
Single and Binary Fe- and Al-hydroxides Affect Potential Phosphorus Mobilization and Transfer from Pools of Different Availability

Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of the Fe- and Al-Hydroxides
2.2. Characterization of the Fe- and Al-Hydroxides
2.3. Desorption Experiments
2.4. Kinetics of P Desorption
3. Results
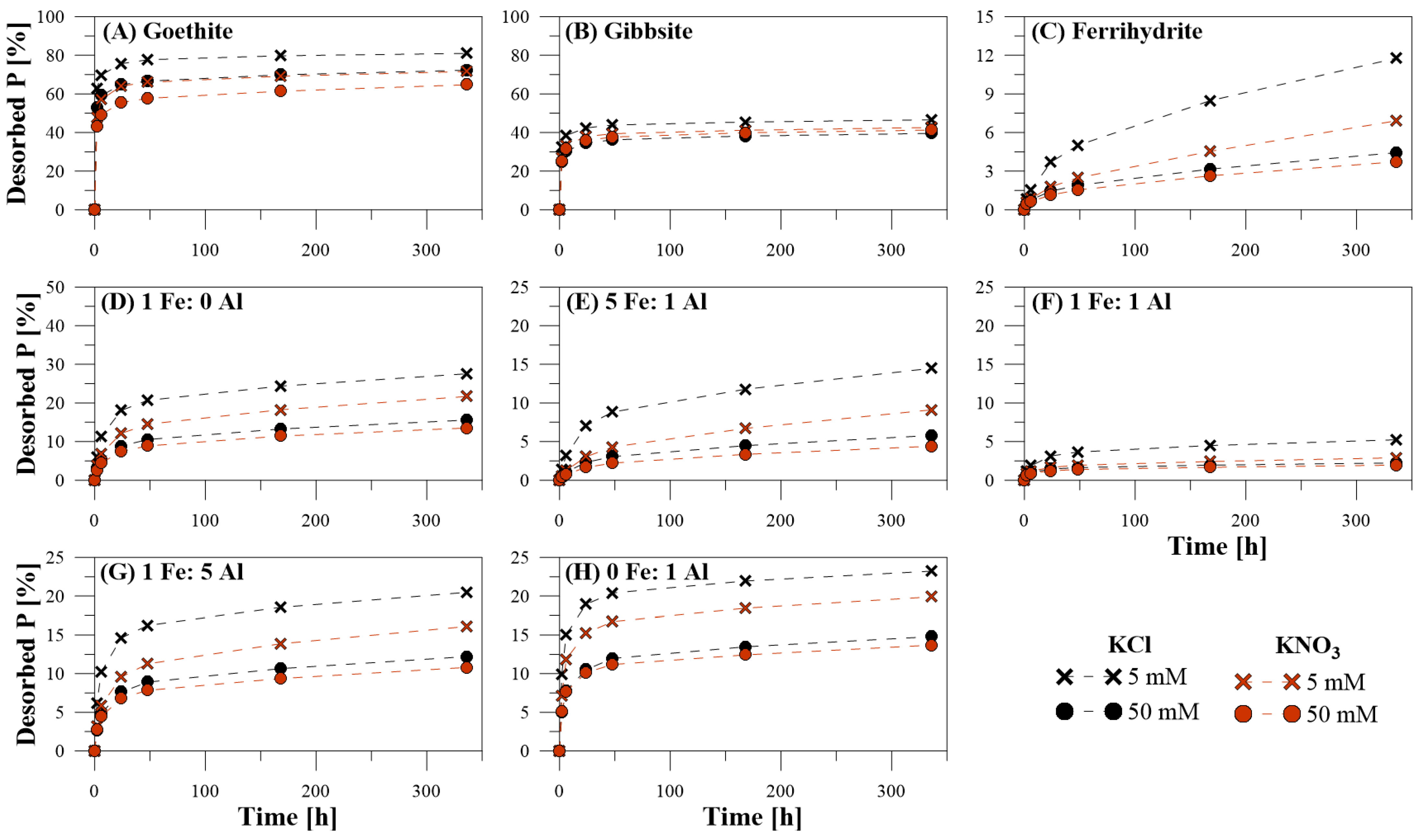
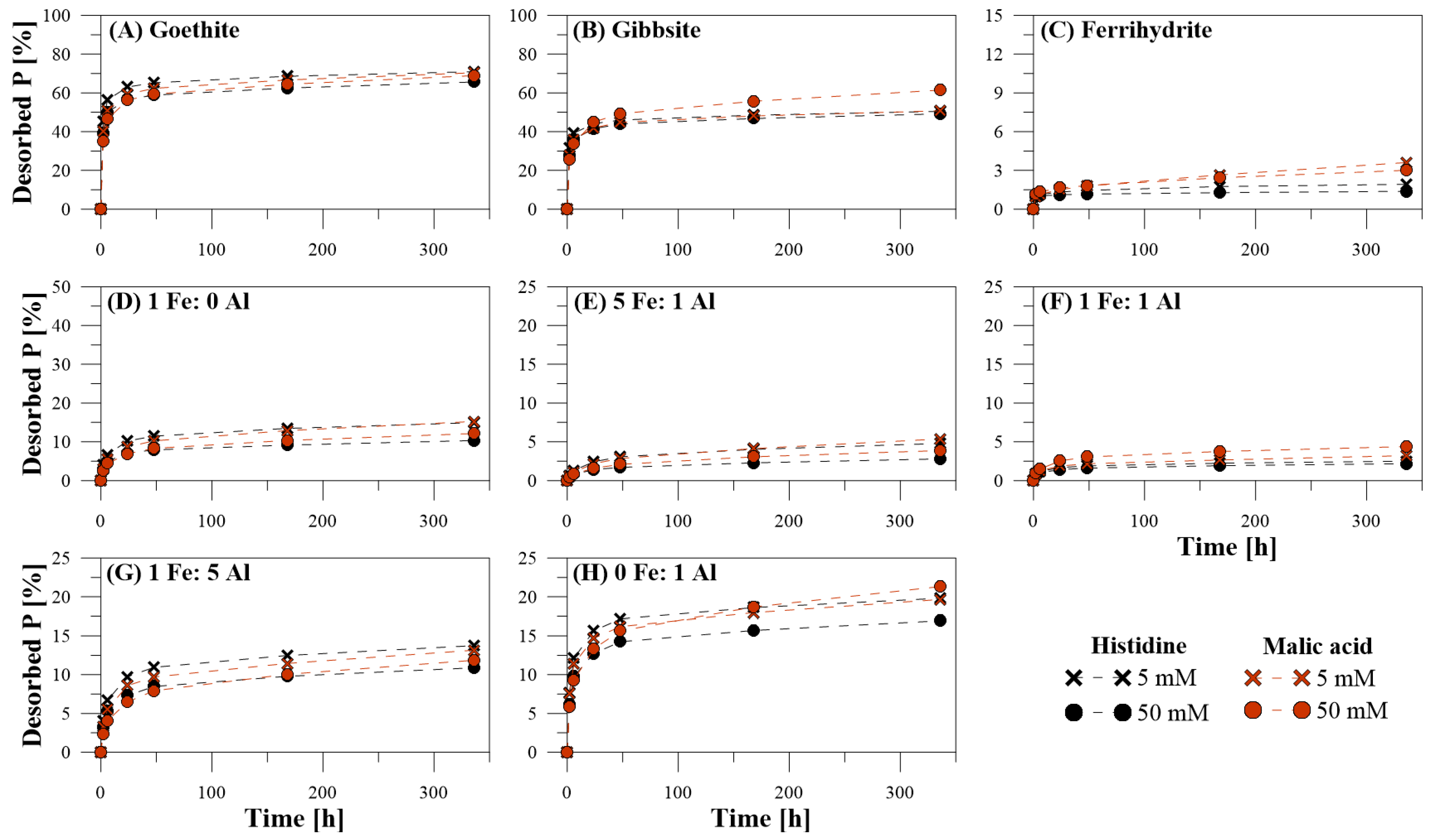
3.1. Efficiency of P Desorption
3.2. Kinetics of P Desorption
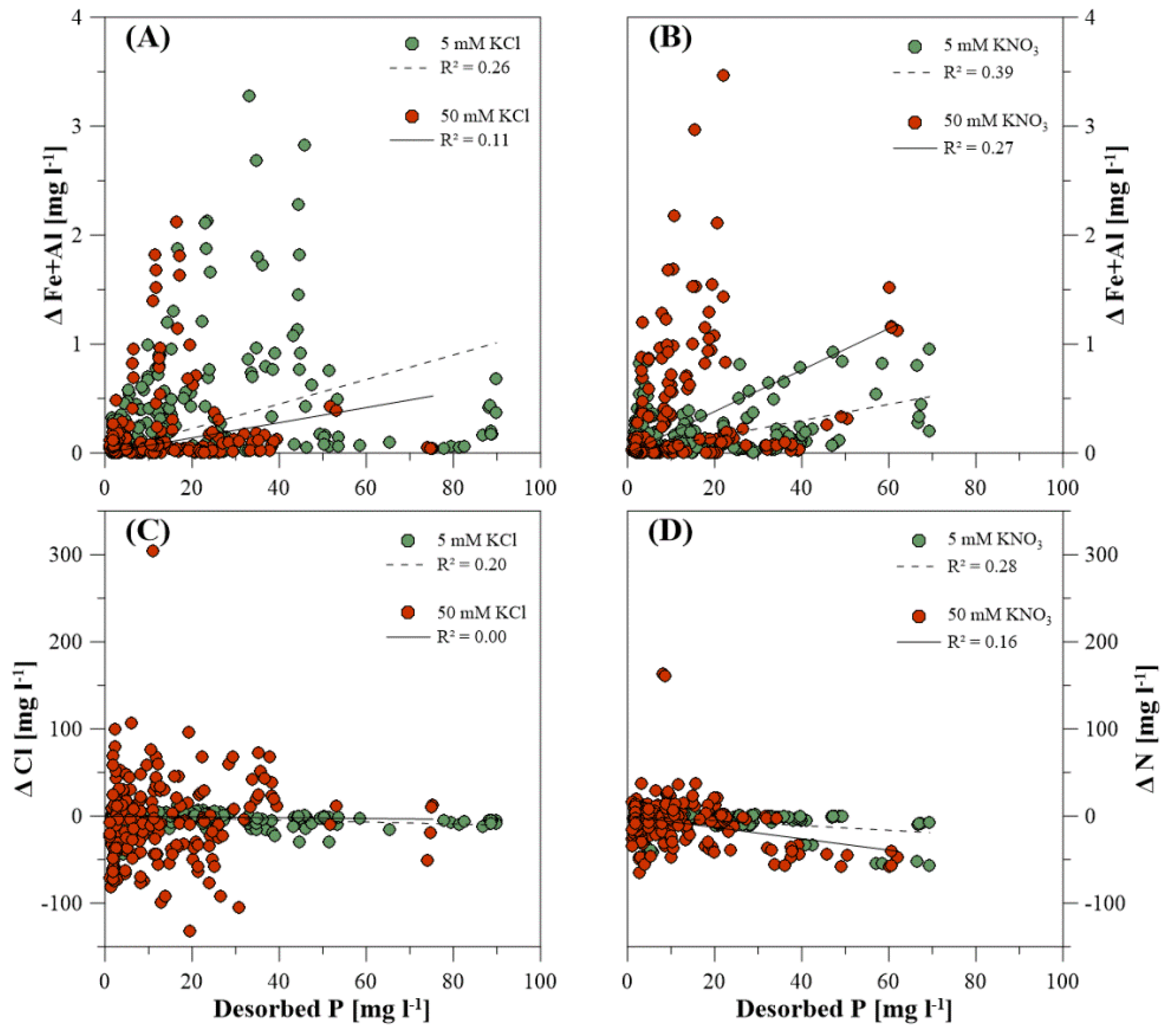
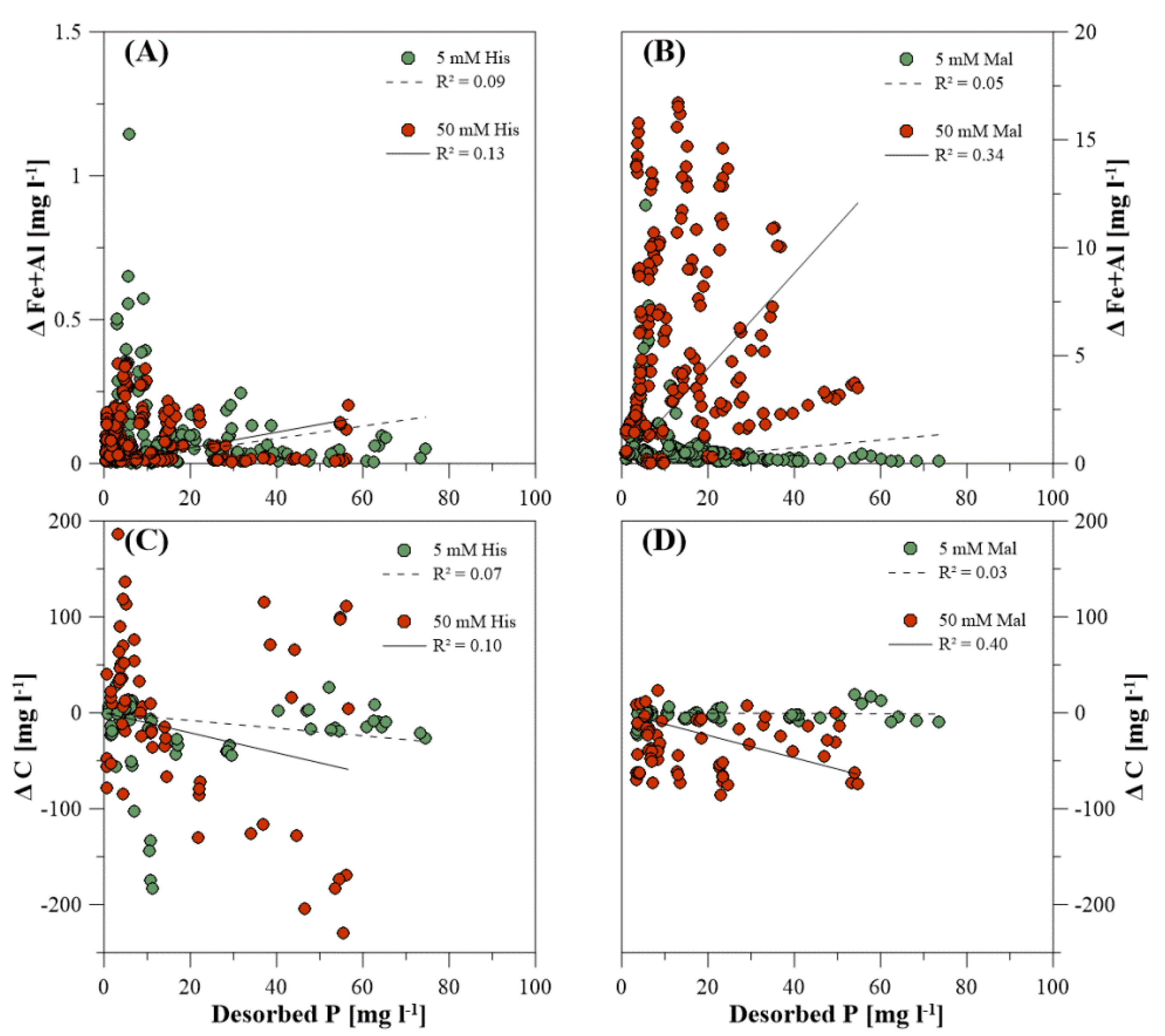
3.3. Dissolved Elemental Composition during P Desorption
3.4. Solution pH
4. Discussion
4.1. Influence of Crystallinity and Fe/Al Content on P Sorption
4.2. Effect of PZC and Electrolytes on P Adsorption
4.3. Desorption Kinetics
4.4. Inorganic Extracting Agents
4.5. Organic Extracting Agents
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kruse, J.; Abraham, M.; Amelung, W.; Baum, C.; Bol, R.; Kühn, O.; Lewandowski, H.; Niederberger, J.; Oelmann, Y.; Rüger, C.; et al. Innovative methods in soil phosphorus research: A review. J. Plant Nutr. Soil Sci. 2015, 178, 43–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celi, L.; Prati, M.; Magnacca, G.; Santoro, V.; Martin, M. Role of crystalline iron oxides on stabilization of inositol phosphates in soil. Geoderma 2020, 374, 114442. [Google Scholar] [CrossRef]
- Jiang, X.; Bol, R.; Willbold, S.; Vereecken, H.; Klumpp, E. Speciation and distribution of P associated with Fe and Al oxides in aggregate-sized fraction of an arable soil. Biogeosciences 2015, 12, 6443–6452. [Google Scholar] [CrossRef] [Green Version]
- Weihrauch, C. Phosphor-Dynamiken in Böden. Grundlagen, Konzepte und Untersuchungen zur räumlichen Verteilung des Nährstoffs; Springer Spektrum: Wiesbaden, Germany, 2018. [Google Scholar]
- Weng, Y.; Vekeman, J.; Zhang, H.; Chou, L.; Elskens, M.; Tielens, F. Unravelling phosphate adsorption on hydrous ferric oxide surfaces at the molecular level. Chemosphere 2020, 261, 127776. [Google Scholar] [CrossRef] [PubMed]
- Blume, H.-P.; Brümmer, G.W.; Horn, R.; Kandeler, E.; Kögel-Knabner, I.; Kretzschmar, R.; Stahr, K.; Wilke, B.-M. Scheffer/Schachtschabel Lehrbuch der Bodenkunde, 16th ed.; Springer Spektrum: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Mnthambala, F.; Maida, J.; Lowole, M.W.; Kabambe, V.H. Phosphorus sorption and external phosphorus requirements of ultisols and oxisols in Malawi. J. Soil Sci. Environ. Manag. 2015, 6, 35–41. [Google Scholar]
- Reddy, K.R.; DeLaune, R.D. Biogeochemistry of Wetlands: Science and Applications; CRC Press Taylor & Francis Group: Boca Raton, FL, USA; London, UK; New York, NY, USA, 2008. [Google Scholar]
- Lang, F.; Bauhus, J.; Frossard, E.; George, E.; Kaiser, K.; Kaupenjohann, M.; Krüger, J.; Matzner, E.; Polle, A.; Prietzel, J.; et al. Phosphorus in forest ecosystems: New insights from an ecosystem nutrition perspective. J. Plant Nutr. Soil Sci. 2016, 179, 129–135. [Google Scholar] [CrossRef]
- Li, L.; Stanforth, R. Distinguishing adsorption and surface precipitation of phosphate on goethite (alpha-FeOOH). J. Colloid Interf. Sci. 2000, 230, 12–21. [Google Scholar] [CrossRef]
- Gypser, S.; Schütze, E.; Freese, D. Crystallization of single and binary iron- and aluminum hydroxides affect phosphorus desorption. J. Plant Nutr. Soil Sci. 2019, 182, 741–750. [Google Scholar] [CrossRef]
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef] [Green Version]
- Basak, B.B. Phosphorus release by low molecular weight organic acids from low-grade indian rock phosphate. Waste Biomass Valor. 2019, 10, 3225–3233. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Comerford, N.B.; Johnston, C.T. Influence of oxalate and soil organic matter on sorption and desorption of phosphate onto a spodic horizon. Soil Sci. Soc. Am. J. 1998, 62, 1089–1095. [Google Scholar] [CrossRef]
