
Hydration and Carbonation of Alternative Binders



Abstract
:1. Introduction
- Which products are formed during hydration/solidification and how does the pore structure develop?
- What is the pH value of the pore solution after hydration/solidification?
- Which phases are formed during carbonation?
- How is the pore structure affected by the carbonation process?
- Is the accelerated carbonation test applicable to the investigated binders?
2. Materials and Methods
2.1. Investigation of the Hydration Progress
2.2. Investigation of Carbonation Depth due to Natural and Accelerated Carbonation
2.3. Investigation of Structural Changes due to Accelerated Carbonation at 1% CO2
2.4. Investigation of the Pore Structures
3. Results
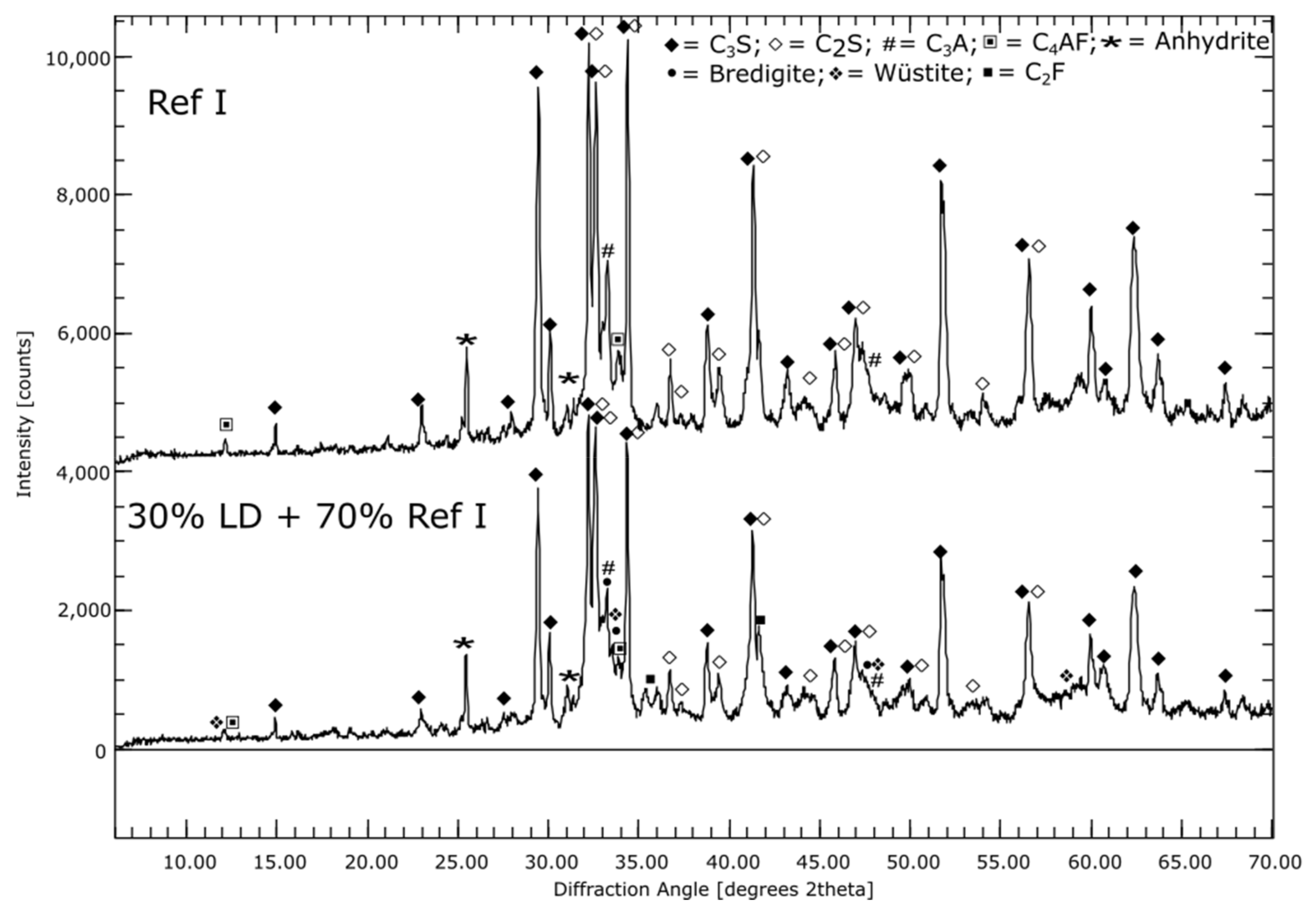
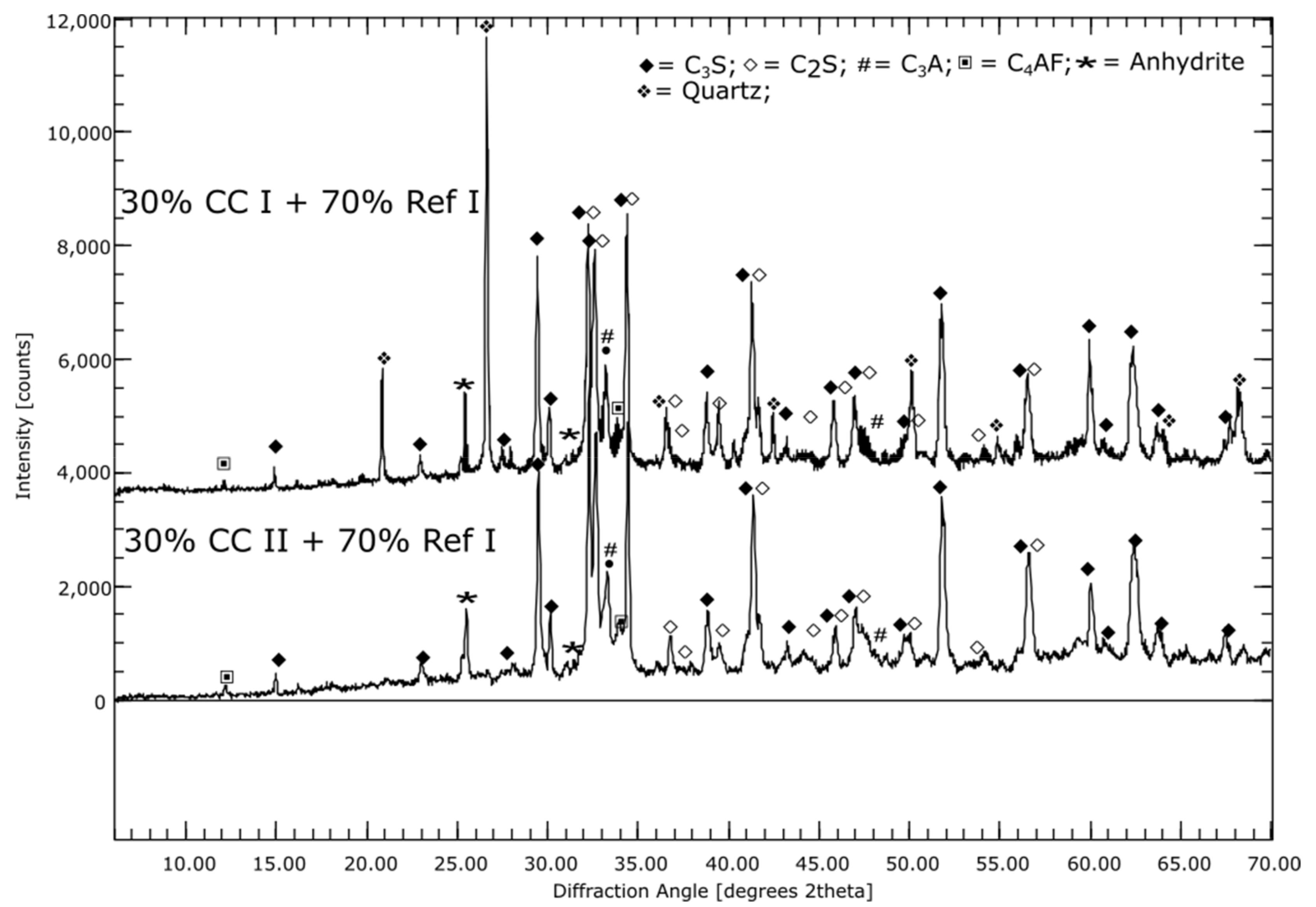
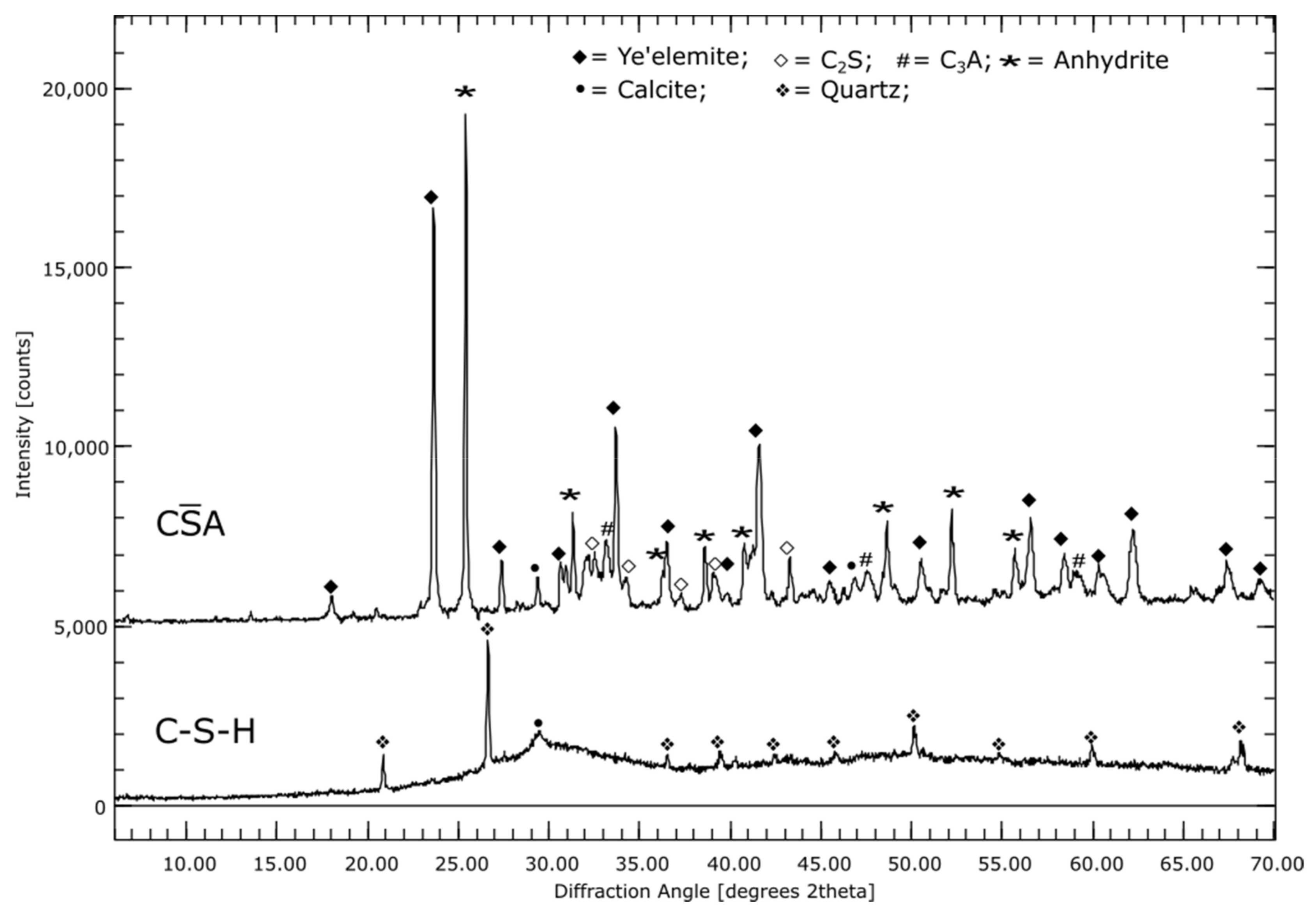
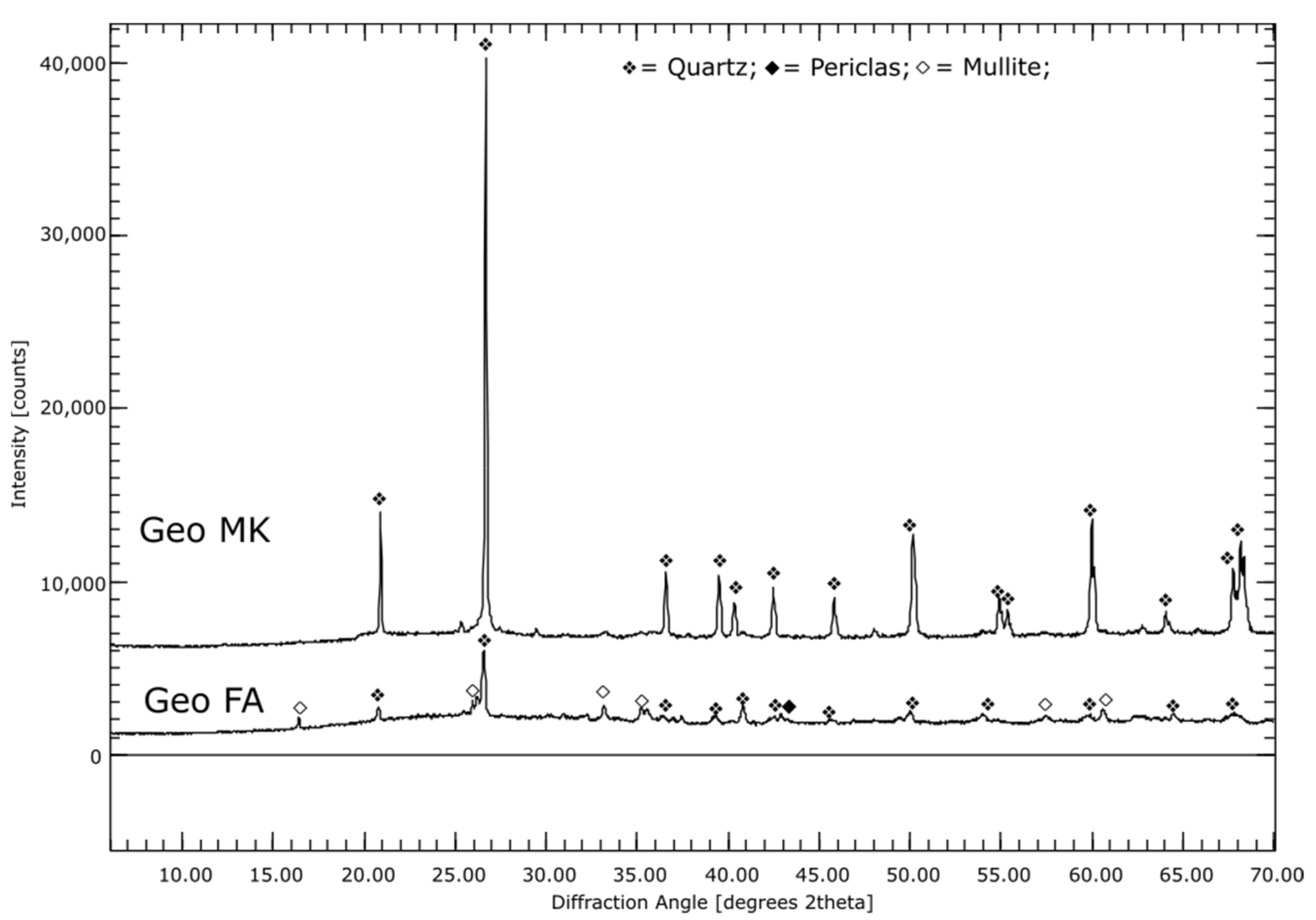
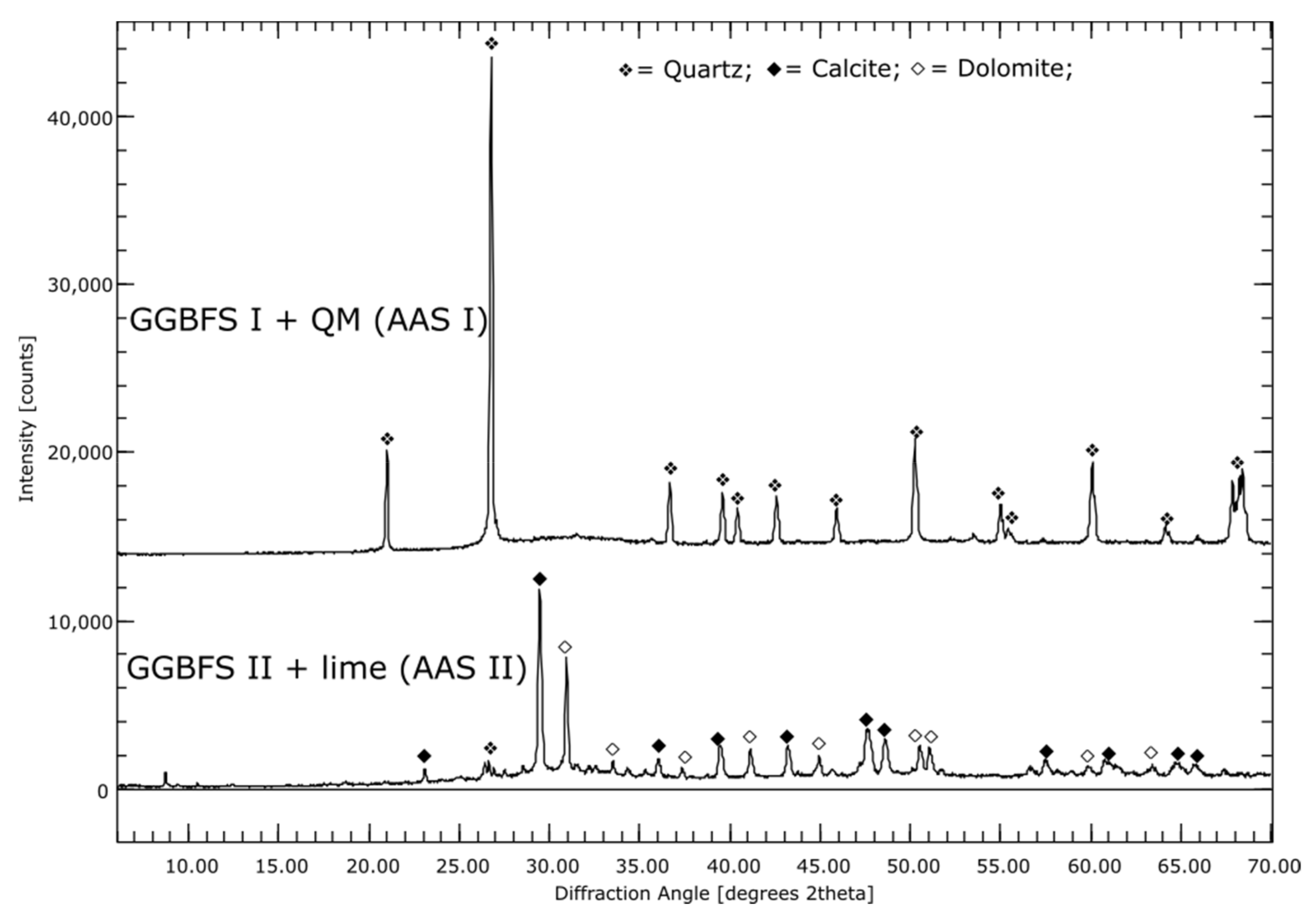
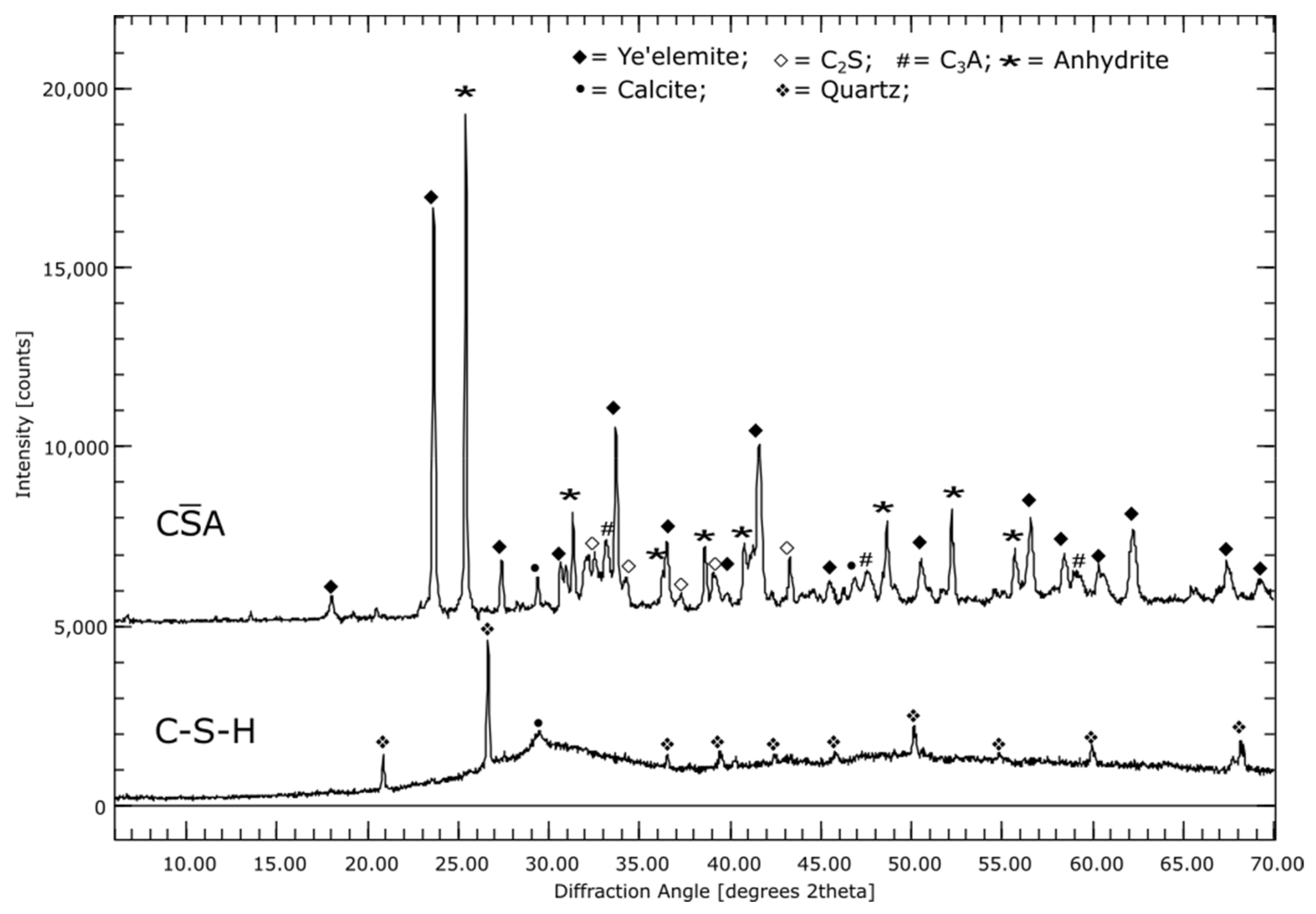
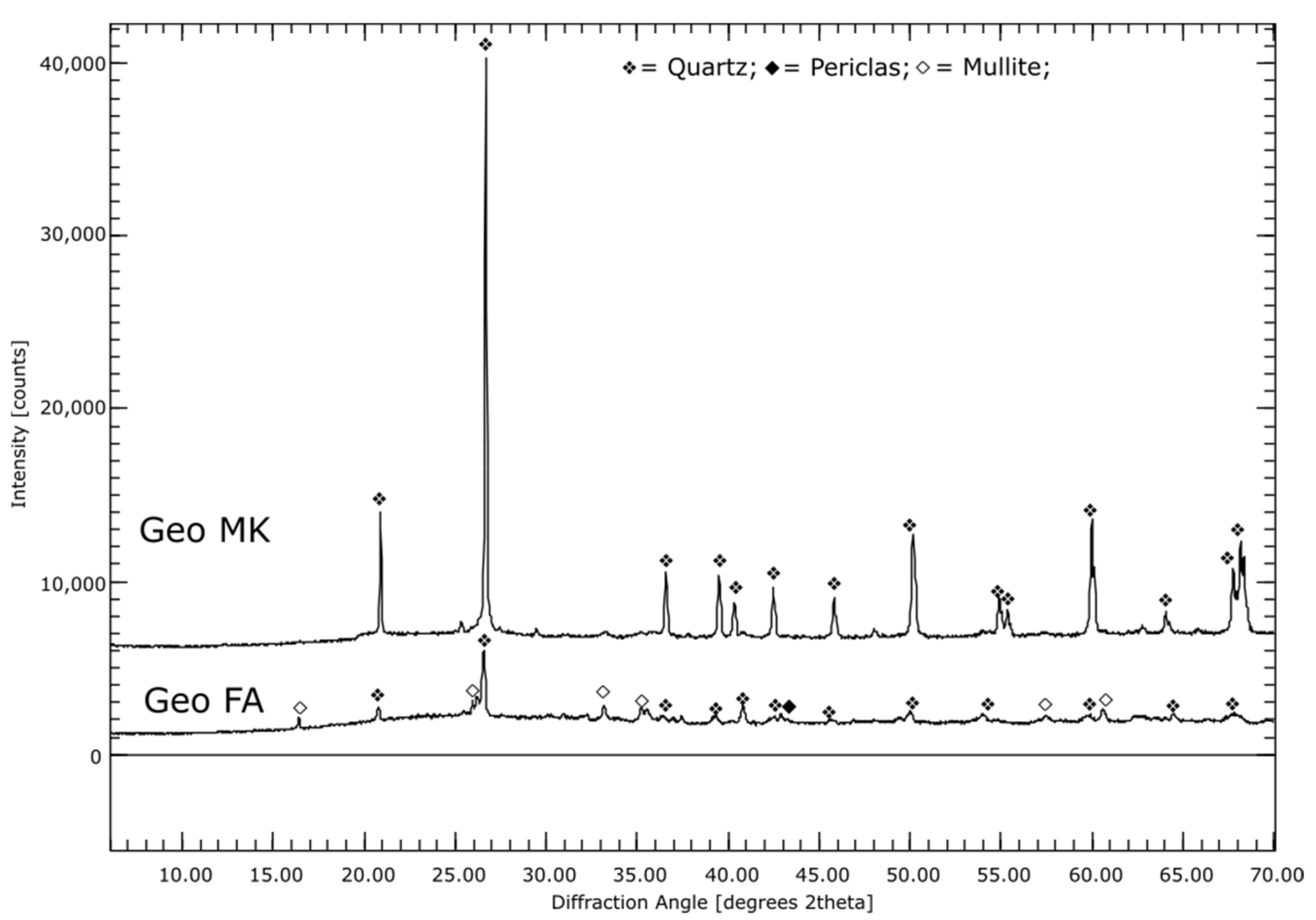
3.1. Phase Compositions of the Initial Materials (XRD)
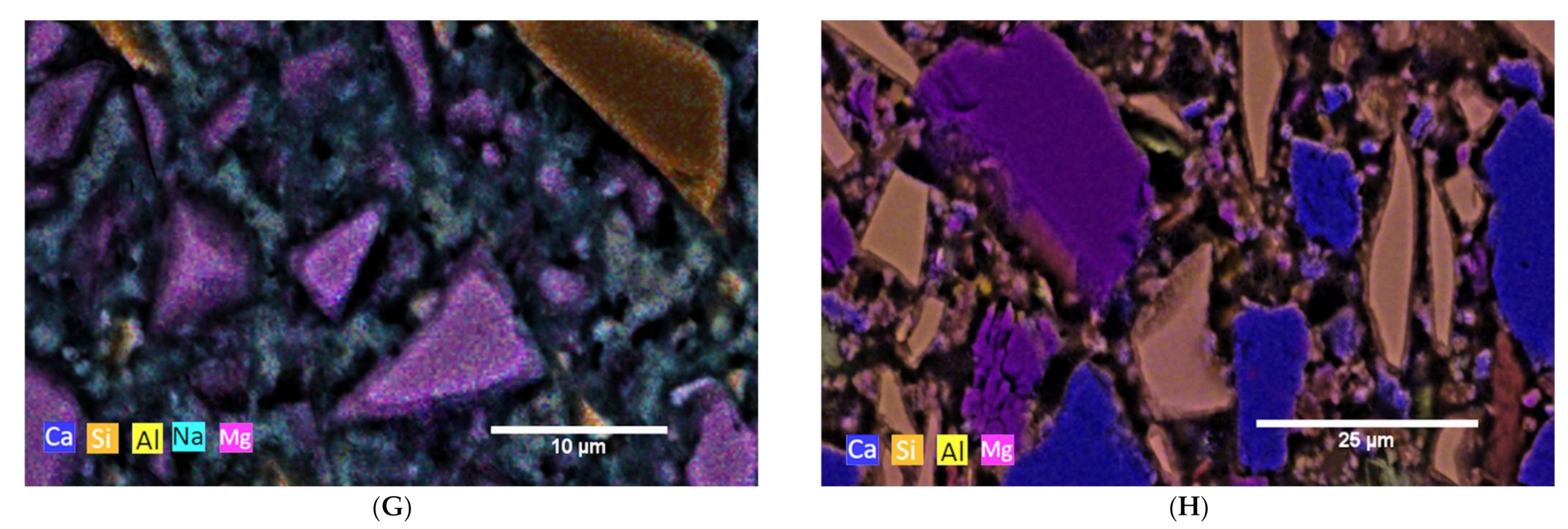
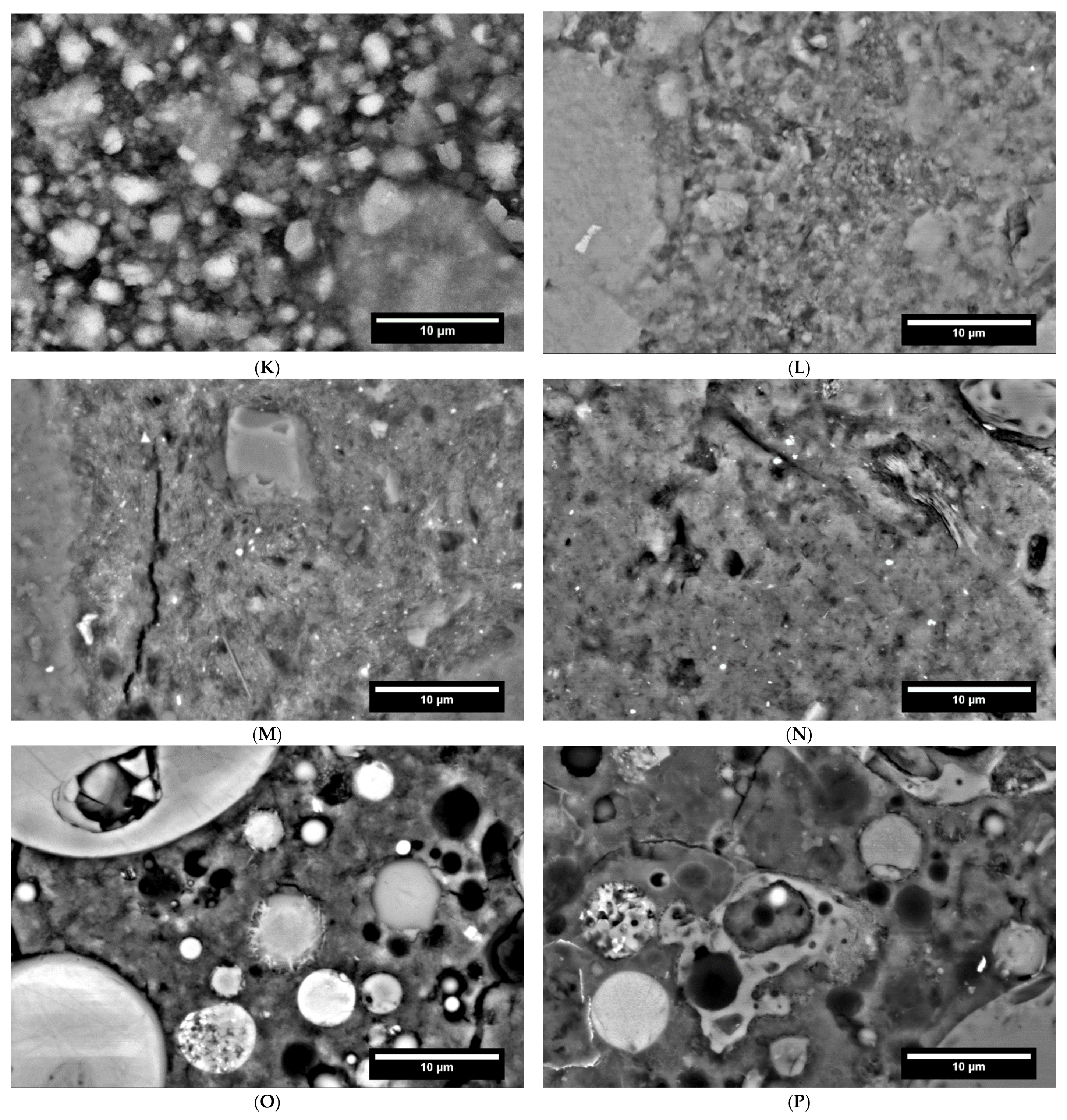
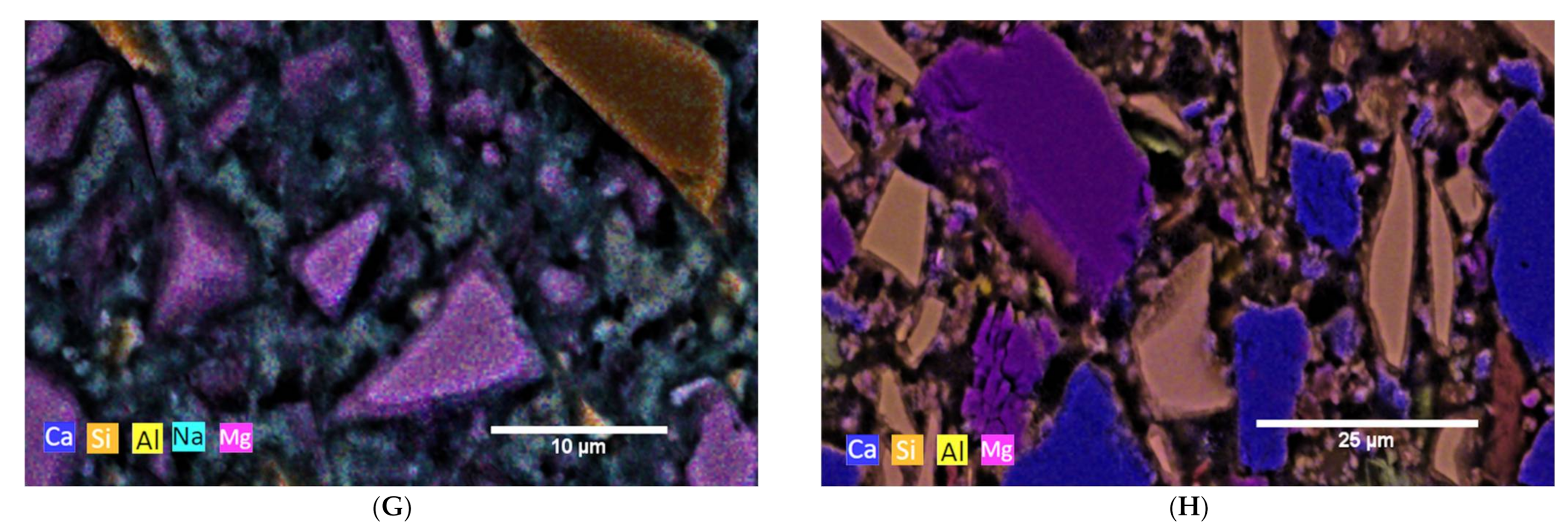
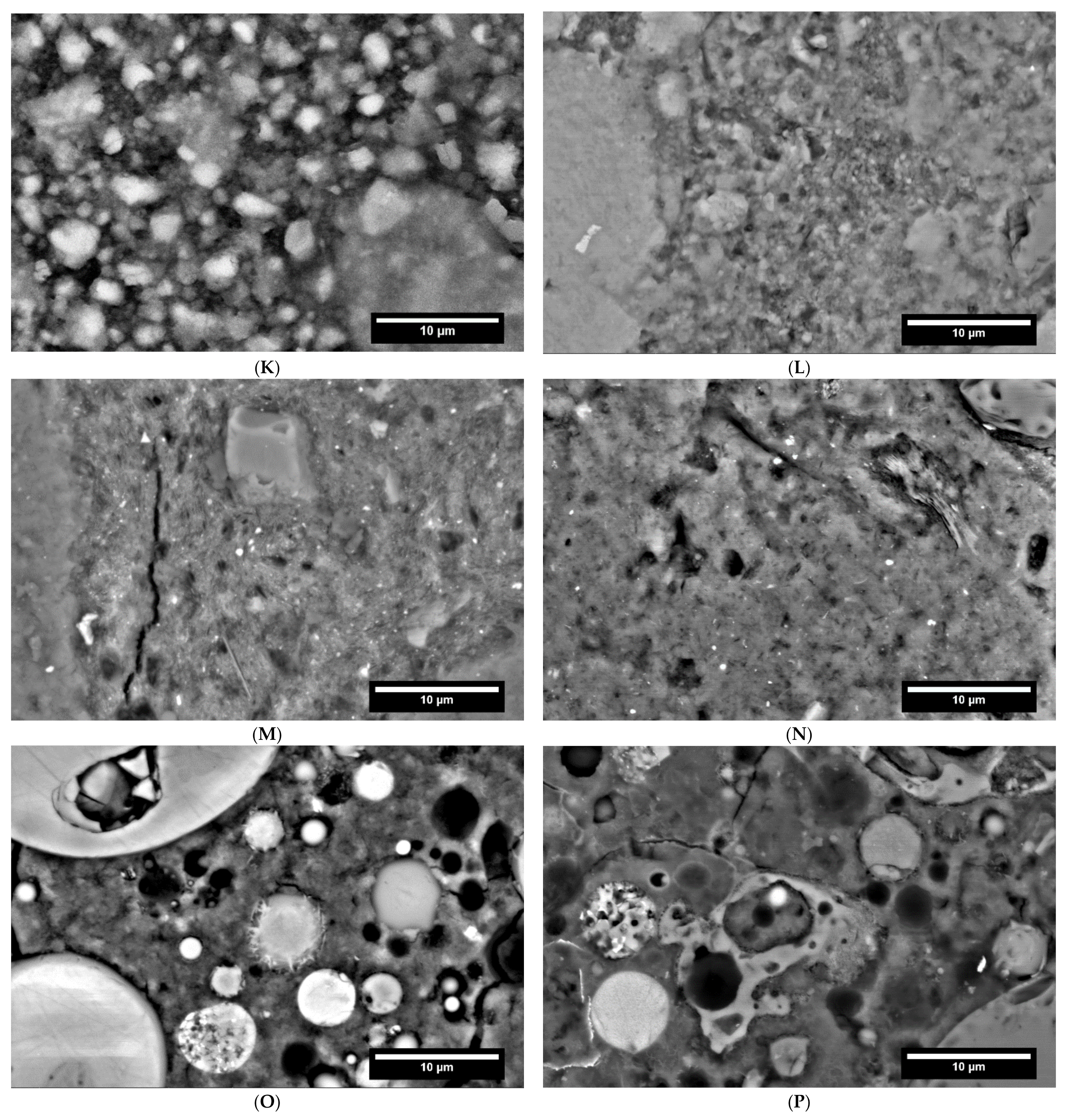
3.2. Hydration Process (SEM and XRD at Cement Paste Samples)
