigating the Landscape of Alzheimer’s Disease: From Epidemiology to Drug Re-purposing. Adv. Pharmacol. Pharm. 2024, 12, 135–148. [Google Scholar] [CrossRef]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front. Cell Infect. Microbiol. 2019, 9, 1–20. [Google Scholar] [CrossRef]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 1584–1590. [Google Scholar] [CrossRef]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- McLarnon, J.G. Microglial Store-operated Calcium Signaling in Health and in Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 17, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, J.L.; Michalski, K.; Chou, T.H.; Grant, T.; Rao, S.; Simorowski, N.; Tucker, S.J.; Grigorieff, N.; Furukawa, H. Structure and assembly of calcium homeostasis modulator proteins. Nat. Struct. Mol. Biol. 2020, 27, 150–159. [Google Scholar] [CrossRef]
- Cheng, J.; Dong, Y.; Ma, J.; Pan, R.; Liao, Y.; Kong, X.; Li, X.; Li, S.; Chen, P.; Wang, L.; et al. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology. Sci. Adv. 2021, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bo, X.; Xie, F.; Zhang, J.; Gu, R.; Li, X.; Li, S.; Yuan, Z.; Cheng, J. Deletion of Calhm2 alleviates MPTP-induced Parkinson’s disease pathology by inhibiting EFHD2-STAT3 signaling in microglia. Theranostics 2023, 13, 1809–1822. [Google Scholar] [CrossRef]
- Graves, N.J.; Gambin, Y.; Sierecki, E. α-Synuclein Strains and Their Relevance to Parkinson’s Disease, Multiple System Atrophy, and Dementia with Lewy Bodies. Int. J. Mol. Sci. 2023, 24, 12134. [Google Scholar] [CrossRef]
- Kaur, S.; Sehrawat, A.; Mastana, S.S.; Kandimalla, R.; Sharma, P.K.; Bhatti, G.K.; Bhatti, J.S. Targeting calcium homeostasis and impaired inter-organelle crosstalk as a potential therapeutic approach in Parkinson’s disease. Life Sci. 2023, 330, 121995. [Google Scholar] [CrossRef]
- Oliveira-Giacomelli, Á.M.; Albino, C.; de Souza, H.D.N.; Corrêa-Velloso, J.; de Jesus Santos, A.P.; Baranova, J.; Ulrich, H. P2Y6 and P2X7 Receptor Antagonism Exerts Neuroprotective/Neuroregenerative Effects in an Animal Model of Parkinson’s Disease. Front. Cell. Neurosci. 2019, 13, 457943. [Google Scholar] [CrossRef]
- Schrank, S.; Barrington, N.; Stutzmann, G.E. Calcium-Handling Defects and Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2019, 12, a035212. [Google Scholar] [CrossRef]
- Kovacs, G.; Reimer, L.; Jensen, P.H. Endoplasmic Reticulum-Based Calcium Dysfunctions in Synucleinopathies. Front. Neurol. 2021, 12, 742625. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Calabresi, P.A. Multiple Sclerosis and Other Acquired Demyelinating Diseases of the Central Nervous System. Cold Spring Harb. Perspect. Biol. 2024, a041374. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Marrodan, M.; Ysrraelit, M.C. Mechanisms of neurodegeneration and axonal dysfunction in progressive multiple sclerosis. Biomedicines 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Airas, L.; Yong, V.W. Microglia in multiple sclerosis—Pathogenesis and imaging. Curr. Opin. Neurol. 2022, 35, 299–306. [Google Scholar] [CrossRef]