- Borggaard, O.K.; Raben-Lange, B.; Gimsing, A.L.; Strobel, B.W. Influence of humic substances on phosphate adsorption by aluminium and iron oxides. Geoderma 2005, 127, 270–279. [Google Scholar] [CrossRef]
- Hinsinger, P. Bioavailability of soil inorganic P in the rhizosphere as affected by root-induced chemical changes: A review. Plant Soil 2001, 237, 173–195. [Google Scholar] [CrossRef]
- Johnson, S.E.; Loeppert, R.H. Role of organic acids in phosphate mobilization from iron oxide. Soil Sci. Soc. Am. J. 2006, 70, 222–234. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.L.; Johnston, A.E. Phosphorus use efficiency and management in agriculture. Resour. Conserv. Recycl. 2015, 105, 275–281. [Google Scholar] [CrossRef]
- Buczko, U.; van Laak, M.; Eichler-Löbermann, B.; Gans, W.; Merbach, I.; Panten, K.; Peiter, E.; Reitz, T.; Spiegel, H.; von Tucher, S. Re-evaluation of the yield response to phosphorus fertilization based on meta-analyses of long-term field experiments. Ambio 2018, 47, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syers, J.K.; Johnston, A.E.; Curtin, D. Efficiency of Soil and Fertilizer Phosphorus Use: Reconciling Changing Concepts of Soil Phosphorus Behaviour with Agronomic Information; FAO Fertilizer and Plant Nutrition No. 18; Food and Agriculture Organization of the United Nations: Rome, Italy, 2008. [Google Scholar]
- van der Bom, F.; McLaren, T.I.; Doolette, A.L.; Magid, J.; Frossard, E.; Oberson, A.; Jensen, L.S. Influence of long-term phosphorus fertilisation history on the availability and chemical nature of soil phosphorus. Geoderma 2019, 355, 113909. [Google Scholar] [CrossRef]
- Antelo, J.; Fiol, S.; Pérez, C.; Mariño, S.; Arce, F.; Gondar, D.; López, R. Analysis of phosphate adsorption onto ferrihydrite using the CD-MUSIC model. J. Colloid Interf. Sci. 2010, 347, 112–119. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Gypser, S.; Leinweber, P.; Freese, D.; Kühn, O. Infrared spectroscopic characterization of phosphate binding at the goethite-water interface. Phys. Chem. Chem. Phys. 2019, 21, 4421–4434. [Google Scholar] [CrossRef] [Green Version]
- Gypser, S.; Hirsch, F.; Schleicher, A.M.; Freese, D. Impact of crystalline and amorphous iron- and aluminum hydroxides on mechanisms of phosphate adsorption and desorption. J. Environ. Sci. 2018, 70, 175–189. [Google Scholar] [CrossRef]
- Khare, N.; Martin, J.D.; Hesterberg, D. Phosphate bonding configuration on ferrihydrite based on molecular orbital calculations and XANES fingerprinting. Geochim. Cosmochim. Acta 2007, 71, 4405–4415. [Google Scholar] [CrossRef]
- Krumina, L.; Kenney, J.P.; Loring, J.S.; Persson, P. Desorption mechanisms of phosphate from ferrihydrite and goethite surfaces. Chem. Geol. 2016, 427, 54–64. [Google Scholar] [CrossRef]
- Kubicki, J.D.; Kwon, K.D.; Paul, K.W.; Sparks, D.L. Surface complex structures modelled with quantum chemical calculations: Carbonate, phosphate, sulphate, arsenate and arsenite. Eur. J. Soil Sci. 2007, 58, 932–944. [Google Scholar] [CrossRef]
- van Emmerik, T.J.; Sandström, D.E.; Antzutkin, O.N.; Angove, M.J.; Johnson, B.B. 31P solid-state nuclear magnetic resonance study of the sorption of phosphate onto gibbsite and kaolinite. Langmuir 2007, 23, 3205–3213. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.-T.; Sun, Z.-X.; Yang, X.-F.; Holmgren, A. Sorption of phosphate onto mesoporous γ-alumina studied with in-situ ATR-FTIR spectroscopy. Chem. Cent. J. 2012, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Sparks, D.L. ATR–FTIR spectroscopic investigation on phosphate adsorption mechanisms at the ferrihydrite–water interface. J. Colloid Interf. Sci. 2001, 241, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Tiessen, H.; Moir, J.O. Characterization of available P by sequential extraction. In Soil Sampling and Methods of Analysis; Carter, M.R., Gregorich, E.G., Eds.; CRC Press: Boca Raton, FL, USA, 2007; ISBN 9780429126222. [Google Scholar]