3.2.1. Reference Cements and SCMs
3.2.2. CA
3.2.3. C-S-H
3.2.4. AAMs
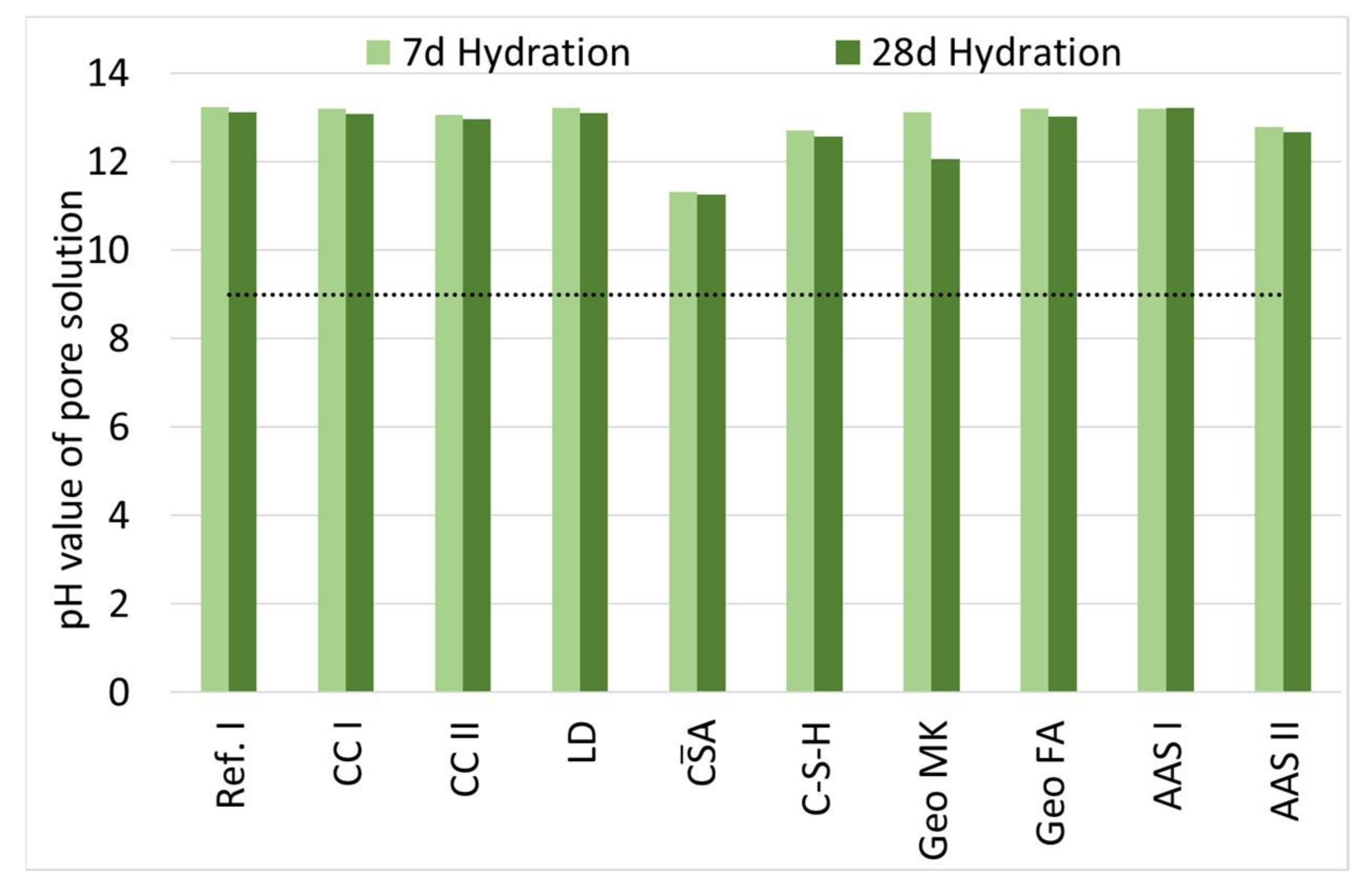
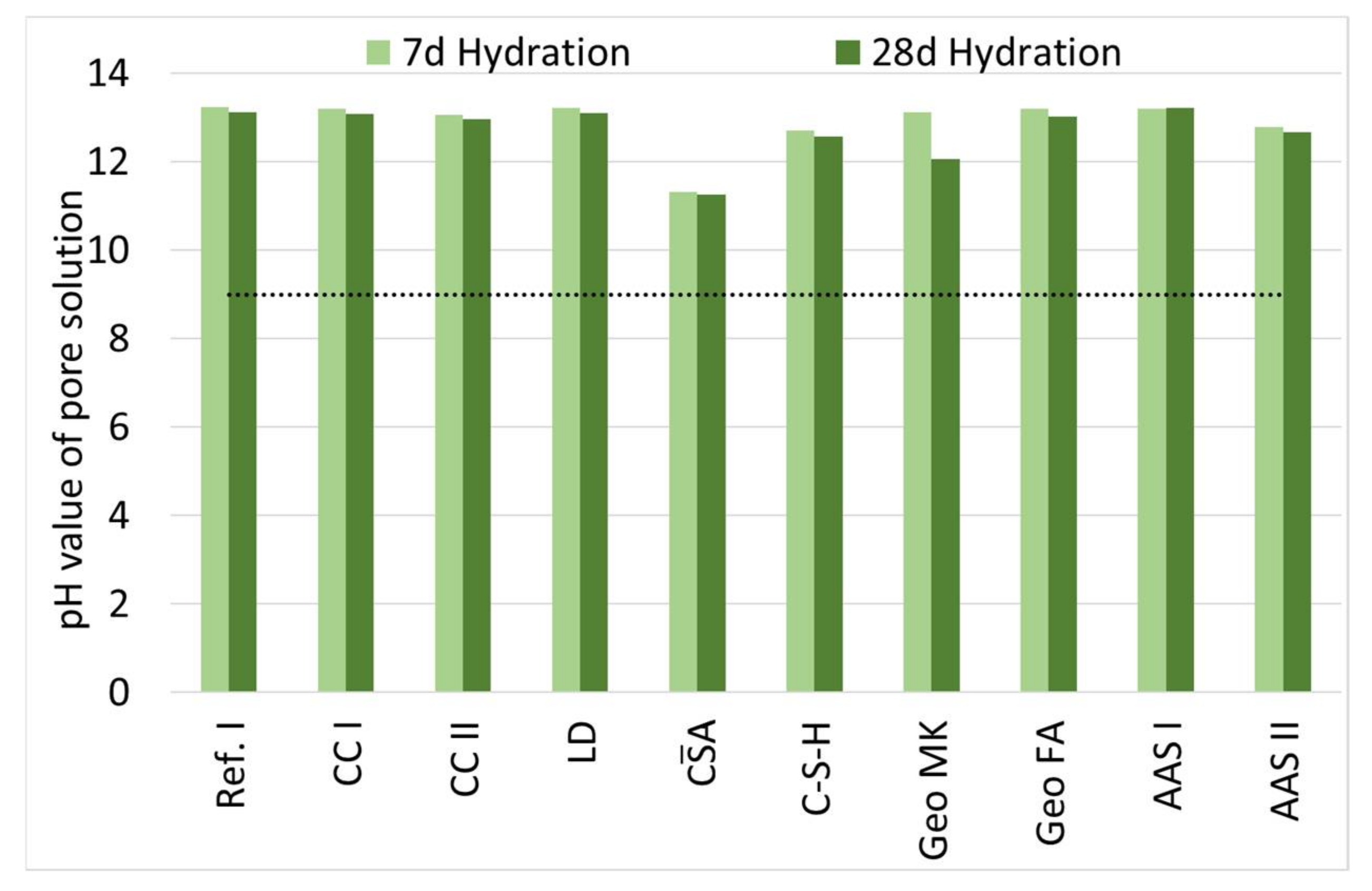
3.3. Influence of Hydration on the pH of the Pore Solution
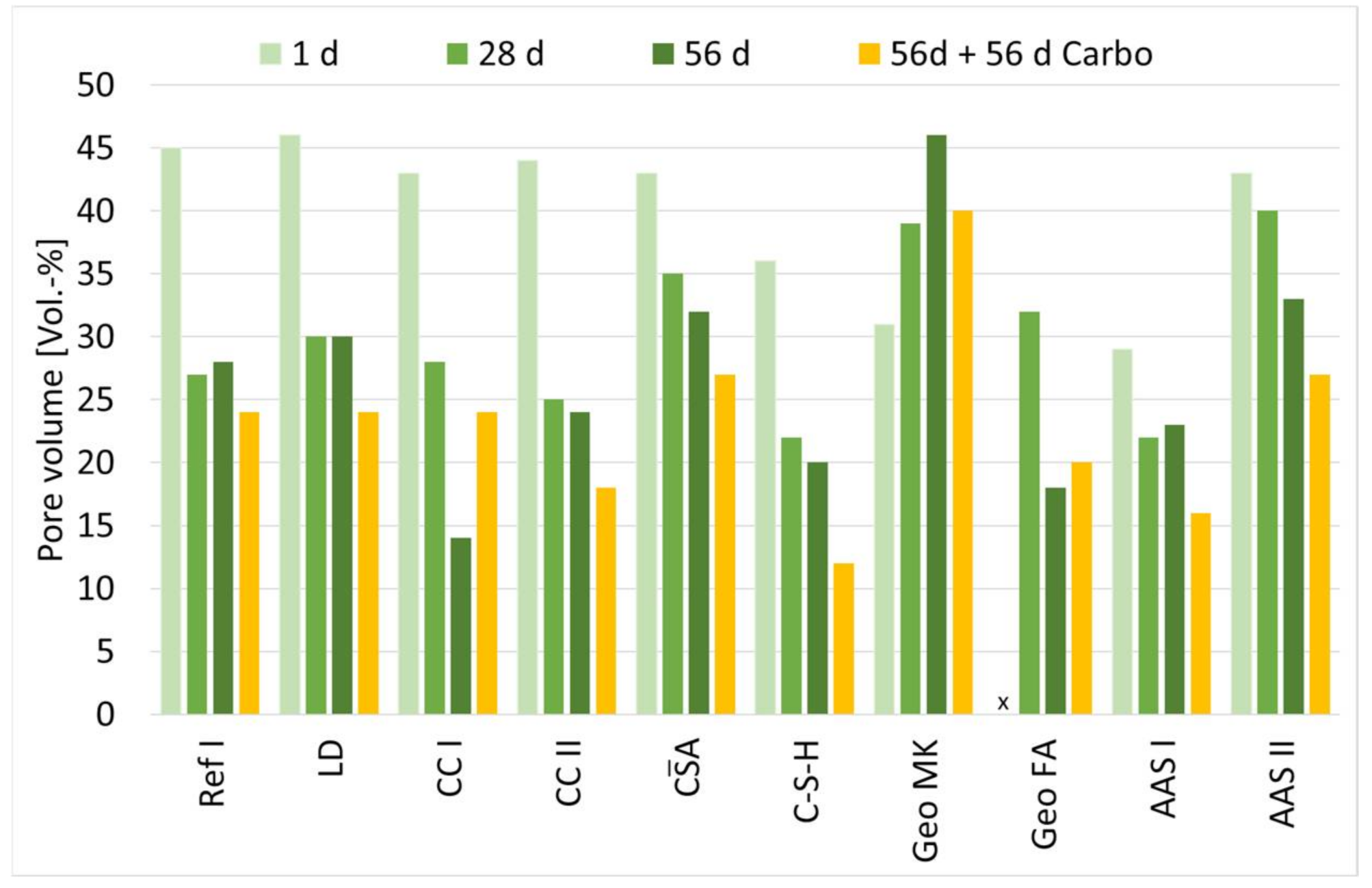
3.4. Influence of Hydration on the Pore Structure
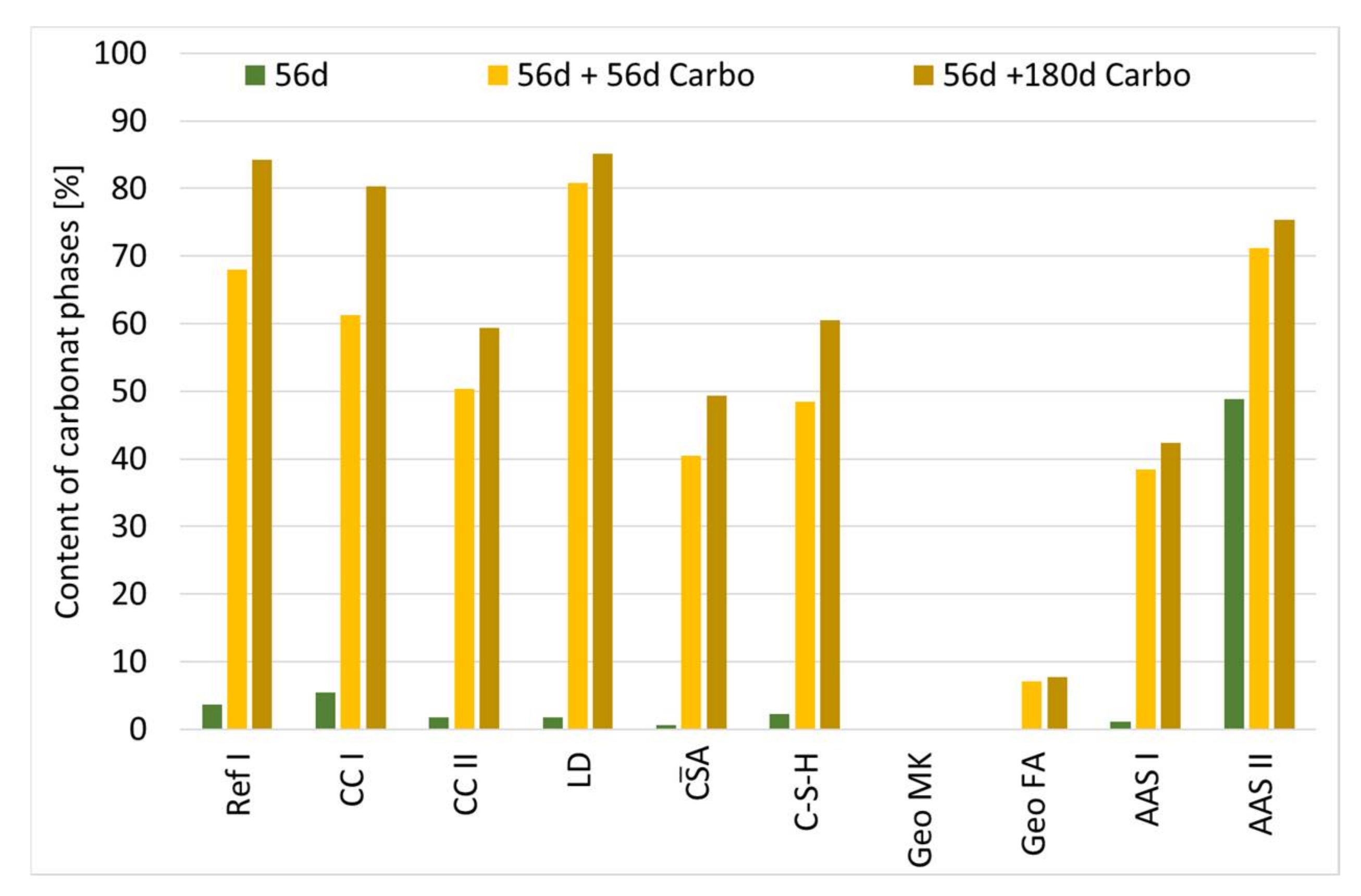
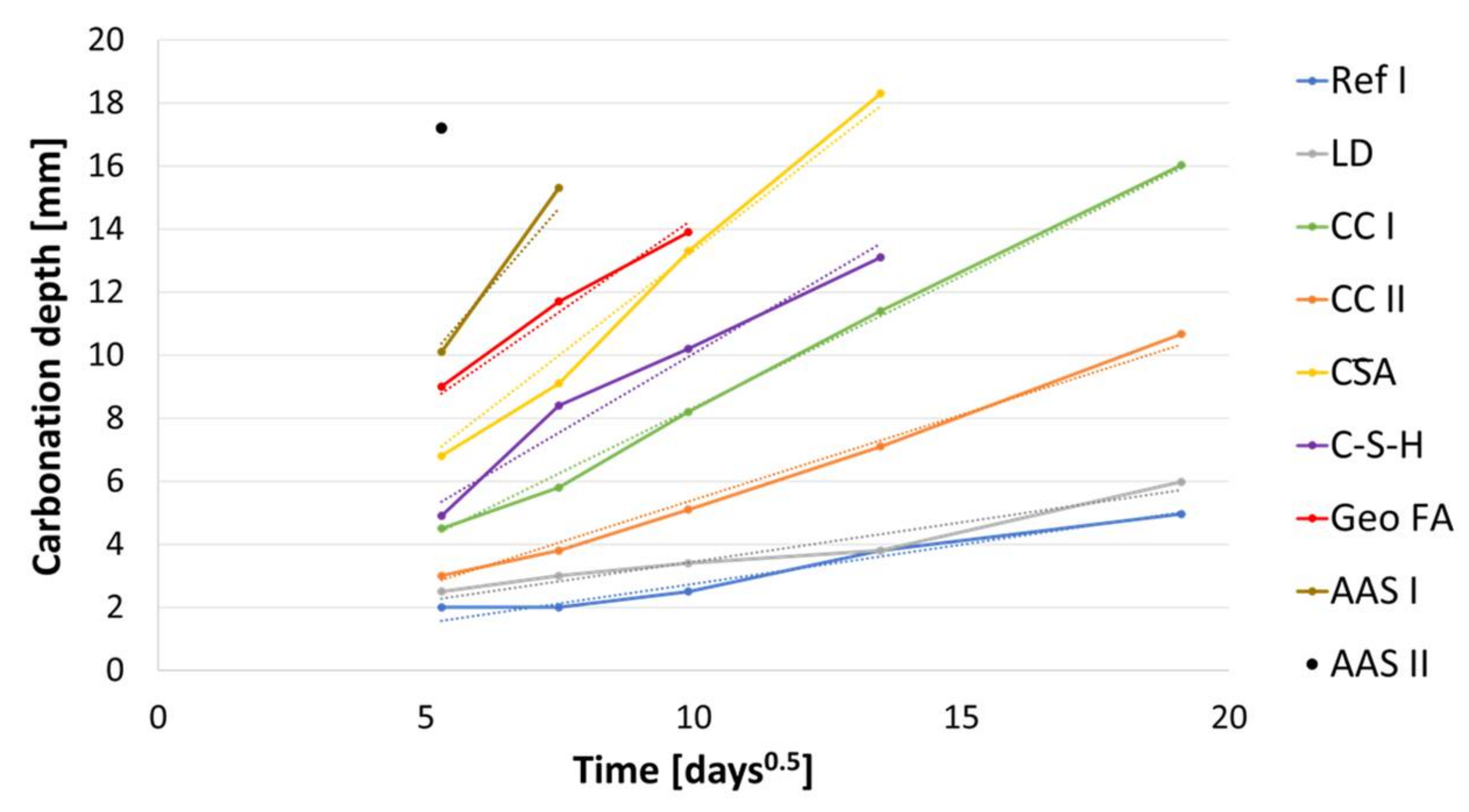
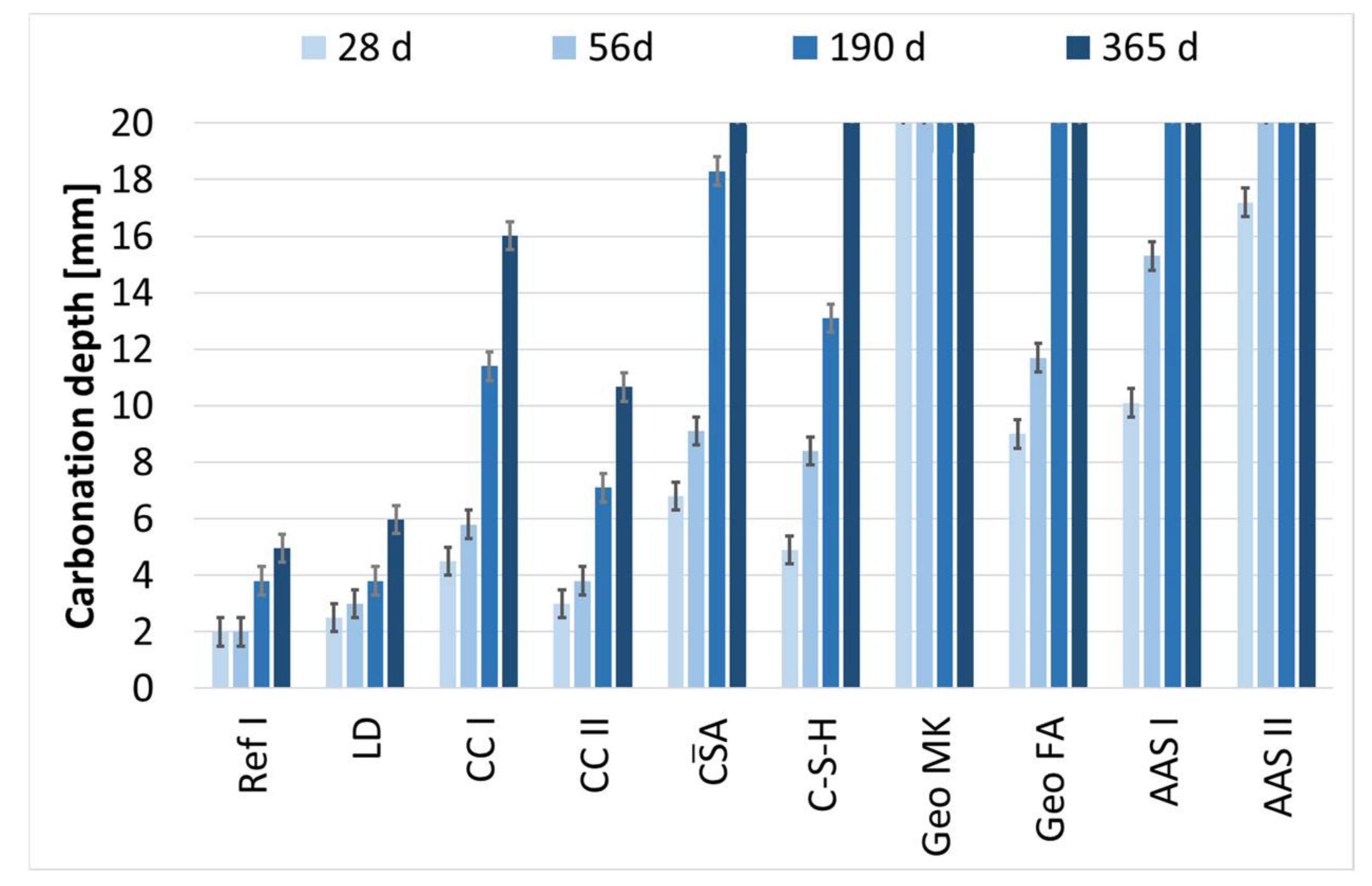
3.5. Carbonation Process at Cement Paste Samples
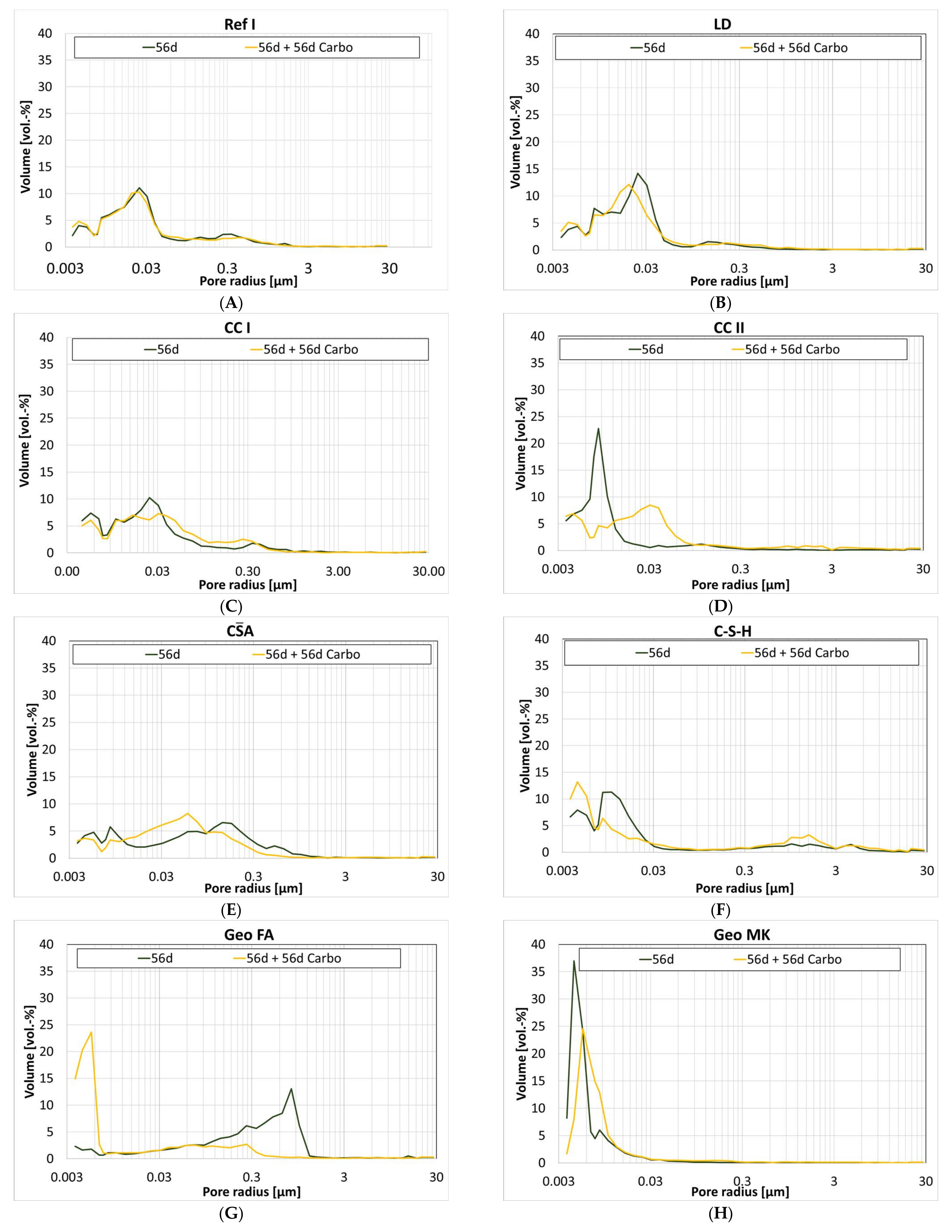
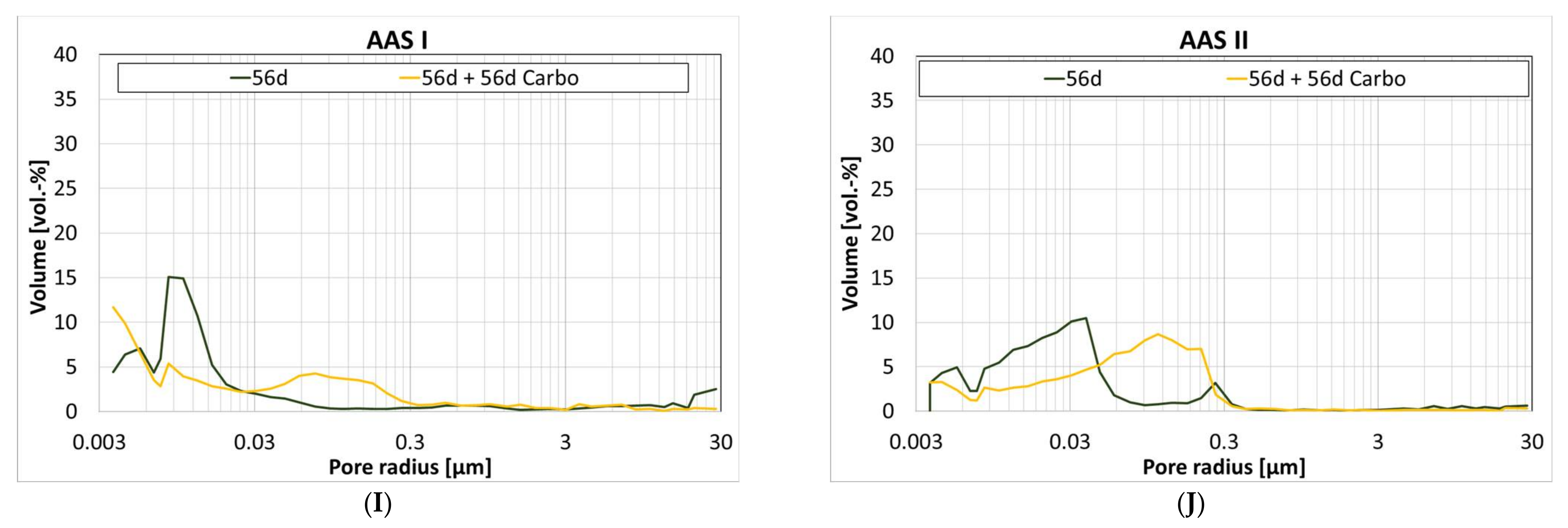
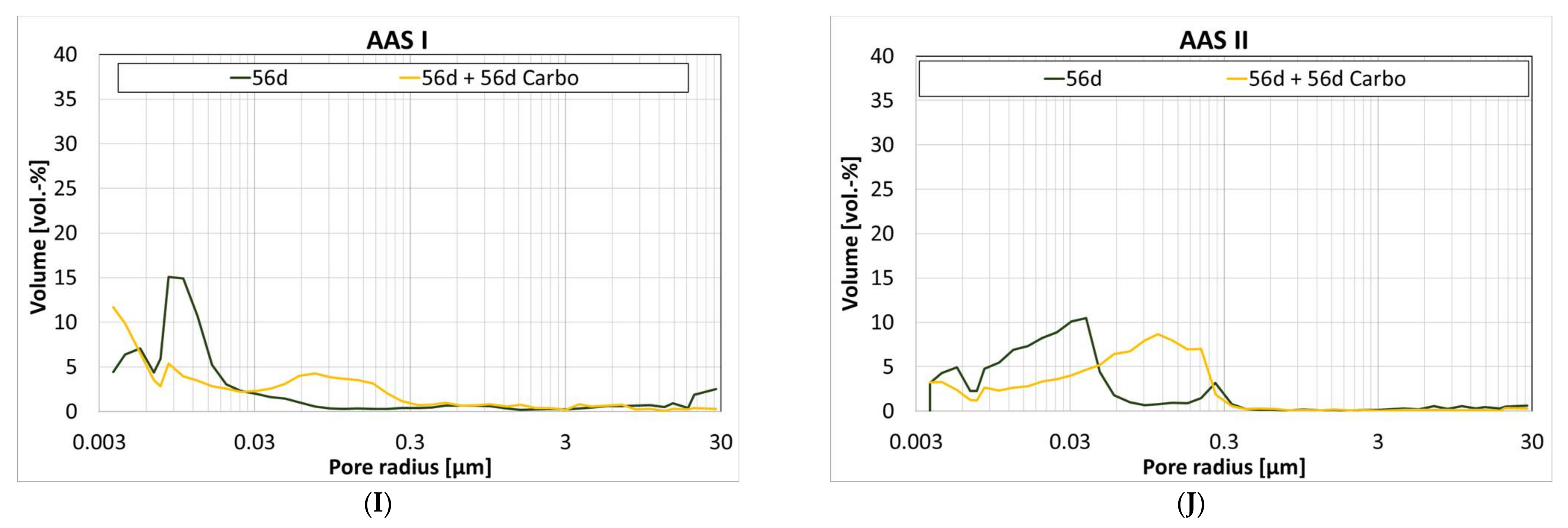
3.6. Influence of Carbonation on the Pore Structure