- Enders, M.; Heider, T.; Ludwig, A.; Kuerten, S. Strategies for neuroprotection in multiple sclerosis and the role of calcium. Int. J. Mol. Sci. 2020, 21, 1663. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; De Vito, F.; Fresegna, D.; Rizzo, F.R.; Bullitta, S.; Guadalupi, L.; Vanni, V.; Buttari, F.; Stampanoni Bassi, M.; Leuti, A.; et al. Peripheral T cells from multiple sclerosis patients trigger synaptotoxic alterations in central neurons. Neuropathol. Appl. Neurobiol. 2020, 46, 160–170. [Google Scholar] [CrossRef]
- Thomas, A.G.; O’Driscoll, C.M.; Bressler, J.; Kaufmann, W.E.; Rojas, C.J.; Slusher, B.S. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem. Biophys. Res. Commun. 2014, 443, 32–36. [Google Scholar] [CrossRef]
- Joshi, D.C.; Tewari, B.P.; Singh, M.; Joshi, P.G.; Joshi, N.B. AMPA receptor activation causes preferential mitochondrial Ca2+ load and oxidative stress in motor neurons. Brain Res. 2015, 1616, 1–9. [Google Scholar] [CrossRef]
- Malko, P.; Mortadza, S.A.S.; McWilliam, J.; Jiang, L.H. TRPM2 Channel in Microglia as a New Player in Neuroinflammation Associated With a Spectrum of Central Nervous System Pathologies. Front. Pharmacol. 2019, 10, 239. [Google Scholar] [CrossRef]
- van den Bosch, A.M.; van der Poel, M.; Fransen, N.L.; Vincenten, M.C.; Bobeldijk, A.M.; Jongejan, A.; Engelenburg, H.J.; Moerland, P.D.; Smolders, J.; Huitinga, I.; et al. Profiling of microglia nodules in multiple sclerosis reveals propensity for lesion formation. Nat. Commun. 2024, 15, 1667. [Google Scholar] [CrossRef]
- Kirdajova, D.B.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Z.; Wang, L.; Zhang, L.; Liu, Y.; Cao, J.; Fu, Q.; Liu, Y.; Li, H.; Lou, J.; et al. Cottonseed oil alleviates ischemic stroke injury by inhibiting the inflammatory activation of microglia and astrocyte. J. Neuroinflamm. 2020, 17, 270. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Qi, Z.; Li, W.; Liang, J.; Zhao, L.; Shi, Y. M1 Microglia Induced Neuronal Injury on Ischemic Stroke via Mitochondrial Crosstalk between Microglia and Neurons. Oxidative Med. Cell. Longev. 2022, 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rahi, V.; Kaundal, R.K. Exploring the intricacies of calcium dysregulation in ischemic stroke: Insights into neuronal cell death and therapeutic strategies. Life Sci. 2024, 347, 122651. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kearns, K.N.; Eli, I.; Sharifi, K.A.; Soldozy, S.; Carlson, E.W.; Scott, K.W.; Sluzewski, M.F.; Acton, S.T.; Stauderman, K.A.; et al. Microglial Calcium Waves During the Hyperacute Phase of Ischemic Stroke. Stroke 2021, 52, 274–283. [Google Scholar] [CrossRef]