- Schwertmann, U.; Cornell, R.M. Iron Oxides in the Laboratory: Preparation and Characterization; Wiley-VCH: Hoboken, NJ, USA, 2008. [Google Scholar]
- Sujana, M.G.; Soma, G.; Vasumathi, N.; Anand, S. Studies on fluoride adsorption capacities of amorphous Fe/Al mixed hydroxides from aqueous solutions. J. Fluor. Chem. 2009, 130, 749–754. [Google Scholar] [CrossRef]
- Harvey, O.R.; Rhue, R.D. Kinetics and energetics of phosphate sorption in a multi-component Al(III)-Fe(III) hydr(oxide) sorbent system. J. Colloid Interf. Sci. 2008, 322, 384–393. [Google Scholar] [CrossRef]
- Johnson, B.B.; Ivanov, A.V.; Antzutkin, O.N.; Forsling, W. 31P nuclear magnetic resonance study of the adsorption of phosphate and phenyl phosphates on γ-Al2O3. Langmuir 2002, 18, 1104–1111. [Google Scholar] [CrossRef]
- Persson, P.; Nilsson, N.; Sjöberg, S. Structure and bonding of orthophosphate ions at the iron oxide-aqueous interface. J. Colloid Interf. Sci. 1996, 177, 263–275. [Google Scholar] [CrossRef]
- Loring, J.S.; Sandström, M.H.; Norén, K.; Persson, P. Rethinking arsenate coordination at the surface of goethite. Chemistry 2009, 15, 5063–5072. [Google Scholar] [CrossRef]
- Luengo, C.; Brigante, M.; Antelo, J.; Avena, M. Kinetics of phosphate adsorption on goethite: Comparing batch adsorption and ATR-IR measurements. J. Colloid Interf. Sci. 2006, 300, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Torrent, J. Fast and slow phosphate sorption by goethite-rich natural materials. Clay. Clay. Miner. 1992, 40, 14–21. [Google Scholar] [CrossRef]
- Lǚ, J.; Liu, H.; Liu, R.; Zhao, X.; Sun, L.; Qu, J. Adsorptive removal of phosphate by a nanostructured Fe–Al–Mn trimetal oxide adsorbent. Powder Technol. 2013, 233, 146–154. [Google Scholar] [CrossRef]
- Arai, Y.; Livi, K.J. Underassessed phosphorus fixation mechanisms in soil sand fraction. Geoderma 2013, 192, 422–429. [Google Scholar] [CrossRef]
- Antelo, J.; Avena, M.; Fiol, S.; López, R.; Arce, F. Effects of pH and ionic strength on the adsorption of phosphate and arsenate at the goethite-water interface. J. Colloid Interf. Sci. 2005, 285, 476–486. [Google Scholar] [CrossRef]
- Strauss, R.; Brümmer, G.W.; Barrow, N.J. Effects of crystallinity of goethite: II. Rates of sorption and desorption of phosphate. Eur. J. Soil Sci. 1997, 48, 101–114. [Google Scholar] [CrossRef]
- Adekola, F.; Fédoroff, M.; Geckeis, H.; Kupcik, T.; Lefèvre, G.; Lützenkirchen, J.; Plaschke, M.; Preocanin, T.; Rabung, T.; Schild, D. Characterization of acid-base properties of two gibbsite samples in the context of literature results. J. Colloid Interf. Sci. 2011, 354, 306–317. [Google Scholar] [CrossRef]
- Jodin, M.-C.; Gaboriaud, F.; Humbert, B. Limitations of potentiometric studies to determine the surface charge of gibbsite gamma-Al(OH)3 particles. J. Colloid Interf. Sci. 2005, 287, 581–591. [Google Scholar] [CrossRef]
- Kosmulski, M. Compilation of PZC and IEP of sparingly soluble metal oxides and hydroxides from literature. Adv. Colloid Interf. Sci. 2009, 152, 14–25. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Lin, C.-L.; Miller, J.D. Significance of particle aggregation in the reverse flotation of kaolinite from bauxite ore. Miner. Eng. 2015, 78, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides. Structure, Properties, Reactions, Occurences and Uses, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Hiemstra, T.; van Riemsdijk, W.H. A surface structural approach to ion adsorption: The charge distribution (CD) model. J. Colloid Interf. Sci. 1996, 179, 488–508. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Gypser, S.; Freese, D.; Leinweber, P.; Kühn, O. Molecular level picture of the interplay between pH and phosphate binding at the goethite-water interface. Phys. Chem. Chem. Phys. 2020, 22, 26509–26524. [Google Scholar] [CrossRef] [PubMed]