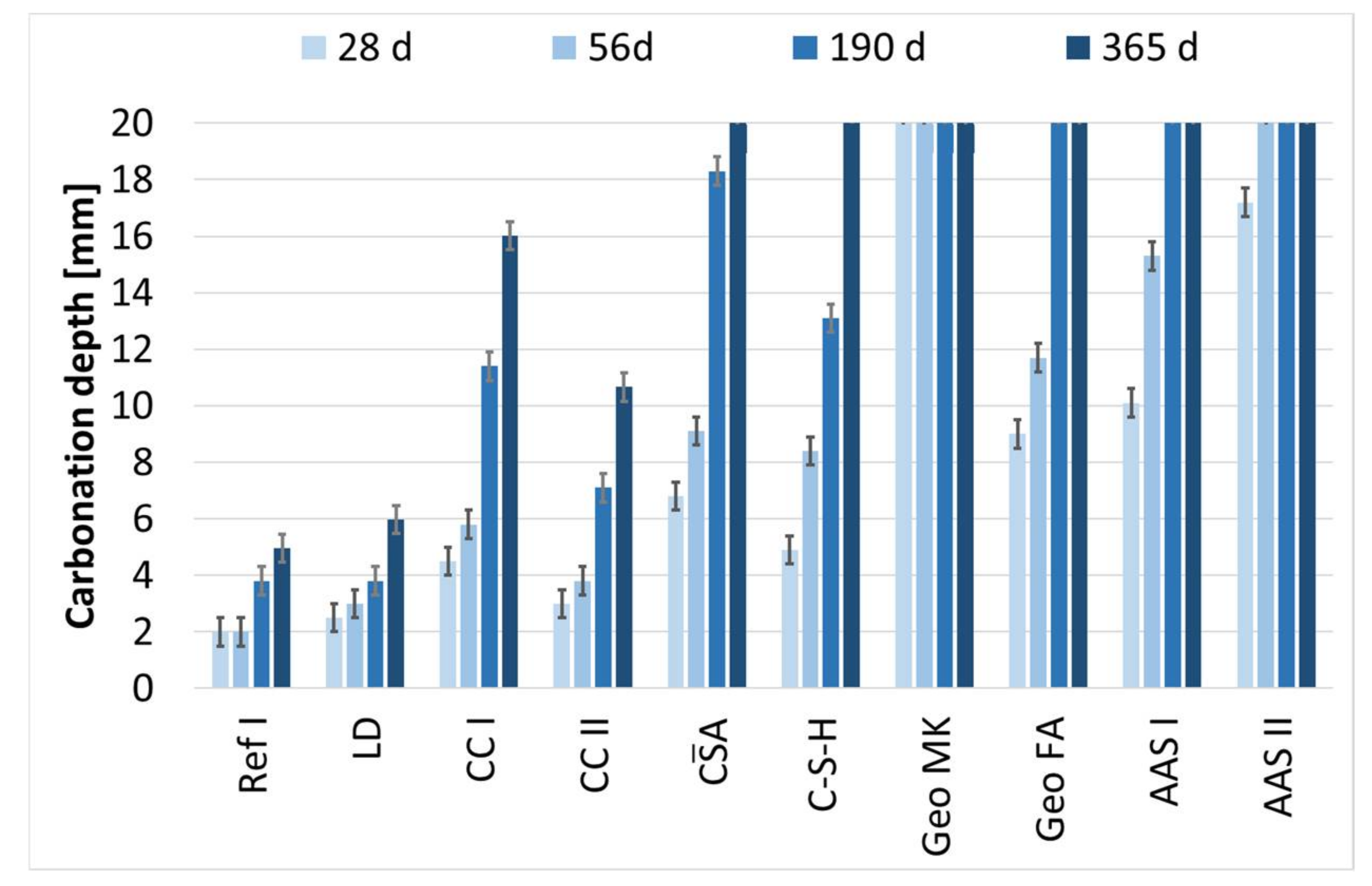
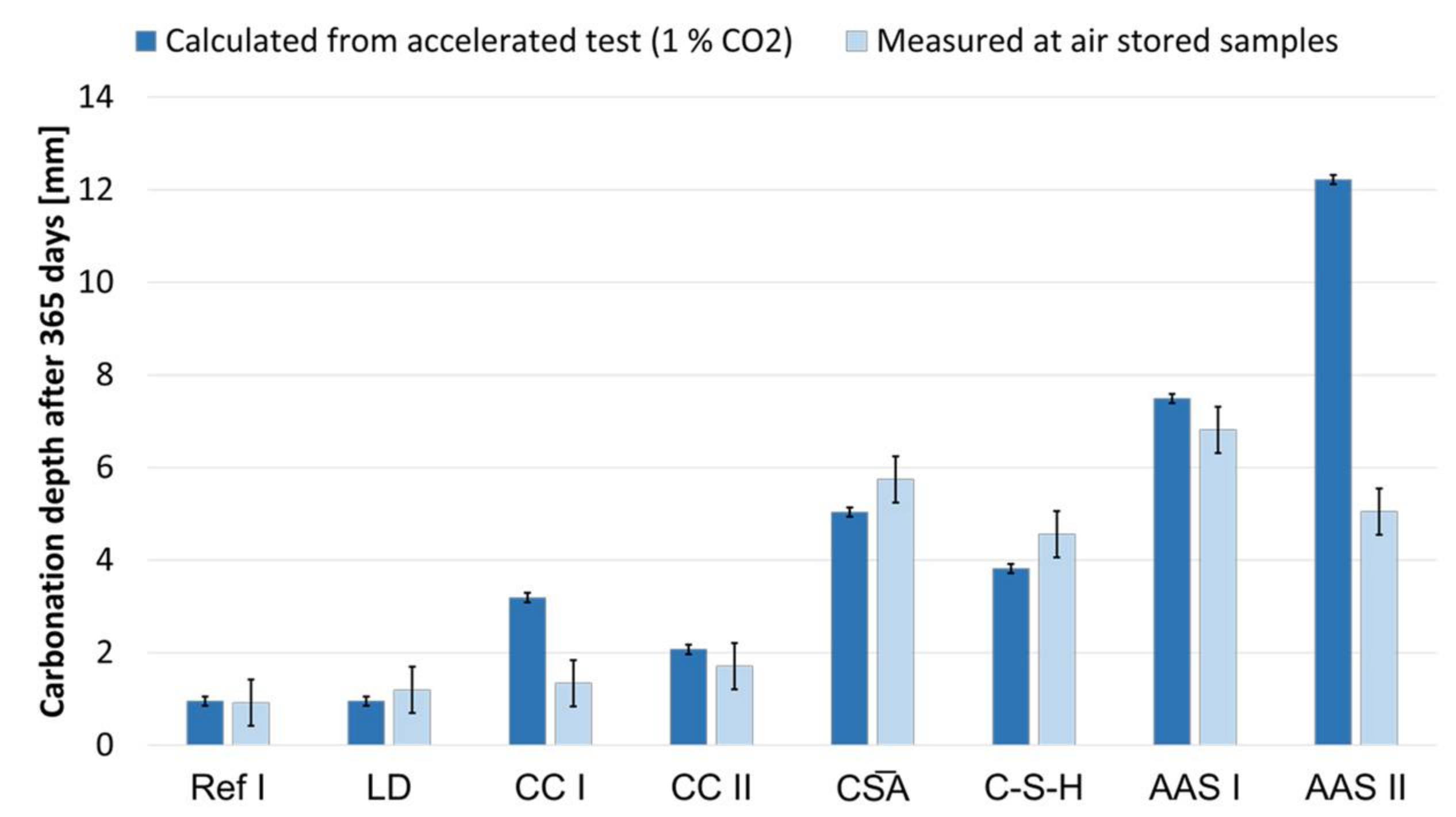
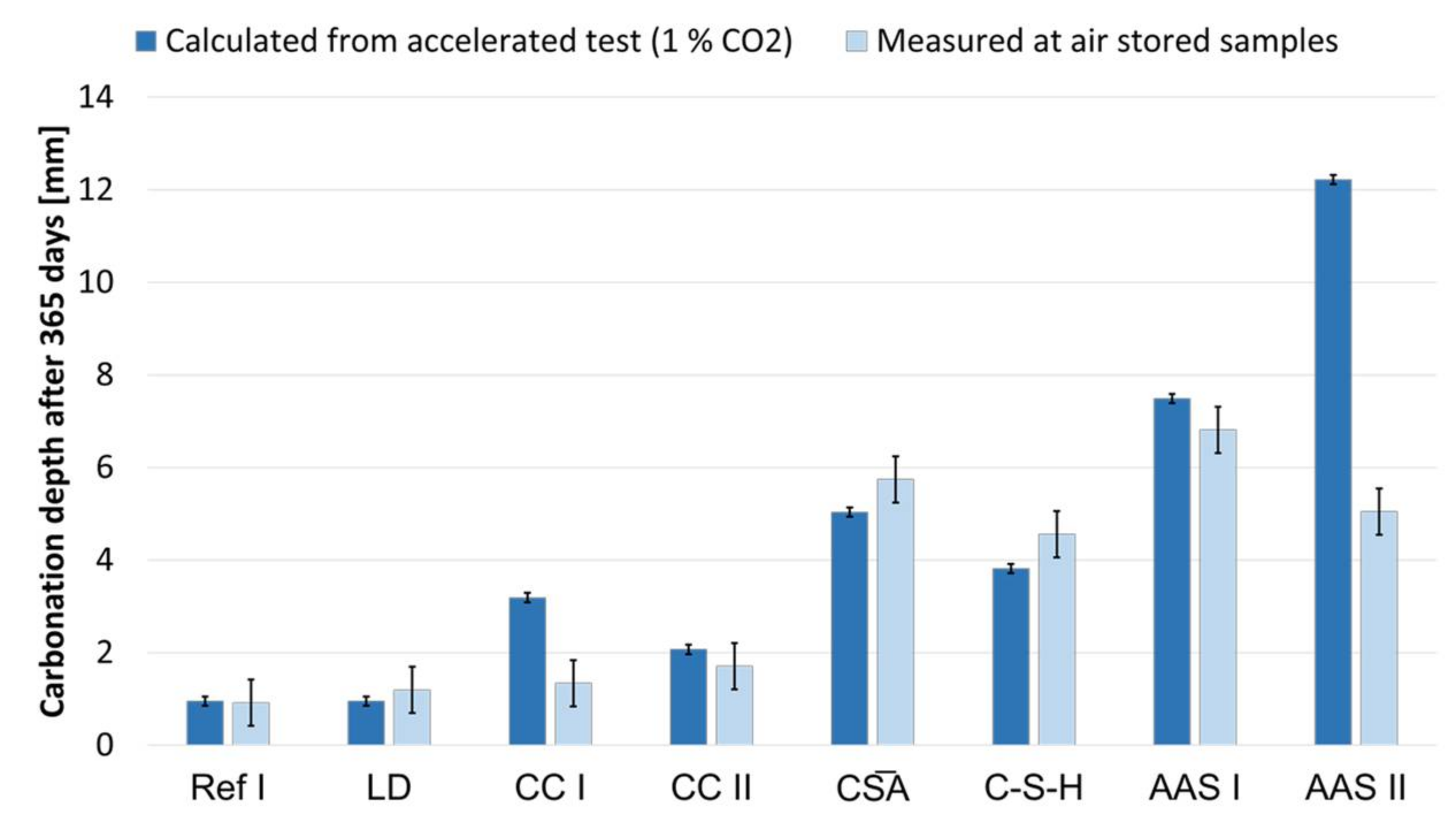
3.7. Carbonation of Mortar Specimens
4. Discussion
4.1. CEM I (Ref I)
4.2. SCM with LD Slag (LD)
4.3. SCM with Metaillite (CC I)
4.4. SCM with Metakaolin (CC II)
4.5. CA
4.6. C-S-H
4.7. Alkali-Activated Metakaolin (Geo MK)
4.8. Alkali-Activated Fly Ash (Geo FA)
4.9. Alkali-Activated Slag; Activator Sodium Silicate (AAS I)
4.10. Hybrid Alkali-Activated Slag; Activator OPC and NaSO4 (AAS II)
5. Conclusions
- The binders investigated differ fundamentally in their hydration and carbonation behavior. Only the composite