- Kearns, K.N.; Liu, L.; Soldozy, S.; Sharifi, K.A.; Shaffrey, M.E.; Park, M.S.; Tvrdik, P. Microglia Modulate Cortical Spreading Depolarizations After Ischemic Stroke: A Narrative Review. Neurocrit. Care 2022, 37, 133–138. [Google Scholar] [CrossRef]
- Stein, D.J.; Phillips, K.A.; Bolton, D.; Fulford KW, M.; Sadler, J.Z.; Kendler, K.S. What is a mental/psychiatric disorder? From DSM-IV to DSM-V. Psychol. Med. 2010, 40, 1759–1765. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Stevens, B. The ‘quad-partite’ synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Deutschenbaur, L.; Beck, J.; Kiyhankhadiv, A.; Mühlhauser, M.; Borgwardt, S.; Walter, M.; Hasler, G.; Sollberger, D.; Lang, U.E. Role of calcium, glutamate and NMDA in major depression and therapeutic application. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 325–333. [Google Scholar] [CrossRef]
- Kang, Y.; Shin, D.; Kim, A.; You, S.H.; Kim, B.; Han, K.M.; Ham, B.J. The effect of inflammation markers on cortical thinning in major depressive disorder: A possible mediator of depression and cortical changes. J. Affect. Disord. 2024, 348, 229–237. [Google Scholar] [CrossRef]
- Jia, X.; Gao, Z.; Hu, H. Microglia in depression: Current perspectives. Sci. China Life Sci. 2021, 64, 911–925. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Enache, D.; Pariante, C.M.; Mondelli, V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 2019, 81, 24–40. [Google Scholar] [CrossRef]
- Muthmainnah, M.; Amris, F. Tinjauan Skizofrenia Secara Psikoneuroimunologi. Termometer J. Ilm. Ilmu Kesehat. Kedokt. 2024, 2, 1–15. [Google Scholar] [CrossRef]
- Doorduin, J.; De Vries, E.F.J.; Willemsen, A.T.M.; De Groot, J.C.; Dierckx, R.A.; Klein, H.C. Neuroinflammation in schizophrenia-related psychosis: A PET study. J. Nucl. Med. 2009, 50, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, Y.; Kato, T.A.; Horikawa, H.; Monji, A. Microglial intracellular Ca2+ signaling as a target of antipsychotic actions for the treatment of schizophrenia. Front. Cell. Neurosci. 2014, 8, 370. [Google Scholar] [CrossRef]
- Kato, T.; Mizoguchi, Y.; Monji, A.; Horikawa, H.; Suzuki, S.O.; Seki, Y.; Iwaki, T.; Hashioka, S.; Kanba, S. Inhibitory effects of aripiprazole on interferon-gamma-induced microglial activation via intracellular Ca2+ regulation in vitro. J. Neurochem. 2008, 106, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.N.; Da Fonseca, M.M.R.; Machado-Viera, R.; Soares, J.C.; Kapczinski, F. Neurophatological and neurochemical abnormalities in bipolar disorder. Rev. Bras. Psiquiatr. 2004, 26, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.H.; Vecera, C.M.; Pinjari, O.F.; Machado-Vieira, R. Inflammatory signaling mechanisms in bipolar disorder. J. Biomed. Sci. 2021, 28, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Mandelli, L.; Wang, S.M.; Han, C.; Lee, S.J.; Patkar, A.A.; Masand, P.S.; Pae, C.U.; Serretti, A. The Impact of a Single Nucleotide Polymorphism in SIGMAR1 on Depressive Symptoms in Major Depressive Disorder and Bipolar Disorder. Adv. Ther. 2017, 34, 713–724. [Google Scholar] [CrossRef]