- Rietra, R.P.; Hiemstra, T.; van Riemsdijk, W.H. Interaction between calcium and phosphate adsorption on goethite. Environ. Sci. Technol. 2001, 35, 3369–3374. [Google Scholar] [CrossRef]
- Wang, X.; Phillips, B.; Boily, J.-F.; Hu, Y.; Hu, Z.; Yang, P.; Feng, X.; Xu, W.; Zhu, M. Phosphate sorption speciation and precipitation mechanisms on amorphous aluminum hydroxide. Soil Syst. 2019, 3, 20. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, X.; Whalen, J.K.; Cao, Y.; Quan, Z.; Lu, C.; Shi, Y. Kinetics of inorganic and organic phosphorus release influenced by low molecular weight organic acids in calcareous, neutral and acidic soils. J. Plant Nutr. Soil Sci. 2015, 178, 555–566. [Google Scholar] [CrossRef]
- Onishi, B.S.D.; dos Reis Ferreira, C.S.; Urbano, A.; Santos, M.J. Modified hydrotalcite for phosphorus slow-release: Kinetic and sorption-desorption processes in clayey and sandy soils from North of Paraná state (Brazil). Appl. Clay Sci. 2020, 197, 105759. [Google Scholar] [CrossRef]
- Wang, X.; Liu, F.; Tan, W.; Li, W.; Feng, X.; Sparks, D.L. Characteristics of phosphate adsorption-desorption onto ferrihydrite. Soil Sci. 2013, 178, 1–11. [Google Scholar] [CrossRef]
- Shariatmadari, H.; Shirvani, M.; Jafari, A. Phosphorus release kinetics and availability in calcareous soils of selected arid and semiarid toposequences. Geoderma 2006, 132, 261–272. [Google Scholar] [CrossRef]
- Steffens, D. Phosphorus release kinetics and extractable phosphorus after long-term fertilization. Soil Sci. Soc. Am. J. 1994, 58, 1702. [Google Scholar] [CrossRef]
- Lammers, A. Phosphatformen und Phosphatfreisetzung in hochgedüngten Böden Europas; Dissertation, Utz: München, Germany, 1997. [Google Scholar]
- Arai, Y.; Sparks, D.L. Phosphate reaction dynamics in soils and soil components: A multiscale approach. Advances Agron. 2007, 94, 135–179. [Google Scholar]
- Gerke, J. Phosphate adsorption by humic/Fe-oxide mixtures aged at pH 4 and 7 and by poorly ordered Fe-oxide. Geoderma 1993, 59, 279–288. [Google Scholar] [CrossRef]
- Borggaard, O.K.; Jørgensen, S.S.; Moberg, J.P.; Raben-Lange, B. Influence of organic matter on phosphate adsorption by aluminium and iron oxides in sandy soils. Eur. J. Soil Sci. 1990, 41, 443–449. [Google Scholar] [CrossRef]
- Jones, D.L. Organic acids in the rhizosphere—A critical review. Plant Soil 1998, 205, 25–44. [Google Scholar] [CrossRef]
- Kpomblekou-A, K.; Tabatabai, M.A. Effect of low-molecular weight organic acids on phosphorus release and phytoavailabilty of phosphorus in phosphate rocks added to soils. Agric. Ecosyst. Environ. 2003, 100, 275–284. [Google Scholar] [CrossRef]
- Chen, C.R.; Condron, L.M.; Davis, M.R.; Sherlock, R.R. Effects of plant species on microbial biomass phosphorus and phosphatase activity in a range of grassland soils. Biol. Fertil. Soils 2004, 40, 313–322. [Google Scholar] [CrossRef]
- Xu, R.; Zhu, Y.; Chittleborough, D. Phosphorus release from phosphate rock and iron phosphate by low-molecular-weight organic acids. J. Environ. Sci. 2004, 16, 5–8. [Google Scholar]
- Taghipour, M.; Jalali, M. Effect of low-molecular-weight organic acids on kinetics release and fractionation of phosphorus in some calcareous soils of western Iran. Environ. Monit. Assess. 2013, 185, 5471–5482. [Google Scholar] [CrossRef]