cements are comparable with the reference cement, whereby in connection with the carbonation of the calcined clay cements, the consumption of portlandite due to the pozzolanic reaction is clearly noticeable. The use of LD slags has no unfavorable effect on the carbonation rate and does not noticeably influence either the hydration or the carbonation reaction.
- The pH of all investigated binders is high enough to allow for a passivation of embedded steel if no corrosive substances are present in the pore solution. Further research of this topic is necessary.
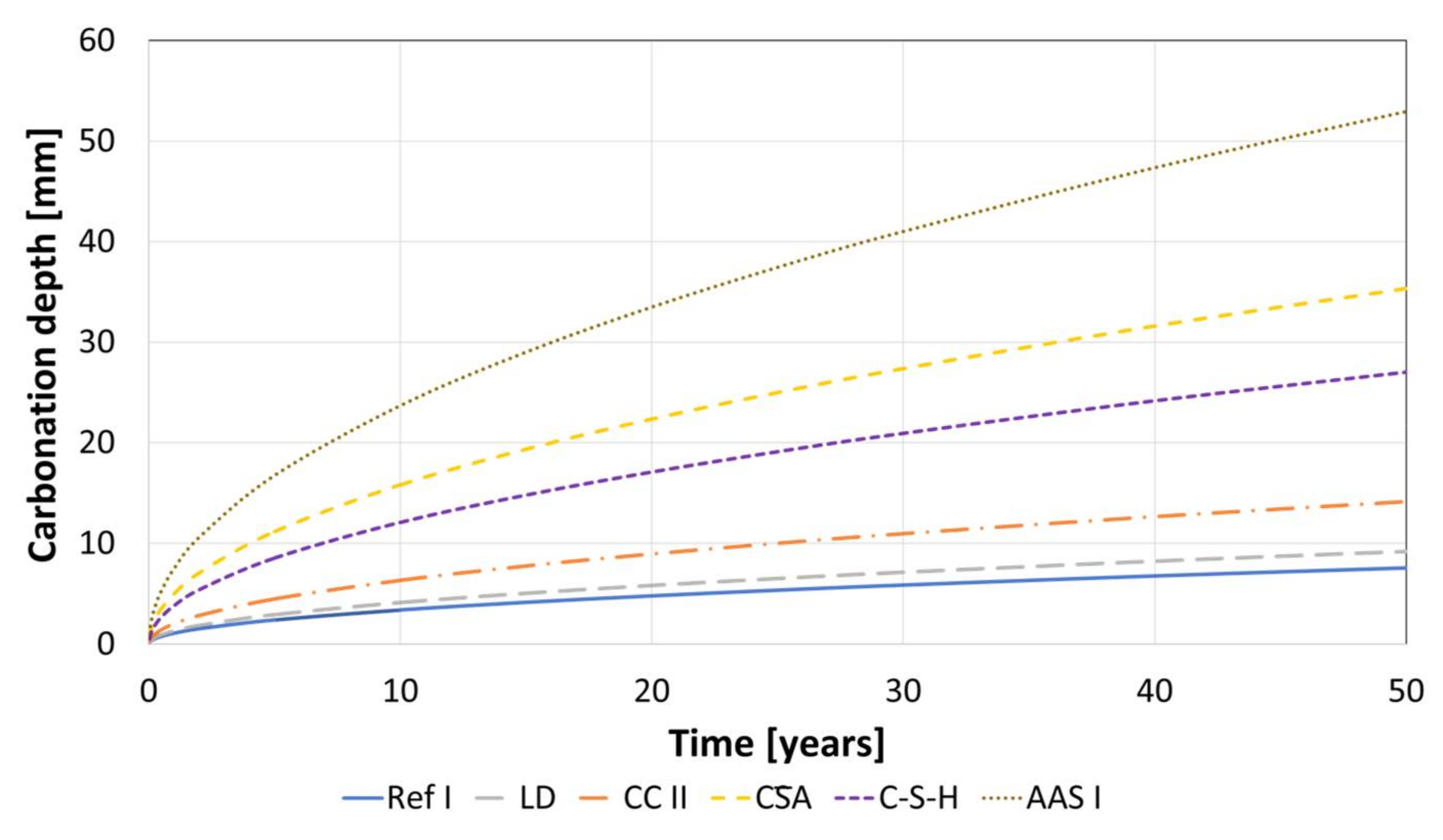
- The accelerated carbonation testing at 1 vol.-% CO2 is applicable for OPC, SCMs with LD slag or metakaolin, CA, C-S-H, and the GGBFS 1 activated by sodium silicate (AAS I).
- The accelerated carbonation testing at 1 vol.-% CO2 is not applicable to gain comparable results to natural carbonation for the SCM with metaillite, the alkali-activated metakaolin and the hybrid alkali-activated slag (AAS II), as the carbonation rates are highly overestimated by the accelerated method.
- The geopolymers show during carbonation little to no change in phase composition by XRD and SEM. The alkali-activated metakaolin showed carbonation depth < 0.1 mm after one year of natural carbonation.