- Jia, J.; Cheng, J.; Wang, C.; Zhen, X. Sigma-1 Receptor-Modulated Neuroinflammation in Neurological Diseases. Front. Cell. Neurosci. 2018, 12, 314. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Hall, N.; Mould, A.; Al-Juffali, N.; Tunbridge, E.M. Cellular calcium in bipolar disorder: Systematic review and meta-analysis. Mol. Psychiatry 2021, 26, 4106–4116. [Google Scholar] [CrossRef] [PubMed]
- Saravanaraman, P.; Chinnadurai, R.K.; Boopathy, R. Why calcium channel blockers could be an elite choice in the treatment of Alzheimer’s disease: A comprehensive review of evidences. Rev. Neurosci. 2014, 25, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lange, F.; Xia, Y.; Chertavian, C.; Cabolis, K.; Sajic, M.; Werring, D.J.; Tachtsidis, I.; Smith, K.J. Nimodipine Protects Vascular and Cognitive Function in an Animal Model of Cerebral Small Vessel Disease. Stroke 2024, 55, 1914–1922. [Google Scholar] [CrossRef]
- Frank, R.; Pesti, I.; Menyhárt, Á.; Szarvas, P.; Gulya, K.; Bari, F.; Farkas, E. Nimodipine exerts a protective effect against spreading depolarization and neuroinflammation. Physiology 2023, 38, 5762940. [Google Scholar] [CrossRef]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef]
- Huang, B.R.; Chang, P.C.; Yeh, W.L.; Lee, C.H.; Tsai, C.F.; Lin, C.; Lin, H.Y.; Liu, Y.S.; Wu, C.Y.J.; Ko, P.Y.; et al. Anti-Neuroinflammatory Effects of the Calcium Channel Blocker Nicardipine on Microglial Cells: Implications for Neuroprotection. PLoS ONE 2014, 9, e91167. [Google Scholar] [CrossRef]
- Sadeghi, M.; Saber, H.; Singh, A.; Hanni, C.; Parker, D.; Desai, A.; Mohamed, W. Nicardipine Associated Risk of Short-Term Mortality in Critically Ill Patients with Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2019, 28, 1168–1172. [Google Scholar] [CrossRef]
- Vahdani, B.; Kian, A.A.; Esmaeilzadeh, A.; Zenoozian, S.; Yousefi, V.; Mazloomzadeh, S. Adjunctive Raloxifene and Isradipine Improve Cognitive Functioning in Patients With Schizophrenia. J. Clin. Psychopharmacol. 2020, 40, 457–463. [Google Scholar] [CrossRef]
- Lovell, M.A.; Fister, S.X.; Lynn, B.C. P3–281: Inhibition of β-amyloid production by nifedipine. Alzheimer’s Dement. 2010, 6, S534. [Google Scholar] [CrossRef]
- Popović, N.; Morales-Delgado, N.; Vidal Mena, D.; Alonso, A.; Pascual Martínez, M.; Caballero Bleda, M.; Popović, M. Verapamil and Alzheimer’s Disease: Past, Present, and Future. Front. Pharmacol. 2020, 11, 535393. [Google Scholar] [CrossRef]
- Mosalam, E.M.; Elberri, A.I.; Sallam, A.S.; Salem, H.R.; Metwally, E.M.; Abdallah, M.S.; Shaldam, M.A.; Mansour, H.E.A. Chronotherapeutic neuroprotective effect of verapamil against lipopolysaccharide-induced neuroinflammation in mice through modulation of calcium-dependent genes. Mol. Med. 2022, 28, 139. [Google Scholar] [CrossRef] [PubMed]