- Zhu, J.; Li, M.; Whelan, M. Phosphorus activators contribute to legacy phosphorus availability in agricultural soils: A review. Sci. Total Environ. 2018, 612, 522–537. [Google Scholar] [CrossRef] [Green Version]
- Vinu, A.; Hossain, K.Z.; Satish Kumar, G.; Ariga, K. Adsorption of l-histidine over mesoporous carbon molecular sieves. Carbon 2006, 44, 530–536. [Google Scholar] [CrossRef]
- Lathrop, E.C. The organic nitrogen compounds of soils and fertilizers. J. Frankl. Inst. 1917, 303–321. [Google Scholar] [CrossRef]
- Mimmo, T.; Ghizzi, M.; Marzadori, C.; Gessa, C.E. Organic acid extraction from rhizosphere soil: Effect of field-moist, dried and frozen samples. Plant Soil 2008, 312, 175–184. [Google Scholar] [CrossRef]
- Schütze, E.; Gypser, S.; Freese, D. Kinetics of phosphorus release from vivianite, hydroxyapatite, and bone char influenced by organic and inorganic compounds. Soil Syst. 2020, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Talebi Atouei, M.; Rahnemaie, R.; Goli Kalanpa, E.; Davoodi, M.H. Competitive adsorption of magnesium and calcium with phosphate at the goethite water interface: Kinetics, equilibrium and CD-MUSIC modeling. Chem. Geol. 2016, 437, 19–29. [Google Scholar] [CrossRef]
- Duputel, M.; van Hoye, F.; Toucet, J.; Gérard, F. Citrate adsorption can decrease soluble phosphate concentration in soil: Experimental and modeling evidence. Appl. Geochem. 2013, 39, 85–92. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydroxide | PZC | SSA | Fe | Al | P | |
|---|---|---|---|---|---|---|
| pH | m² g−1 | mg g−1 | mg g−1 | mg g−1 | mg m−2 | |
| Goethite | 8.8 | 17.2 ± 0.4 | 564.17 ± 9.74 | - | 7.07 ± 0.24 | 0.41 ± 0.01 |
| Gibbsite | 8.5 | 0.9 ± 0.0 | - | 337.39 ± 6.05 | 7.39 ± 0.06 | 8.21 ± 0.06 |
| Ferrihydrite | 7.1 | 251.8 ± 2.7 | 520.99 ± 12.27 | - | 30.92 ± 2.63 | 0.12 ± 0.01 |
| 1 Fe: 0 Al | 6.0 | 297.3 ± 10.4 | 494.30 ± 25.04 | - | 36.50 ± 2.74 | 0.07 ± 0.09 |
| 5 Fe: 1 Al | 7.1 | 203.8 ± 0.9 | 424.59 ±2.51 | 32.10 ± 1.19 | 43.47 ± 5.23 | 0.21 ± 0.03 |
| 1 Fe: 1 Al | 7.6 | 73.7 ± 8.0 | 275.44 ± 1.13 | 137.22 ± 0.87 | 31.34 ± 0.69 | 0.43 ± 0.01 |
| 1 Fe: 5 Al | 9.8 | 0.8 ± 0.0 | 58.55 ± 0.65 | 162.27 ± 1.57 | 64.65 ± 0.64 | 80.82 ± 0.80 |
| 0 Fe: 1 Al | 9.8 | 1.1 ± 0.0 | - | 131.44 ± 2.69 | 44.27 ± 0.33 | 40.25 ± 0.30 |
| Kinetic Model | Linearized Equation | Declaration |
|---|---|---|
| Elovich | Qt—amount of desorbed P in mg P m−2 Hydroxide at time t α/a—initial P release constants in mg P m−2 Hydroxide min−1 β/b—P release rate constants in mg P m−2 Hydroxide min−1 kp—diffusion rate constant in m s−1 Q0—equals value of 0 at the beginning of desorption | |
| Exponential | ||
| Parabolic |
| Hydroxide | Desorbed P [%] | |||||||
|---|---|---|---|---|---|---|---|---|
| KCl | KNO3 | His | Mal | |||||
| 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | |
| Goethite | 80.95 ± 0.39 | 72.24 ± 0.40 | 71.58 ± 0.51 | 64.72 ± 0.33 | 70.94 ± 0.46 | 65.61 ± 0.40 | 70.38 ± 0.32 | 68.91 ± 0.81 |
| Gibbsite | 46.58 ± 2.96 | 39.53 ± 1.85 | 42.60 ± 2.12 | 41.26 ± 2.84 | 50.48 ± 3.62 | 49.17 ± 3.56 | 50.74 ± 2.51 | 61.43 ± 3.33 |
| Ferrihydrite | 11.77 ± 0.46 | 4.44 ± 0.22 | 6.91 ± 0.23 | 3.72 ± 0.11 | 1.92 ± 0.09 | 1.38 ± 0.21 | 3.61 ± 0.12 | 3.01 ± 0.29 |
| 1 Fe: 0 Al | 27.55 ± 0.23 | 15.57 ± 0.14 | 21.68 ± 0.17 | 13.52 ± 0.07 | 14.93 ± 0.19 | 10.33 ± 0.11 | 15.25 ± 0.13 | 12.14 ± 0.06 |
| 5 Fe: 1 Al | 14.49 ± 0.32 | 5.79 ± 0.13 | 9.11 ± 0.23 | 4.40 ± 0.08 | 4.78 ± 0.10 | 2.79 ± 0.15 | 5.36 ± 0.10 | 3.87 ± 0.13 |
| 1 Fe: 1 Al | 5.23 ± 0.29 | 2.24 ± 0.16 | 2.90 ± 0.20 | 1.95 ± 0.28 | 2.51 ± 0.15 | 2.18 ± 0.13 | 3.22 ± 0.27 | 4.38 ± 0.35 |