- To establish reliable correlations between the hydration, carbonation, and corrosion behavior of these binders, various studies on corrosion behavior must be carried out.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Explanation |
|---|---|
| AAM | alkali-activated material |
| AAS | alkali-activated slag |
| BOF | basic oxygen furnace |
| CC | calcined clay |
| A | calcium sulfoaluminate |
| C-S-H | calcium silicate hydrate |
| EDX | energy dispersive X-ray spectroscopy |
| FA | fly ash |
| Geo FA | geopolymer with fly ash |
| Geo MK | geopolymer with metakaolin |
| GGBFS | ground granulated blast furnace slag |
| LD | Linz-Donawitz |
| MK | metakaolin |
| OPC | ordinary portland cement |
| QXRD | quantitative X-ray diffraction |
| SCM | supplementary cementitious material |
| SEM | scanning electron microscope |
| XRD | X-ray diffraction |



References
- Bertolini, L.; Elsener, B.; Pedeferri, P.; Polder, R. Corrosion of Steel in Concrete: Prevention, Diagnosis, Repair; Wiley-VCH: Weinheim, Germany, 2005; ISBN 3-527-30800-8. [Google Scholar]
- Gouda, V.K. Corrosion and Corrosion Inhibition of Reinforcing Steel: I. Immersed in Alkaline Solutions. Br. Corros. J. 1970, 5, 198–203. [Google Scholar] [CrossRef]
- Mundra, S. Corrosion of Steel in Alkali-Activated Materials. Ph.D. Thesis, University of Sheffield, Sheffield, UK, 2018. [Google Scholar]
- Beverskog, B.; Puigdomenech, I. Revised pourbaix diagrams for iron at 25–300 °C. Corros. Sci. 1996, 38, 2121–2135. [Google Scholar] [CrossRef]
- Criado, M. The corrosion behaviour of reinforced steel embedded in alkali-activated mortar. In Handbook of Alkali-Activated Cements, Mortars and Concretes; Elsevier: Cambridge, UK, 2015; pp. 333–372. ISBN 9781782422761. [Google Scholar]
- von Greve-Dierfeld, S.; Lothenbach, B.; Vollpracht, A.; Wu, B.; Huet, B.; Andrade, C.; Medina, C.; Thiel, C.; Gruyaert, E.; Vanoutrive, H.; et al. Understanding the carbonation of concrete with supplementary cementitious materials: A critical review by RILEM TC 281-CCC. Matériaux Constr. 2020, 53. [Google Scholar] [CrossRef]
- Pu, Q.; Jiang, L.; Xu, J.; Chu, H.; Xu, Y.; Zhang, Y. Evolution of pH and chemical composition of pore solution in carbonated concrete. Constr. Build. Mater. 2012, 28, 519–524. [Google Scholar] [CrossRef]
- Stefanoni, M.; Angst, U.; Elsener, B. Corrosion rate of carbon steel in carbonated concrete—A critical review. Cem. Concr. Res. 2018, 103, 35–48. [Google Scholar] [CrossRef]
- Kern, D.M. The hydration of carbon dioxide. J. Chem. Educ. 1960, 37, 14. [Google Scholar] [CrossRef]
- Kitamura, M. Strategy for control of crystallization of polymorphs. CrystEngComm 2009, 11, 949. [Google Scholar] [CrossRef]
- Ashraf, W. Carbonation of cement-based materials: Challenges and opportunities. Constr. Build. Mater. 2016, 120, 558–570. [Google Scholar] [CrossRef]
- Deng, H.; Wang, S.; Wang, X.; Du, C.; Shen, X.; Wang, Y.; Cui, F. Two competitive nucleation mechanisms of calcium carbonate biomineralization in response to surface functionality in low calcium ion concentration solution. Regen. Biomater. 2015, 2, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, W.; Xing, F.; Wang, S.; Tang, L.; Lin, S.; Dong, Z. Carbonation of the synthetic calcium silicate hydrate (C-S-H) under different concentrations of CO2: Chemical phases analysis and kinetics. J. CO2 Util. 2020, 35, 303–313. [Google Scholar] [CrossRef]
- Black, L.; Breen, C.; Yarwood, J.; Garbev, K.; Stemmermann, P.; Gasharova, B. Structural Features of C-S-H(I) and Its Carbonation in Air—A Raman Spectroscopic Study. Part II: Carbonated Phases. J. Am. Ceram. Soc. 2007, 90, 908–917. [Google Scholar] [CrossRef]
- Shah, V.; Scrivener, K.; Bhattacharjee, B.; Bishnoi, S. Changes in microstructure characteristics of cement paste on carbonation. Cem. Concr. Res. 2018, 109, 184–197. [Google Scholar] [CrossRef]
- Sevelsted, T.F.; Skibsted, J. Carbonation of C–S–H and C–A–S–H samples studied by 13 C, 27 Al and 29 Si MAS NMR spectroscopy. Cem. Concr. Res. 2015, 71, 56–65. [Google Scholar] [CrossRef]
- Ortaboy, S.; Li, J.; Geng, G.; Myers, R.J.; Monteiro, P.J.M.; Maboudian, R.; Carraro, C. Effects of CO2 and temperature on the structure and chemistry of C–(A–)S–H investigated by Raman spectroscopy. RSC Adv. 2017, 7, 48925–48933. [Google Scholar] [CrossRef] [Green Version]
- de Weerdt, K.; Plusquellec, G.; Belda Revert, A.; Geiker, M.R.; Lothenbach, B. Effect of carbonation on the pore solution of mortar. Cem. Concr. Res. 2019, 118, 38–56. [Google Scholar] [CrossRef]
- Hargis, C.W.; Lothenbach, B.; Müller, C.J.; Winnefeld, F. Carbonation of calcium sulfoaluminate mortars. Cem. Concr. Compos. 2017, 80, 123–134. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kim, J.; Kadoya, K.; Hama, Y. Physical and Chemical Relationships in Accelerated Carbonation Conditions of Alkali-Activated Cement Based on Type of Binder and Alkali Activator. Polymers 2021, 13, 671. [Google Scholar] [CrossRef]
- Bernal, S.A. Microstructural Changes Induced by CO2 Exposure in Alkali-Activated Slag/Metakaolin Pastes. Front. Mater. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Lothenbach, B.; Geiker, M.R.; Kaufmann, J.; Leemann, A.; Ferreiro, S.; Skibsted, J. Experimental studies and thermodynamic modeling of the carbonation of Portland cement, metakaolin and limestone mortars. Cem. Concr. Res. 2016, 88, 60–72. [Google Scholar] [CrossRef]
- Ngala, V.T.; Page, C.L. Effects of Carbonation on Pore Structure and Diffusional Properties of Hydrated Cement Pastes. Cem. Concr. Res. 1997, 27, 995–1007. [Google Scholar] [CrossRef]
- Arandigoyen, M.; Bicer-Simsir, B.; Alvarez, J.I.; Lange, D.A. Variation of microstructure with carbonation in lime and blended pastes. Appl. Surf. Sci. 2006, 252, 7562–7571. [Google Scholar] [CrossRef] [Green Version]
- Lagerblad, B. Carbon Dioxide Uptake during Concrete Life Cycle: State of the Art; Swedish Cement and Concrete Research Institute: Stockholm, Sweden, 2005; ISBN 91-976070-0-2. [Google Scholar]
- Morandeau, A.; Thiéry, M.; Dangla, P. Investigation of the carbonation mechanism of CH and C-S-H in terms of kinetics, microstructure changes and moisture properties. Cem. Concr. Res. 2014, 56, 153–170. [Google Scholar] [CrossRef] [Green Version]
- Gruyaert, E.; van den Heede, P.; De Belie, N. Carbonation of slag concrete: Effect of the cement replacement level and curing on the carbonation coefficient—Effect of carbonation on the pore structure. Cem. Concr. Compos. 2013, 35, 39–48. [Google Scholar] [CrossRef]
- Chen, J.J.; Thomas, J.J.; Jennings, H.M. Decalcification shrinkage of cement paste. Cem. Concr. Res. 2006, 36, 801–809. [Google Scholar] [CrossRef]
- Ludwig, H.M. Zur Rolle von Phasenumwandlungen bei Frost- und Frost-Tausalz-Belastung von Beton. Ph.D. Thesis, Universitat (HAB) Weimar, Weimar, Germany, 1996. [Google Scholar]
- Park, S.-M.; Jang, J.-G.; Kim, G.-M.; Lee, H.-K. Strength Development of Alkali-Activated Fly Ash Exposed to a Carbon Dioxide-Rich Environment at an Early Age. J. Korean Ceram. Soc 2016, 53, 18–23. [Google Scholar] [CrossRef]
- Puertas, F.; Palacios, M.; Vázquez, T. Carbonation process of alkali-activated slag mortars. J. Mater. Sci. 2006, 41, 3071–3082. [Google Scholar] [CrossRef]
- Hunkeler, F.; von Greve-Dierfeld, S. Karbonatisierung von Beton und Korrosionsgeschwindigkeit der Bewehrung im Karbonatisierten Beton: Forschungsprojekt AGB 2013/005 auf Antrag der Arbeitsgruppe Brückenforschung (AGB), Schweiz. 2019. Available online: http://www.mobilityplatform.ch (accessed on 28 December 2021).
- von Greve-Dierfeld, S. Bemessungsregeln zur Sicherstellung der Dauerhaftigkeit XC-Exponierter Stahlbetonbauteile. Ph.D. Thesis, Technische Universität München, München, Germany, 2015. [Google Scholar]
- Hunkeler, F.; Lammar, L. Anfoderungen an den Karbonatisierungswiderstand von Betonen: Forschungsprojekt AGB 2008/012 auf Antrag der Arbeitsgruppe Brückenforschung (AGB), Schweiz. 2012. Available online: http://www.mobilityplatform.ch (accessed on 28 December 2021).
- Malhotra, V.M. Reducing CO2 emissions. Concr. Int. 2006, 28, 42–45. [Google Scholar]
- Scrivener, K. Issues in sustainability in cements and concrete. Am. Ceram. Soc. Bull. 2012, 91, 47–50. [Google Scholar]
- Trümer, A. Calcinierte Tone als Puzzolane der Zukunft: Von den Rohstoffen bis zur Wirkung im Beton; Universität Weimar: Weimar, Germany, 2019; ISBN 978-3-00-065011-6. [Google Scholar]
- Scrivener, K.; Avet, F.; Maraghechi, H.; Zunino, F.; Ston, J.; Hanpongpun, W.; Favier, A. Impacting factors and properties of limestone calcined clay cements (LC 3). Green Mater. 2019, 7, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Bucher, R.; Diederich, P.; Escadeillas, G.; Cyr, M. Service life of metakaolin-based concrete exposed to carbonation. Cem. Concr. Res. 2017, 99, 18–29. [Google Scholar] [CrossRef]
- Homayoonmehr, R.; Ramezanianpour, A.A.; Mirdarsoltany, M. Influence of metakaolin on fresh properties, mechanical properties and corrosion resistance of concrete and its sustainability issues: A review. J. Build. Eng. 2021, 44, 103011. [Google Scholar] [CrossRef]
- Newlands, M.D. Development of a Simulated Natural Carbonation Test and Durability of Selected CEM II Concretes. Ph.D. Thesis, University of Dundee, Dundee, UK, 2001. [Google Scholar]
- Cordoba, G.P.; Zito, S.; Tironi, A.; Rahhal, V.F.; Irassar, E.F. Durability of Concrete Containing Calcined Clays: Comparison of Illite and Low-Grade Kaolin. In Calcined Clays for Sustainable Concrete, Proceedings of the 3rd International Conference on Calcined Clays for Sustainable Concrete, Singapore, 14 April 2020; Bishnoi, S., Ed.; Springer: Singapore, 2020; pp. 631–640. ISBN 978-981-15-2806-4. [Google Scholar]
- Achenbach, R.; Kraft, B.; Ludwig, H.-M.; Raupach, M. Dauerhaftigkeitseigenschaften von alternativen Bindemitteln. Beton- Und Stahlbetonbau 2021, 116, 775–785. [Google Scholar] [CrossRef]
- Yi, H.; Xu, G.; Cheng, H.; Wang, J.; Wan, Y.; Chen, H. An Overview of Utilization of Steel Slag. Procedia Environ. Sci. 2012, 16, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yan, P.; Mi, G. Effect of blended steel slag–GBFS mineral admixture on hydration and strength of cement. Constr. Build. Mater. 2012, 35, 8–14. [Google Scholar] [CrossRef]
- Wulfert, H.; Keyssner, M.; Ludwig, H.M.; Adamczyk, B. Metal recovery and conversion of steel slag into highly reactive cement components [Metallgewinnung und Umwandlung von LD-Schlacke in hochreaktive Zementkomponenten]. ZKG Int. 2013, 66, 34–40. [Google Scholar]
- Santos, R.M.; Ling, D.; Sarvaramini, A.; Guo, M.; Elsen, J.; Larachi, F.; Beaudoin, G.; Blanpain, B.; van Gerven, T. Stabilization of basic oxygen furnace slag by hot-stage carbonation treatment. Chem. Eng. J. 2012, 203, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Vollpracht, A.; Brameshuber, W. Einbindung von Schwermetallen in Portlandzementstein: Lehrstuhl für Baustoffkunde. Ph.D. Thesis, RWTH Aachen University, Aachen, Germany, 2012. [Google Scholar]
- Jiang, Y.; Ling, T.-C.; Shi, C.; Pan, S.-Y. Characteristics of steel slags and their use in cement and concrete—A review. Resour. Conserv. Recycl. 2018, 136, 187–197. [Google Scholar] [CrossRef]
- Kaja, A.M.; Delsing, A.; van der Laan, S.R.; Brouwers, H.; Yu, Q. Effects of carbonation on the retention of heavy metals in chemically activated BOF slag pastes. Cem. Concr. Res. 2021, 148, 106534. [Google Scholar] [CrossRef]
- Aranda, M.A.G.; De La Torre, A.G. Sulfoaluminate cement. In Eco-Efficient Concrete; Elsevier: Amsterdam, The Netherlands, 2013; pp. 488–522. ISBN 9780857094247. [Google Scholar]
- Winnefeld, F.; Lothenbach, B. Hydration of calcium sulfoaluminate cements—Experimental findings and thermodynamic modelling. Cem. Concr. Res. 2010, 40, 1239–1247. [Google Scholar] [CrossRef]
- Telesca, A.; Marroccoli, M.; Pace, M.L.; Tomasulo, M.; Valenti, G.L.; Monteiro, P. A hydration study of various calcium sulfoaluminate cements. Cem. Concr. Compos. 2014, 53, 224–232. [Google Scholar] [CrossRef]
- Li, L.; Wang, R.; Zhang, S. Effect of curing temperature and relative humidity on the hydrates and porosity of calcium sulfoaluminate cement. Constr. Build. Mater. 2019, 213, 627–636. [Google Scholar] [CrossRef]