- Boboc, I.K.S.; Cojocaru, A.; Nedelea, G.; Catalin, B.; Bogdan, M.; Calina, D. Chronic Administration of Ion Channel Blockers Impact Microglia Morphology and Function in a Murine Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 14474. [Google Scholar] [CrossRef] [PubMed]
- Brand-Schieber, E.; Werner, P. Calcium channel blockers ameliorate disease in a mouse model of multiple sclerosis. Exp. Neurol. 2004, 189, 5–9. [Google Scholar] [CrossRef]
- Kerkhofs, D.; Helgers, R.; Hermes, D.; Steinbusch, H.P.; Van Essen, H.; Leenders, P.; Prickaerts, J.; Staals, J.; Biessen, E.A.; Van Oostenbrugge, R.J.; et al. Amlodipine limits microglia activation and cognitive dysfunction in aged hypertensive mice. J. Hypertens. 2023, 41, 1159–1167. [Google Scholar] [CrossRef]
- Kalar, I.; Xu, H.; Secnik, J.; Schwertner, E.; Kramberger, M.G.; Winblad, B.; von Euler, M.; Eriksdotter, M.; Garcia-Ptacek, S. Calcium channel blockers, survival and ischaemic stroke in patients with dementia: A Swedish registry study. J. Intern. Med. 2021, 289, 508–522. [Google Scholar] [CrossRef]
- Alluri, R.; Kilari, E.K.; Pasala, P.K.; Kopalli, S.R.; Koppula, S. Repurposing Diltiazem for Its Neuroprotective Anti-Dementia Role against Intra-Cerebroventricular Streptozotocin-Induced Sporadic Alzheimer’s Disease-Type Rat Model. Life 2023, 13, 1688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Li, D.; Zhang, C.; Liu, M. Calcium antagonists for acute ischemic stroke. Cochrane Database Syst. Rev. 2019, 2019, CD001928. [Google Scholar] [CrossRef]
- Colbourne, L.; Harrison, P.J. Brain-penetrant calcium channel blockers are associated with a reduced incidence of neuropsychiatric disorders. Mol. Psychiatry 2022, 27, 3904–3912. [Google Scholar] [CrossRef]
- Mitterreiter, S.; Page, R.M.; Kamp, F.; Hopson, J.; Winkler, E.; Ha, H.R.; Hamid, R.; Herms, J.; Mayer, T.U.; Nelson, D.J.; et al. Bepridil and Amiodarone Simultaneously Target the Alzheimer’s Disease-and-Secretase via Distinct Mechanisms. J. Neurosci. 2010, 30, 8974–8983. [Google Scholar] [CrossRef]
- Lintunen, J.; Lähteenvuo, M.; Tanskanen, A.; Tiihonen, J.; Taipale, H. Allopurinol, dipyridamole and calcium channel blockers in the treatment of bipolar disorder—A nationwide cohort study. J. Affect. Disord. 2022, 313, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Chavali, V.D.; Agarwal, M.; Vyas, V.K.; Saxena, B. Neuroprotective Effects of Ethyl Pyruvate against Aluminum Chloride-Induced Alzheimer’s Disease in Rats via Inhibiting Toll-Like Receptor 4. J. Mol. Neurosci. 2020, 70, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Kim, I.D.; Kim, S.W.; Lee, H.K.; Jin, Y.; Park, J.H.; Kim, T.K.; Suh, C.K.; Kwak, J.; Lee, K.H.; et al. Ethyl Pyruvate Inhibits HMGB1 Phosphorylation and Release by Chelating Calcium. Mol. Med. 2014, 20, 649–657. [Google Scholar] [CrossRef]