| 1 Fe: 5 Al | 20.48 ± 1.02 | 12.20 ± 0.22 | 16.10 ± 0.09 | 10.80 ± 0.12 | 13.75 ± 0.06 | 10.89 ± 0.30 | 13.16 ± 0.32 | 11.86 ± 0.52 |
| 0 Fe: 1 Al | 23.22 ± 0.39 | 14.79 ± 0.44 | 19.90 ± 0.54 | 13.69 ± 0.30 | 19.82 ± 0.35 | 16.94 ± 0.12 | 19.66 ± 0.44 | 21.33 ± 0.33 |
| Desorbed P [mg m−2] | ||||||||
| Goethite | 0.33 ± 0.00 | 0.30 ± 0.00 | 0.29 ± 0.00 | 0.27 ± 0.00 | 0.29 ± 0.00 | 0.27 ± 0.00 | 0.29 ± 0.00 | 0.28 ± 0.00 |
| Gibbsite | 3.96 ± 0.25 | 3.36 ± 0.16 | 3.62 ± 0.18 | 3.51 ± 0.24 | 4.29 ± 0.31 | 4.18 ± 0.30 | 4.31 ± 0.21 | 5.22 ± 0.28 |
| Ferrihydrite | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| 1 Fe: 0 Al | 0.03 ± 0.00 | 0.02 ± 0.00 | 0.03 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.01 ± 0.00 |
| 5 Fe: 1 Al | 0.03 ± 0.00 | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 |
| 1 Fe: 1 Al | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.02 ± 0.00 |
| 1 Fe: 5 Al | 16.71 ± 0.83 | 9.96 ± 0.18 | 13.13± 0.07 | 8.81 ± 0.10 | 11.21± 0.05 | 8.88 ± 0.25 | 10.73 ± 0.26 | 9.67 ± 0.43 |
| 0 Fe: 1 Al | 9.19 ± 0.15 | 5.85 ± 0.17 | 7.87 ± 0.22 | 5.42 ± 0.12 | 7.84 ± 0.14 | 6.70 ± 0.05 | 7.78 ± 0.17 | 8.44 ± 0.13 |
| Hydroxide | Elovich | KCl | KNO3 | His | Mal | ||||
|---|---|---|---|---|---|---|---|---|---|
| 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | ||
| Goethite | R² | 0.93 | 0.97 | 0.94 | 0.98 | 0.93 | 0.94 | 0.96 | 0.97 |
| S.E. | 0.01 | 0.00 | 0.01 | 0.00 | 0.01 | 0.01 | 0.01 | 0.01 | |
| α | 1.47 × 106 | 2.40 × 104 | 973.69 | 440.79 | 348.89 | 75.12 | 26.16 | 5.25 | |
| β | 71.32 | 66.86 | 55.46 | 59.67 | 52.10 | 51.00 | 43.47 | 38.44 | |
| Gibbsite | R² | 0.74 | 0.90 | 0.87 | 0.89 | 0.93 | 0.38 | 0.87 | 0.80 |
| S.E. | 0.24 | 0.17 | 0.19 | 0.24 | 0.30 | 0.27 | 0.23 | 0.26 | |
| α | 6.62 × 104 | 1.62 × 103 | 3.03 × 103 | 1.17 × 103 | 2.84 × 103 | 337.64 | 211.40 | 14.21 | |
| β | 4.54 | 4.25 | 4.09 | 3.95 | 3.42 | 2.97 | 2.77 | 1.72 | |
| Ferrihydrite | R² | 0.92 | 0.87 | 0.85 | 0.80 | 0.77 | 0.84 | 0.89 * | 0.94 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| α | 1.03 × 10−3 | 4.86 × 10−4 | 5.45 × 10−4 | 3.91 × 10−4 | 0.02 | 5.74 | 1.12 × 10−3 | 3.78 × 10−3 | |
| β | 392.88 | 1.12 × 103 | 696.87 | 1.33 × 103 | 4.58 × 103 | 1.02 × 104 | 1.69 × 103 | 2.40 × 103 | |
| 1 Fe: 0 Al | R² | 0.99 | 1.00 | 1.00 | 0.99 | 0.99 | 0.99 | 1.00 | 1.00 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| α | 0.01 | 4.98 × 10−3 | 0.01 | 3.90 × 10−3 | 0.01 | 0.01 | 0.01 | 4.13 × 10−3 | |
| β | 197.39 | 339.08 | 236.18 | 386.40 | 385.73 | 591.45 | 350.24 | 439.41 | |
| 5 Fe: 1 Al | R² | 0.99 | 0.97 | 0.95 | 0.96 | 0.99 | 0.98 | 0.97 | 0.97 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| α | 3.82 × 10−3 | 1.19 × 10−3 | 1.47 × 10−3 | 8.59 × 10−4 | 1.64 × 10−3 | 1.04 × 10−3 | 1.22 × 10−3 | 9.32 × 10−4 | |
| β | 184.08 | 459.54 | 287.03 | 608.12 | 581.63 | 1.04 × 103 | 510.68 | 708.58 | |
| 1 Fe: 1 Al | R² | 0.98 | 0.93 | 0.96 | 0.82 | 0.96 | 0.95 | 0.93 | 0.95 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| α | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | |
| β | 299.90 | 805.25 | 596.31 | 937.29 | 719.88 | 826.31 | 531.43 | 354.14 | |
| 1 Fe: 5 Al | R² | 0.97 | 0.99 | 1.00 | 0.99 | 0.98 | 0.97 | 0.98 | 0.99 |
| S.E. | 0.72 | 0.33 | 0.18 | 0.18 | 0.40 | 0.38 | 0.39 | 0.34 | |
| α | 14.62 | 3.59 | 3.69 | 3.87 | 8.78 | 6.31 | 5.02 | 2.37 | |
| β | 0.45 | 0.67 | 0.49 | 0.79 | 0.66 | 0.83 | 0.65 | 0.67 | |
| 0 Fe: 1 Al | R² | 0.94 | 0.94 | 0.95 | 0.96 | 0.94 | 0.97 | 0.97 | 0.99 |
| S.E. | 0.45 | 0.35 | 0.41 | 0.23 | 0.43 | 0.26 | 0.31 | 0.16 | |
| α | 52.56 | 7.14 | 16.61 | 10.76 | 23.30 | 13.45 | 18.73 | 4.45 | |
| β | 1.02 | 1.36 | 1.07 | 1.57 | 1.11 | 1.25 | 1.12 | 0.85 | |
| Hydroxide | Exponential | KCl | KNO3 | His | Mal | ||||
|---|---|---|---|---|---|---|---|---|---|