- Tan, B.; Okoronkwo, M.U.; Kumar, A.; Ma, H. Durability of calcium sulfoaluminate cement concrete. J. Zhejiang Univ. Sci. A 2020, 21, 118–128. [Google Scholar] [CrossRef]
- Möller, H. A Novel Cements Based on Hydraulic Calcium Hydrosilicates; Celitement GmbH: Paris, France, 2017. [Google Scholar]
- Provis, J.L.; van Deventer, J.S.J. (Eds.) Alkali Activated Materials: State-of-the-Art Report, RILEM TC 224-AAM; Springer: Dordrecht, The Netherlands, 2014; ISBN 978-94-007-7672-2. [Google Scholar]
- Garcia-Lodeiro, I.; Palomo, A.; Fernández-Jiménez, A.; Macphee, D.E. Compatibility studies between N-A-S-H and C-A-S-H gels. Study in the ternary diagram Na2O–CaO–Al2O3–SiO2–H2O. Cem. Concr. Res. 2011, 41, 923–931. [Google Scholar] [CrossRef]
- Provis, J.L.; van Deventer, J.S.J. Alkali Activated Materials; Springer: Dordrecht, The Netherlands, 2014; ISBN 978-94-007-7671-5. [Google Scholar]
- Garcia-Lodeiro, I.; Palomo, A.; Fernández-Jiménez, A. An overview of the chemistry of alkali-activated cement-based binders. In Handbook of Alkali-Activated Cements, Mortars and Concretes; Elsevier: Amsterdam, The Netherlands, 2015; pp. 19–47. ISBN 9781782422761. [Google Scholar]
- Bernal, S.A.; Provis, J.L.; Fernández-Jiménez, A.; Krivenko, P.V.; Kavalerova, E.; Palacios, M.; Shi, C. Binder Chemistry—High-Calcium Alkali-Activated Materials. In Alkali Activated Materials: State-of-the-Art Report, RILEM TC 224-AAM; Provis, J.L., van Deventer, J.S.J., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 59–91. ISBN 978-94-007-7672-2. [Google Scholar]
- Zhang, J.; Shi, C.; Zhang, Z.; Ou, Z. Durability of alkali-activated materials in aggressive environments: A review on recent studies. Constr. Build. Mater. 2017, 152, 598–613. [Google Scholar] [CrossRef]
- Bernal, S.A.; Provis, J.L.; Brice, D.G.; Kilcullen, A.; Duxson, P.; van Deventer, J.S. Accelerated carbonation testing of alkali-activated binders significantly underestimates service life: The role of pore solution chemistry. Cem. Concr. Res. 2012, 42, 1317–1326. [Google Scholar] [CrossRef]
- Gluth, G.J.G.; Arbi, K.; Bernal, S.A.; Bondar, D.; Castel, A.; Chithiraputhiran, S.; Dehghan, A.; Dombrowski-Daube, K.; Dubey, A.; Ducman, V.; et al. RILEM TC 247-DTA round robin test: Carbonation and chloride penetration testing of alkali-activated concretes. Mater. Struct. 2020, 53, 131. [Google Scholar] [CrossRef]
- Pouhet, R.; Cyr, M. Studies of Natural and Accelerated Carbonation in Metakaolin-Based Geopolymer. AST 2014, 92, 38–43. [Google Scholar] [CrossRef]
- Pouhet, R.; Cyr, M. Carbonation in the pore solution of metakaolin-based geopolymer. Cem. Concr. Res. 2016, 88, 227–235. [Google Scholar] [CrossRef]
- Gökçe, H.S.; Tuyan, M.; Nehdi, M.L. Alkali-activated and geopolymer materials developed using innovative manufacturing techniques: A critical review. Constr. Build. Mater. 2021, 303, 124483. [Google Scholar] [CrossRef]
- Amran, M.; Abdelgader, H.S.; Onaizi, A.M.; Fediuk, R.; Ozbakkaloglu, T.; Rashid, R.S.; Murali, G. 3D-printable alkali-activated concretes for building applications: A critical review. Constr. Build. Mater. 2022, 319, 126126. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, Z.; Wang, H. Hydration mechanisms and durability of hybrid alkaline cements (HACs): A review. Constr. Build. Mater. 2021, 266, 121039. [Google Scholar] [CrossRef]
- Wang, L.; Zhan, S.; Tang, X.; Xu, Q.; Qian, K. Pore Solution Chemistry of Calcium Sulfoaluminate Cement and Its Effects on Steel Passivation. Appl. Sci. 2019, 9, 1092. [Google Scholar] [CrossRef] [Green Version]
- Plusquellec, G.; Geiker, M.R.; Lindgård, J.; Duchesne, J.; Fournier, B.; De Weerdt, K. Determination of the pH and the free alkali metal content in the pore solution of concrete: Review and experimental comparison. Cem. Concr. Res. 2017, 96, 13–26. [Google Scholar] [CrossRef]
- Granizo, M.L.; Blanco-Varela, M.T.; Martínez-Ramírez, S. Alkali activation of metakaolins: Parameters affecting mechanical, structural and microstructural properties. J. Mater. Sci. 2007, 42, 2934–2943. [Google Scholar] [CrossRef]
- Carsana, M.; Canonico, F.; Bertolini, L. Corrosion resistance of steel embedded in sulfoaluminate-based binders. Cem. Concr. Compos. 2018, 88, 211–219. [Google Scholar] [CrossRef]
- Setzer, M.J. Entwicklung und Präzision eines Prüfverfahrens zum Frost-Tausalz-Widerstand. Weimar 1994, 40, 87–93. [Google Scholar]
- Bernal, S.A. The resistance of alkali-activated cement-based binders to carbonation. In Handbook of Alkali-Activated Cements, Mortars and Concretes; Elsevier: Amsterdam, The Netherlands, 2015; pp. 319–332. ISBN 9781782422761. [Google Scholar]
- Bernal, S.A.; Provis, J.L.; Walkley, B.; San Nicolas, R.; Gehman, J.D.; Brice, D.G.; Kilcullen, A.R.; Duxson, P.; van Deventer, J.S. Gel nanostructure in alkali-activated binders based on slag and fly ash, and effects of accelerated carbonation. Cem. Concr. Res. 2013, 53, 127–144. [Google Scholar] [CrossRef]
- Kourounis, S.; Tsivilis, S.; Tsakiridis, P.E.; Papadimitriou, G.D.; Tsibouki, Z. Properties and hydration of blended cements with steelmaking slag. Cem. Concr. Res. 2007, 37, 815–822. [Google Scholar] [CrossRef]
- Palod, R.; Deo, S.V.; Ramtekkar, G.D. Sustainable Approach for Linz-Donawitz Slag Waste as a Replacement of Cement in Concrete: Mechanical, Microstructural, and Durability Properties. Adv. Civ. Eng. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Ma, J.; Yu, Z.; Ni, C.; Shi, H.; Shen, X. Effects of limestone powder on the hydration and microstructure development of calcium sulphoaluminate cement under long-term curing. Constr. Build. Mater. 2019, 199, 688–695. [Google Scholar] [CrossRef]
- Gastaldi, D.; Bertola, F.; Canonico, F.; Buzzi, L.; Mutke, S.; Irico, S.; Paul, G.; Marchese, L.; Boccaleri, E. A chemical/mineralogical investigation of the behavior of sulfoaluminate binders submitted to accelerated carbonation. Cem. Concr. Res. 2018, 109, 30–41. [Google Scholar] [CrossRef]
- Si, R.; Dai, Q.; Guo, S.; Wang, J. Mechanical property, nanopore structure and drying shrinkage of metakaolin-based geopolymer with waste glass powder. J. Clean. Prod. 2020, 242, 118502. [Google Scholar] [CrossRef]
- Perera, D.S.; Uchida, O.; Vance, E.R.; Finnie, K.S. Influence of curing schedule on the integrity of geopolymers. J. Mater. Sci. 2007, 42, 3099–3106. [Google Scholar] [CrossRef]
- Law, D.W.; Adam, A.A.; Molyneaux, T.K.; Patnaikuni, I.; Wardhono, A. Long term durability properties of class F fly ash geopolymer concrete. Matériaux Constr. 2015, 48, 721–731. [Google Scholar] [CrossRef]
- Criado, M.; Palomo, A.; Fernandezjimenez, A. Alkali activation of fly ashes. Part 1: Effect of curing conditions on the carbonation of the reaction products. Fuel 2005, 84, 2048–2054. [Google Scholar] [CrossRef]
- Sufian Badar, M.; Kupwade-Patil, K.; Bernal, S.A.; Provis, J.L.; Allouche, E.N. Corrosion of steel bars induced by accelerated carbonation in low and high calcium fly ash geopolymer concretes. Constr. Build. Mater. 2014, 61, 79–89. [Google Scholar] [CrossRef]
- Pasupathy, K.; Sanjayan, J.; Rajeev, P. Evaluation of alkalinity changes and carbonation of geopolymer concrete exposed to wetting and drying. J. Build. Eng. 2021, 35, 102029. [Google Scholar] [CrossRef]
- Myers, R.J.; Bernal, S.A.; Gehman, J.D.; van Deventer, J.S.; Provis, J.L. The Role of Al in Cross-Linking of Alkali-Activated Slag Cements. J. Am. Ceram. Soc. 2015, 98, 996–1004. [Google Scholar] [CrossRef]
- Myers, R.J.; Bernal, S.A.; Provis, J.L. Phase diagrams for alkali-activated slag binders. Cem. Concr. Res. 2017, 95, 30–38. [Google Scholar] [CrossRef]
- Li, N.; Shi, C.; Wang, Q.; Zhang, Z.; Ou, Z. Composition design and performance of alkali-activated cements. Mater. Struct. 2017, 50. [Google Scholar] [CrossRef]
- Collins, F.; Sanjayan, J. Effect of pore size distribution on drying shrinking of alkali-activated slag concrete. Cem. Concr. Res. 2000, 30, 1401–1406. [Google Scholar] [CrossRef]
- Puertas, F.; Fernández-Jiménez, A.; Blanco-Varela, M. Pore solution in alkali-activated slag cement pastes. Relation to the composition and structure of calcium silicate hydrate. Cem. Concr. Res. 2004, 34, 139–148. [Google Scholar] [CrossRef]
- Bernal, S.A.; Ke, X.; Criado, M.; Mundra, S.; Provis, J.L. Factors controlling carbonation resistance of alkali-activated materials. In Special Publication 320, Proceedings of the 10th ACI/RILEM International Conference on Cementitious Materials and Alternative Binders for Sustainable Concrete, Montreal, QC, Canada, 2–4 October 2017; Tagnit-Hamou, A., Ed.; American Concrete Institute: Farmington Hills, MI, USA, 2017; pp. 361–3610. ISBN 0193-2527. [Google Scholar]
- Li, N.; Farzadnia, N.; Shi, C. Microstructural changes in alkali-activated slag mortars induced by accelerated carbonation. Cem. Concr. Res. 2017, 100, 214–226. [Google Scholar] [CrossRef]









| Group | Identifier | Description | w/b |
|---|---|---|---|
| Reference | Ref I | CEM I 42.5 N | 0.50 |
| SCMs | CC I | 30% Metaillite, 70% Ref I | 0.50 |
| CC II | 30% Metakaolin, 70% Ref I | 0.50 | |
| LD | 30% LD slag, 70% Ref I | 0.49 | |
| CA | CA | CA with tartaric acid as retarder | 0.50 |
| C-S-H | C-S-H | Celitement® | 0.40 |
| AAMs | Geo MK | Metakaolin activated by potassium silicate | 0.50 |
| Geo FA | Fly ash activated by NaOH and sodium silicate | 0.34 | |
| AAS I | GGBFS 1 activated by sodium silicate | 0.38 | |
| AAS II | GGBFS 2 activated by CEM I and Na2SO4 | 0.39 |
| Portlandite (wt.-%) | Ettringite (wt.-%) | X-ray Amorphous (wt.-%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 d | 7 d | 28 d | 56 d | 1 d | 7 d | 28 d | 56 d | 1 d | 7 d | 28 d | 56 d | |
| Ref I | 18 | 24 | 26 | 20 | 7 | 8 | 7 | 5 | 15 | 32 | 34 | 50 |
| CC I | 18 | 14 | 13 | 11 | 7 | 2 | 3 | 4 | 17 | 52 | 50 | 53 |
| CC II | 14 | 7 | 6 | 4 | 4 | 2 | 3 | 4 | 48 | 70 | 71 | 74 |
| LD | 13 | 19 | 21 | 19 | 6 | 4 | 3 | 2 | 25 | 36 | 41 | 52 |
| A | <1 | <1 | <1 | <1 | 34 | 50 | 52 | 40 | 15 | 2 | 0 | 26 |
| C-S-H | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 85 | 85 | 90 | 88 |
| Geo MK | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 50 | 50 | 49 | 54 |
| Geo FA | - | n.d. | n.d. | n.d. | - | n.d. | n.d. | n.d. | - | 75 | 80 | 78 |
| AAS I | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 57 | 52 | 51 | 54 |
| AAS II | n.d. | n.d. | n.d. | n.d. | 8 | 8 | 10 | 13 | 13 | 19 | 22 | 32 |
| Percentage (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gel Pores (<0.03 µm) | Capillary Pores (0.03 µm to 30 µm) | |||||||
| 1 d | 28 d | 56 d | 56 d + 56 d Carbo | 1 d | 28 d | 56 d | 56 d + 56 d Carbo | |
| Ref I | 14 | 62 | 49 | 53 | 86 | 38 | 51 | 47 |
| CC I | 17 | 56 | 63 | 52 | 83 | 44 | 37 | 48 |
| CC II | 21 | 91 | 87 | 51 | 79 | 9 | 13 | 49 |
| LD | 15 | 45 | 55 | 62 | 85 | 55 | 45 | 38 |
| A | 29 | 35 | 34 | 32 | 71 | 65 | 66 | 68 |
| C-S-H | 60 | 74 | 74 | 62 | 40 | 26 | 26 | 38 |
| Geo MK | 91 | 89 | 95 | 92 | 9 | 11 | 5 | 8 |
| Geo FA | n.d. | 79 | 12 | 68 | n.d. | 21 | 88 | 32 |
| AAS I | 81 | 72 | 77 | 53 | 19 | 28 | 23 | 47 |
| AAS II | 11 | 23 | 50 | 25 | 89 | 77 | 50 | 75 |
| KACC | R2 | KNAC | |
|---|---|---|---|
| - | |||
| Ref I | 4.8 | 0.97 | 1.0 |
| LD | 4.7 | 0.96 | 0.9 |
| CC I | 15.9 | 1.0 | 3.2 |
| CC II | 10.3 | 1.0 | 2.1 |
| A | 25.2 | 0.99 | 5.0 |
| C-S-H | 19.1 | 0.98 | 3.8 |
| Geo MK | - | - | - |
| Geo FA | 22.5 | 0.99 | 4.5 |
| AAS I | 37.4 | 1.0 | 7.5 |
| AAS II | 61.1 | - | 12.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraft, B.; Achenbach, R.; Ludwig, H.-M.; Raupach, M. Hydration and Carbonation of Alternative Binders. Corros. Mater. Degrad. 2022, 3, 19-52. https://doi.org/10.3390/cmd3010003
Kraft B, Achenbach R, Ludwig H-M, Raupach M. Hydration and Carbonation of Alternative Binders. Corrosion and Materials Degradation. 2022; 3(1):19-52. https://doi.org/10.3390/cmd3010003
Chicago/Turabian StyleKraft, Bettina, Rebecca Achenbach, Horst-Michael Ludwig, and Michael Raupach. 2022. "Hydration and Carbonation of Alternative Binders" Corrosion and Materials Degradation 3, no. 1: 19-52. https://doi.org/10.3390/cmd3010003
APA StyleKraft, B., Achenbach, R., Ludwig, H.-M., & Raupach, M. (2022). Hydration and Carbonation of Alternative Binders. Corrosion and Materials Degradation, 3(1), 19-52. https://doi.org/10.3390/cmd3010003