- Olcum, M.; Tufekci, K.U.; Durur, D.Y.; Tastan, B.; Gokbayrak, I.N.; Genc, K.; Genc, S. Ethyl Pyruvate Attenuates Microglial NLRP3 Inflammasome Activation via Inhibition of HMGB1/NF-κB/miR-223 Signaling. Antioxidants 2021, 10, 745. [Google Scholar] [CrossRef]
- Gao, H.-M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.-S. HMGB1 Acts on Microglia Mac1 to Mediate Chronic Neuroinflammation That Drives Progressive Neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Pena-Altamira, E.; Prati, F.; Massenzio, F.; Virgili, M.; Contestabile, A.; Bolognesi, M.L.; Monti, B. Changing paradigm to target microglia in neurodegenerative diseases: From anti-inflammatory strategy to active immunomodulation. Expert. Opin. Ther. Targets 2016, 20, 627–640. [Google Scholar] [CrossRef]
- Stopschinski, B.E.; Weideman, R.A.; McMahan, D.; Jacob, D.A.; Little, B.B.; Chiang, H.S.; Saez Calveras, N.; Stuve, O. Microglia as a cellular target of diclofenac therapy in Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2023, 16, 17562864231156674. [Google Scholar] [CrossRef]
- Singh, A.; Tripathi, P.; Singh, S. Neuroinflammatory responses in Parkinson’s disease: Relevance of Ibuprofen in therapeutics. Inflammopharmacology 2021, 29, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Zeh, H.J.; Lotze, M.T.; Tang, D. HMGB1 in Cancer: Good, Bad, or Both? Clin. Cancer Res. 2023, 19, 4046. [Google Scholar] [CrossRef]
- Georgiou, C.D.; Margaritis, L.H. Oxidative Stress and NADPH Oxidase: Connecting Electromagnetic Fields, Cation Channels and Biological Effects. Int. J. Mol. Sci. 2021, 22, 10041. [Google Scholar] [CrossRef]
- Anjum, F.; Zahra, A.; Alvi, T.N.; Singha, S.P.; Derick, A.; Qazi, A.F. The role of oxidative stress in neurodegenerative diseases: Mechanisms and therapeutic implications. J. Popul. Ther. Clin. Pharmacol. 2024, 31, 238–246. [Google Scholar] [CrossRef]
- Houldsworth, A. Role of oxidative stress in neurodegenerative disorders: A review of reactive oxygen species and prevention by antioxidants. Brain Commun. 2024, 6, fcad356. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Bhattacharya, N.; Mishra, S.; Bhattacharya, A.; Banerjee, P.; Senapati, S.; Mishra, R. Microglia Specific Drug Targeting Using Natural Products for the Regulation of Redox Imbalance in Neurodegeneration. Front. Pharmacol. 2021, 12, 654489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ren, W.; Zhao, F.; Han, Y.; Liu, C.; Jia, K. Curcumin amends Ca2+ dysregulation in microglia by suppressing the activation of P2X7 receptor. Mol. Cell. Biochem. 2020, 465, 65–73. [Google Scholar] [CrossRef]
- Tang, M.; Taghibiglou, C. The Mechanisms of Action of Curcumin in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1003–1016. [Google Scholar] [CrossRef]
- Tufekci, K.U.; Eltutan, B.I.; Isci, K.B.; Genc, S. Resveratrol Inhibits NLRP3 Inflammasome-Induced Pyroptosis and miR-155 Expression in Microglia Through Sirt1/AMPK Pathway. Neurotox. Res. 2021, 39, 1812–1829. [Google Scholar] [CrossRef]