| 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | ||
| Goethite | R² | 0.91 | 0.96 | 0.91 | 0.97 | 0.89 | 0.90 | 0.92 | 0.92 |
| S.E. | 0.03 | 0.02 | 0.04 | 0.03 | 0.05 | 0.06 | 0.06 | 0.07 | |
| a | 0.26 | 0.22 | 0.20 | 0.17 | 0.19 | 0.17 | 0.17 | 0.14 | |
| b | 0.05 | 0.06 | 0.07 | 0.08 | 0.08 | 0.09 | 0.10 | 0.12 | |
| Gibbsite | R² | 0.72 | 0.84 | 0.83 | 0.77 | 0.75 | 0.82 | 0.87 | 0.92 |
| S.E. | 0.08 | 0.07 | 0.07 | 0.09 | 0.09 | 0.09 | 0.08 | 0.09 | |
| a | 2.80 | 2.12 | 2.34 | 2.17 | 2.72 | 2.39 | 2.38 | 2.09 | |
| b | 0.07 | 0.09 | 0.08 | 0.09 | 0.08 | 0.10 | 0.11 | 0.17 | |
| Ferrihydrite | R² | 0.99 | 0.99 | 1.00 | 1.00 | 0.95 | 0.38 | 0.95 | 0.82 |
| S.E. | 0.09 | 0.06 | 0.06 | 0.05 | 0.06 | 0.17 | 0.10 | 0.16 | |
| a | 7.88 × 10−4 | 4.78 × 10−4 | 4.58 × 10−4 | 3.88 × 10−4 | 1.11 × 10−3 | 1.09 × 10−3 | 9.15 × 10−4 | 1.19 × 10−3 | |
| b | 0.51 | 0.41 | 0.49 | 0.42 | 0.13 | 0.07 | 0.25 | 0.18 | |
| 1 Fe: 0 Al | R² | 0.92 | 0.96 | 0.96 | 0.97 | 0.93 | 0.93 | 0.96 | 0.96 |
| S.E. | 0.15 | 0.11 | 0.13 | 0.11 | 0.13 | 0.11 | 0.11 | 0.12 | |
| a | 0.01 | 3.61 × 10−3 | 4.44 × 10−3 | 2.96 × 10−3 | 4.89 × 10−3 | 3.73 × 10−3 | 3.61 × 10−3 | 2.91 × 10−3 | |
| b | 0.28 | 0.30 | 0.33 | 0.31 | 0.25 | 0.23 | 0.30 | 0.30 | |
| 5 Fe: 1 Al | R² | 0.94 | 0.97 | 0.98 | 0.98 | 0.95 | 0.97 | 0.98 | 0.98 |
| S.E. | 0.20 | 0.15 | 0.15 | 0.12 | 0.15 | 0.12 | 0.12 | 0.11 | |
| a | 2.85 × 10−3 | 9.23 × 10−4 | 9.98 × 10−4 | 6.84 × 10−4 | 1.32 × 10−3 | 8.61 × 10−4 | 1.03 × 10−3 | 8.04 × 10−4 | |
| b | 0.44 | 0.47 | 0.53 | 0.47 | 0.38 | 0.35 | 0.43 | 0.42 | |
| 1 Fe: 1 Al | R² | 0.95 | 0.90 | 0.96 | 0.83 | 0.92 | 0.92 | 0.92 | 0.92 |
| S.E. | 0.12 | 0.14 | 0.09 | 0.18 | 0.12 | 0.12 | 0.13 | 0.15 | |
| a | 4.71 × 10−3 | 2.79 × 10−3 | 3.14 × 10−3 | 2.41 × 10−3 | 3.27 × 10−3 | 2.77 × 10−3 | 3.32 × 10−3 | 3.77 × 10−3 | |
| b | 0.28 | 0.22 | 0.24 | 0.22 | 0.21 | 0.22 | 0.25 | 0.29 | |
| 1 Fe: 5 Al | R² | 0.92 | 0.92 | 0.94 | 0.95 | 0.92 | 0.91 | 0.93 | 0.96 |
| S.E. | 0.12 | 0.15 | 0.14 | 0.11 | 0.13 | 0.14 | 0.13 | 0.12 | |
| a | 5.15 | 2.18 | 2.53 | 2.15 | 3.34 | 2.56 | 2.65 | 1.76 | |
| b | 0.22 | 0.28 | 0.30 | 0.26 | 0.23 | 0.23 | 0.26 | 0.31 | |
| 0 Fe: 1 Al | R² | 0.88 | 0.87 | 0.87 | 0.89 | 0.87 | 0.91 | 0.91 | 0.96 |
| S.E. | 0.10 | 0.15 | 0.13 | 0.12 | 0.13 | 0.11 | 0.10 | 0.10 | |
| a | 4.13 | 1.96 | 2.97 | 2.03 | 3.18 | 2.54 | 3.04 | 2.24 | |
| b | 0.15 | 0.20 | 0.18 | 0.18 | 0.17 | 0.18 | 0.17 | 0.24 | |
| Hydroxide | Parabolic | KCl | KNO3 | His | Mal | ||||
|---|---|---|---|---|---|---|---|---|---|
| 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | ||
| Goethite | R² | 0.69 | 0.79 | 0.71 | 0.82 | 0.70 | 0.73 | 0.76 | 0.77 |
| S.E. | 0.02 | 0.01 | 0.02 | 0.01 | 0.02 | 0.02 | 0.02 | 0.02 | |
| Q0 | 0.28 | 0.23 | 0.22 | 0.19 | 0.22 | 0.19 | 0.19 | 0.17 | |
| kp | 3.58 × 10−3 | 3.99 × 10−3 | 4.67 × 10−3 | 4.55 × 10−3 | 4.94 × 10−3 | 0.01 | 0.01 | 0.01 | |
| Gibbsite | R² | 0.55 | 0.67 | 0.64 | 0.61 | 0.59 | 0.65 | 0.71 | 0.81 |
| S.E. | 0.32 | 0.27 | 0.30 | 0.33 | 0.39 | 0.40 | 0.38 | 0.48 | |
| Q0 | 3.09 | 2.41 | 2.65 | 2.48 | 3.09 | 2.78 | 2.79 | 2.59 | |
| kp | 0.06 | 0.06 | 0.06 | 0.07 | 0.08 | 0.09 | 0.10 | 0.16 | |
| Ferrihydrite | R² | 0.99 | 0.99 | 0.99 | 1.00 | 0.92 | 0.37 | 0.99 | 0.88 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Q0 | 2.85 × 10−4 | 3.22 × 10−4 | −6.07 × 10−5 | 2.26 × 10−4 | 1.26 × 10−3 | 1.19 × 10−3 | 9.30 × 10−4 | 1.33 × 10−3 | |
| kp | 7.83 × 10−4 | 2.79 × 10−4 | 4.56 × 10−4 | 2.36 × 10−4 | 6.44 × 10−5 | 2.89 × 10−5 | 1.87 × 10−4 | 1.30 × 10−4 | |
| 1 Fe: 0 Al | R² | 0.84 | 0.91 | 0.91 | 0.92 | 0.83 | 0.84 | 0.91 | 0.91 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Q0 | 0.01 | 0.01 | 0.01 | 4.12 × 10−3 | 0.01 | 0.01 | 0.01 | 4.09 × 10−3 | |
| kp | 1.38 × 10−3 | 8.35 × 10−4 | 1.20 × 10−3 | 7.38 × 10−4 | 7.04 × 10−4 | 4.61 × 10−4 | 8.09 × 10−4 | 6.43 × 10−4 | |
| 5 Fe: 1 Al | R² | 0.92 | 0.97 | 0.98 | 0.98 | 0.92 | 0.94 | 0.97 | 0.97 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Q0 | 4.44 × 10−3 | 1.06 × 10−3 | 6.39 × 10−4 | 6.87 × 10−4 | 1.95 × 10−3 | 1.18 × 10−3 | 1.20 × 10−3 | 9.50 × 10−4 | |
| kp | 1.56 × 10−3 | 6.45 × 10−4 | 1.05 × 10−3 | 4.93 × 10−4 | 4.90 × 10−4 | 2.78 × 10−4 | 5.82 × 10−4 | 4.19 × 10−4 | |
| 1 Fe: 1 Al | R² | 0.88 | 0.84 | 0.91 | 0.76 | 0.84 | 0.85 | 0.87 | 0.86 |
| S.E. | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Q0 | 0.01 | 3.65 × 10−3 | 3.95 × 10−3 | 3.11 × 10−3 | 4.29 × 10−3 | 3.60 × 10−3 | 4.31 × 10−3 | 0.01 | |
| kp | 9.38 × 10−4 | 3.49 × 10−4 | 4.86 × 10−4 | 3.04 × 10−4 | 3.86 × 10−4 | 3.39 × 10−4 | 5.41 × 10−4 | 7.99 × 10−4 | |
| 1 Fe: 5 Al | R² | 0.80 | 0.86 | 0.88 | 0.88 | 0.81 | 0.82 | 0.85 | 0.91 |
| S.E. | 1.87 | 1.05 | 1.28 | 0.83 | 1.25 | 0.96 | 1.12 | 0.85 | |
| Q0 | 7.07 | 3.17 | 3.70 | 2.97 | 4.65 | 3.53 | 3.74 | 2.46 | |
| kp | 0.60 | 0.41 | 0.57 | 0.35 | 0.41 | 0.33 | 0.42 | 0.43 | |
| 0 Fe: 1 Al | R² | 0.71 | 0.78 | 0.74 | 0.79 | 0.72 | 0.77 | 0.79 | 0.88 |
| S.E. | 1.00 | 0.66 | 0.91 | 0.55 | 0.91 | 0.72 | 0.77 | 0.74 | |
| Q0 | 5.25 | 2.63 | 3.96 | 2.63 | 4.17 | 3.33 | 3.89 | 2.99 | |
| kp | 0.25 | 0.20 | 0.25 | 0.17 | 0.23 | 0.21 | 0.24 | 0.33 | |
| Hydroxide | Δ c(H+) (10−7 mol L−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| KCl | KNO3 | His | Mal | |||||
| 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | 5 mM | 50 mM | |
| Goethite | −0.79 ± 0.00 | 1.18 ± 0.07 | −3.36 ± 0.04 | −2.91 ± 0.00 | −3.44 ± 0.04 | 0.45 ± 0.64 | −2.43 ± 0.00 | −3.38 ± 0.46 |
| Gibbsite | −2.25 ± 0.04 | −0.48 ± 0.01 | −4.24 ± 0.01 | −4.11 ± 0.03 | −3.84 ± 0.26 | 0.00 ± 0.24 | −2.73 ± 0.04 | −4.44 ± 0.39 |
| Ferrihydrite | −1.87 ± 0.11 | 0.26 ± 0.30 | −3.47 ± 0.16 | −2.85 ± 0.20 | −4.30 ± 0.24 | −0.57 ± 0.11 | −3.27 ± 0.01 | −3.61 ± 0.78 |
| 1 Fe: 0 Al | −2.30 ± 0.03 | −0.44 ± 0.01 | −4.18 ± 0.02 | −4.06 ± 0.01 | −5.71 ± 0.01 | −1.72 ± 0.09 | −3.05 ± 0.01 | −7.08 ± 0.22 |
| 5 Fe: 1 Al | −2.07 ± 0.00 | 0.17 ± 0.21 | −3.58 ± 0.07 | −3.39 ± 0.08 | −5.39 ± 0.03 | −1.52 ± 0.19 | −2.89 ± 0.01 | −5.34 ± 0.17 |
| 1 Fe: 1 Al | −1.96 ± 0.11 | 0.00 ± 0.09 | −3.40 ± 0.15 | −2.75 ± 0.38 | −4.77 ± 0.16 | −1.17 ± 0.30 | −2.88 ± 0.02 | −3.86 ± 0.43 |
| 1 Fe: 5 Al | −2.44 ± 0.01 | −0.57 ± 0.06 | −4.36 ± 0.06 | −4.31 ± 0.02 | −5.88 ± 0.02 | −2.51 ± 0.32 | −3.18 ± 0.04 | −6.53 ± 0.19 |
| 0 Fe: 1 Al | −2.44 ± 0.01 | −0.63 ± 0.04 | −4.46 ± 0.00 | −4.33 ± 0.03 | −5.78 ± 0.18 | −3.78 ± 0.24 | −3.21 ± 0.01 | −8.88 ± 0.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gypser, S.; Schütze, E.; Freese, D. Single and Binary Fe- and Al-hydroxides Affect Potential Phosphorus Mobilization and Transfer from Pools of Different Availability. Soil Syst. 2021, 5, 33. https://doi.org/10.3390/soilsystems5020033
Gypser S, Schütze E, Freese D. Single and Binary Fe- and Al-hydroxides Affect Potential Phosphorus Mobilization and Transfer from Pools of Different Availability. Soil Systems. 2021; 5(2):33. https://doi.org/10.3390/soilsystems5020033
Chicago/Turabian StyleGypser, Stella, Elisabeth Schütze, and Dirk Freese. 2021. "Single and Binary Fe- and Al-hydroxides Affect Potential Phosphorus Mobilization and Transfer from Pools of Different Availability" Soil Systems 5, no. 2: 33. https://doi.org/10.3390/soilsystems5020033
APA StyleGypser, S., Schütze, E., & Freese, D. (2021). Single and Binary Fe- and Al-hydroxides Affect Potential Phosphorus Mobilization and Transfer from Pools of Different Availability. Soil Systems, 5(2), 33. https://doi.org/10.3390/soilsystems5020033