- Zeini, S.; Davoodian, N.; Kazemi, H.; Brojeni, M.S.; Ghani, E.; Firouzjaei, M.A.; Atashabparvar, A. Resveratrol prevents cognitive impairment and hippocampal inflammatory response induced by lipopolysaccharide in a mouse model of chronic neuroinflammation. Physiol. Behav. 2024, 278, 114508. [Google Scholar] [CrossRef]
- Yang, S.; Du, Y.; Zhao, X.; Tang, Q.; Su, W.; Hu, Y.; Yu, P. Cannabidiol Enhances Microglial Beta-Amyloid Peptide Phagocytosis and Clearance via Vanilloid Family Type 2 Channel Activation. Int. J. Mol. Sci. 2022, 23, 5367. [Google Scholar] [CrossRef]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.H.; Kendall, D.A. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef]
- Madhi, I.; Kim, J.H.; Shin, J.E.; Kim, Y. Ginsenoside Re exhibits neuroprotective effects by inhibiting neuroinflammation via CAMK/MAPK/NF-κB signaling in microglia. Mol. Med. Rep. 2021, 24, 698. [Google Scholar] [CrossRef] [PubMed]
- Minocha, T.; Birla, H.; Obaid, A.A.; Rai, V.; Sushma, P.; Shivamallu, C.; Moustafa, M.; Al-Shehri, M.; Al-Emam, A.; Tikhonova, M.A.; et al. Flavonoids as Promising Neuroprotectants and Their Therapeutic Potential against Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2022, 2022, 6038996. [Google Scholar] [CrossRef] [PubMed]
- Cornell, J.; Salinas, S.; Huang, H.-Y.; Zhou, M. Microglia regulation of synaptic plasticity and learning and memory. Neural Regen. Res. 2022, 17, 705. [Google Scholar] [CrossRef]
- León-Rodríguez, A.; Fernández-Arjona, M.d.M.; Grondona, J.M.; Pedraza, C.; López-Ávalos, M.D. Anxiety-like behavior and microglial activation in the amygdala after acute neuroinflammation induced by microbial neuraminidase. Sci. Rep. 2022, 12, 11581. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, G.; Mango, D.; Saidi, A.; Corbo, M.; Nisticò, R. Targeting Microglia-Synapse Interactions in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2342. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 87. [Google Scholar] [CrossRef]
- Qu, W.; Li, L. Microglial TREM2 at the intersection of brain aging and Alzheimer’s disease. Neuroscientist 2023, 29, 302. [Google Scholar] [CrossRef]
- Hu, B.; Duan, S.; Wang, Z.; Li, X.; Zhou, Y.; Zhang, X.; Zhang, Y.W.; Xu, H.; Zheng, H. Insights Into the Role of CSF1R in the Central Nervous System and Neurological Disorders. Front. Aging Neurosci. 2021, 13, 789834. [Google Scholar] [CrossRef]
- Young, A.P.; Denovan-Wright, E.M. The Dynamic Role of Microglia and the Endocannabinoid System in Neuroinflammation. Front. Pharmacol. 2022, 12, 806417. [Google Scholar] [CrossRef]
- Saw, G.; Krishna, K.; Gupta, N.; Soong, T.W.; Mallilankaraman, K.; Sajikumar, S.; Dheen, S.T.S. Epigenetic regulation of microglial phosphatidylinositol 3-kinase pathway involved in long-term potentiation and synaptic plasticity in rats. Glia 2020, 68, 656. [Google Scholar] [CrossRef]
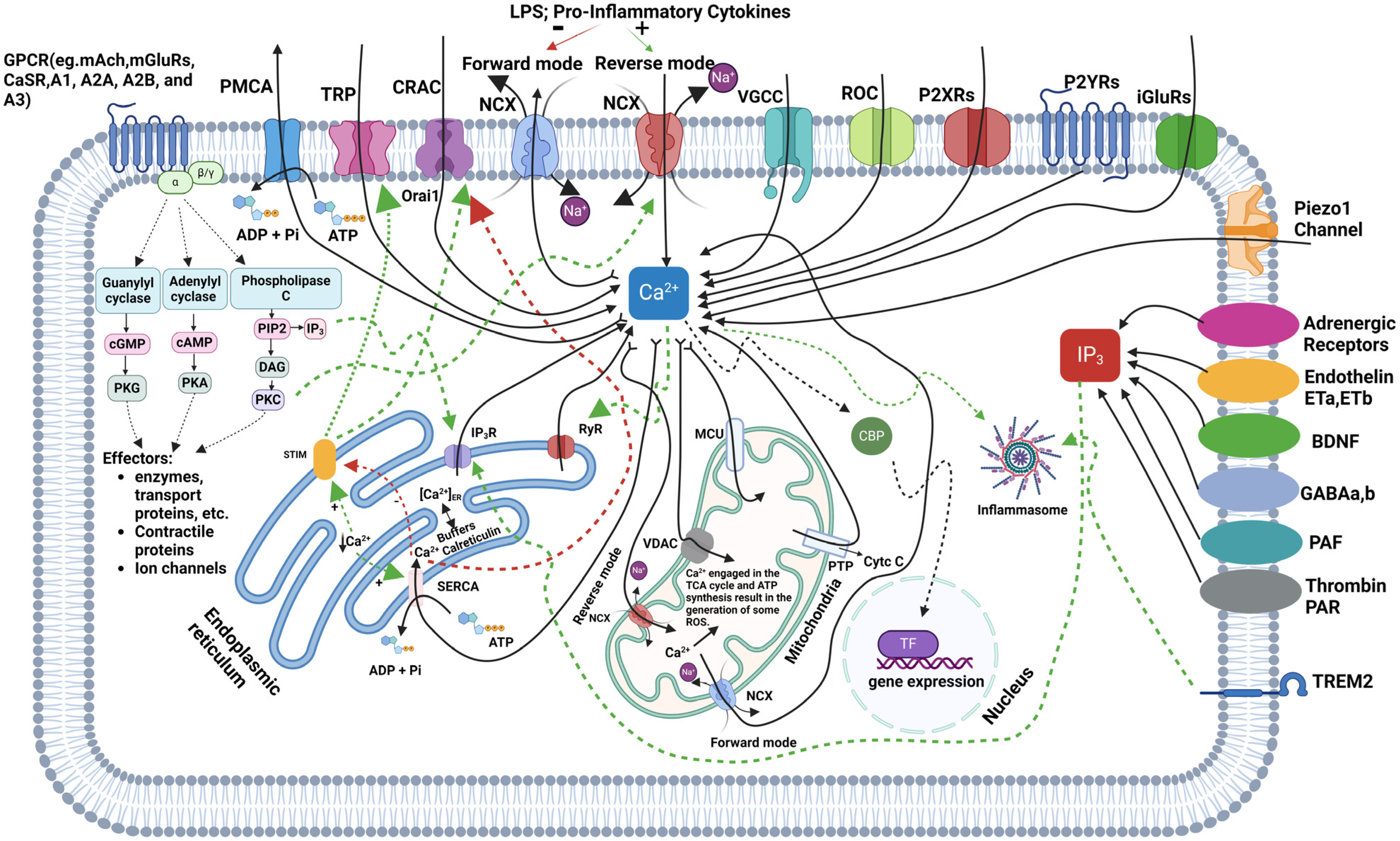
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).