
The Alkyne Zipper Reaction: A Useful Tool in Synthetic Chemistry

 and
and
Abstract
:1. Introduction
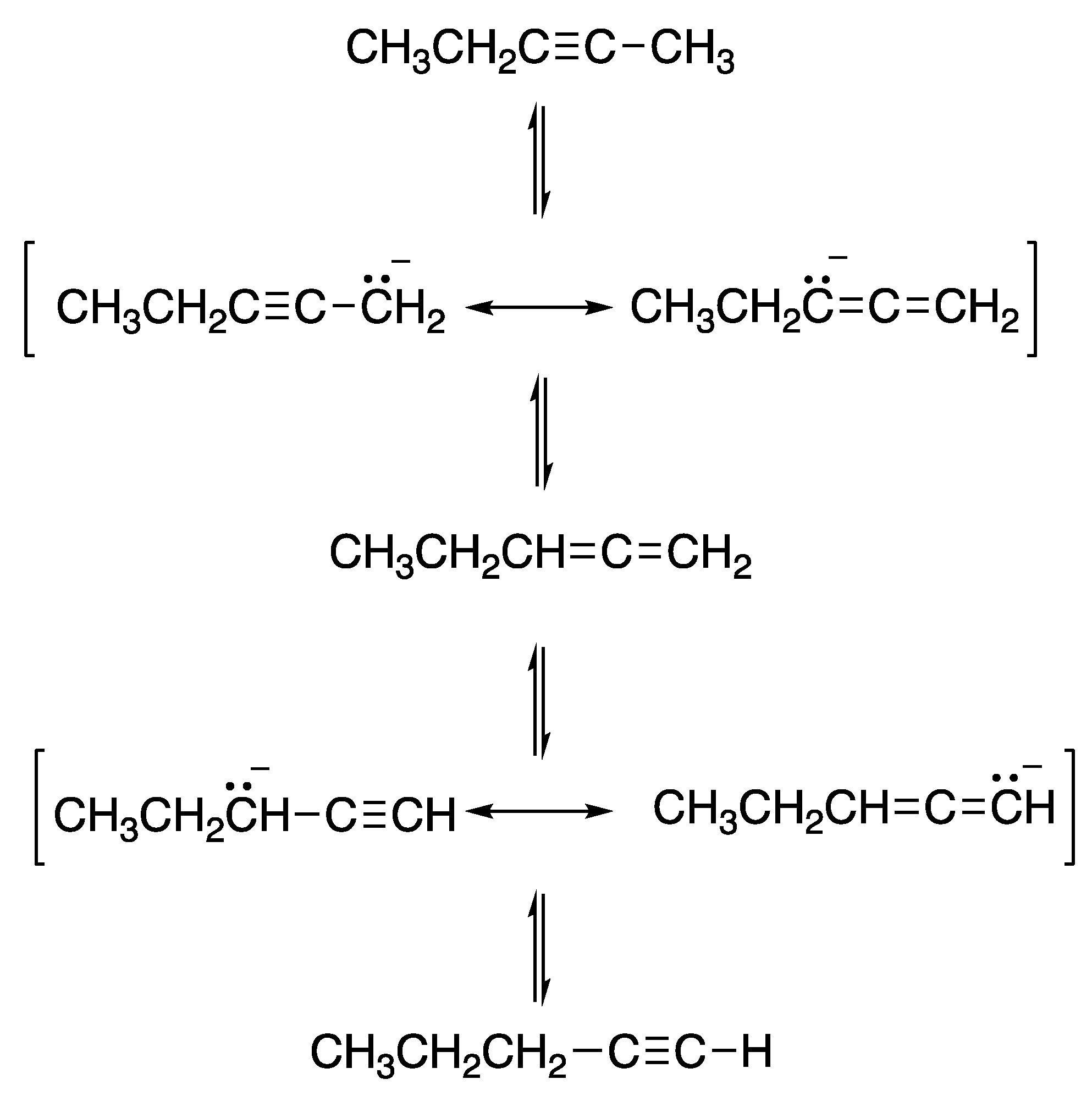
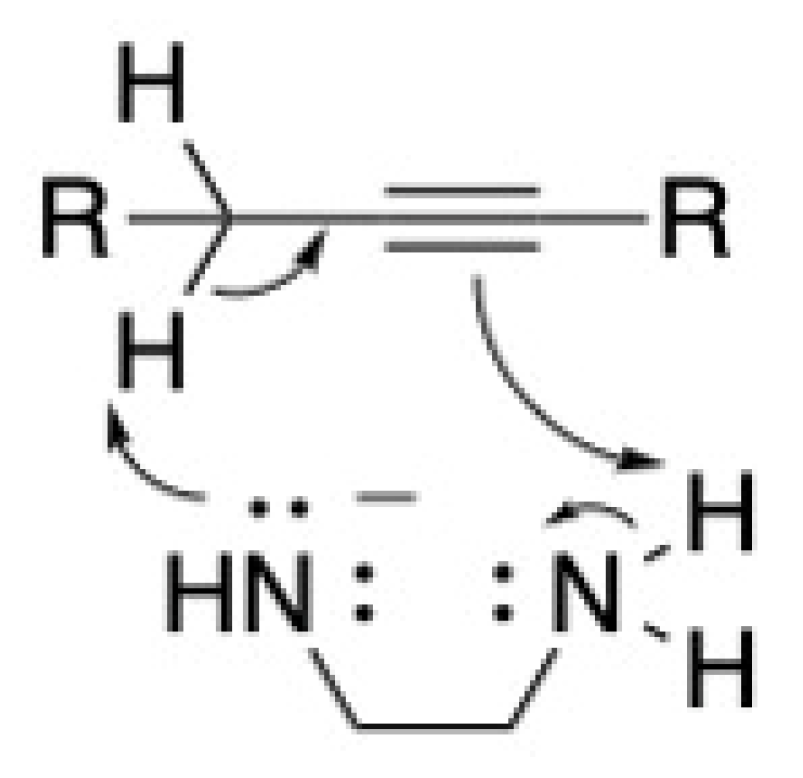
2. The Alkyne Zipper Reaction Mechanism
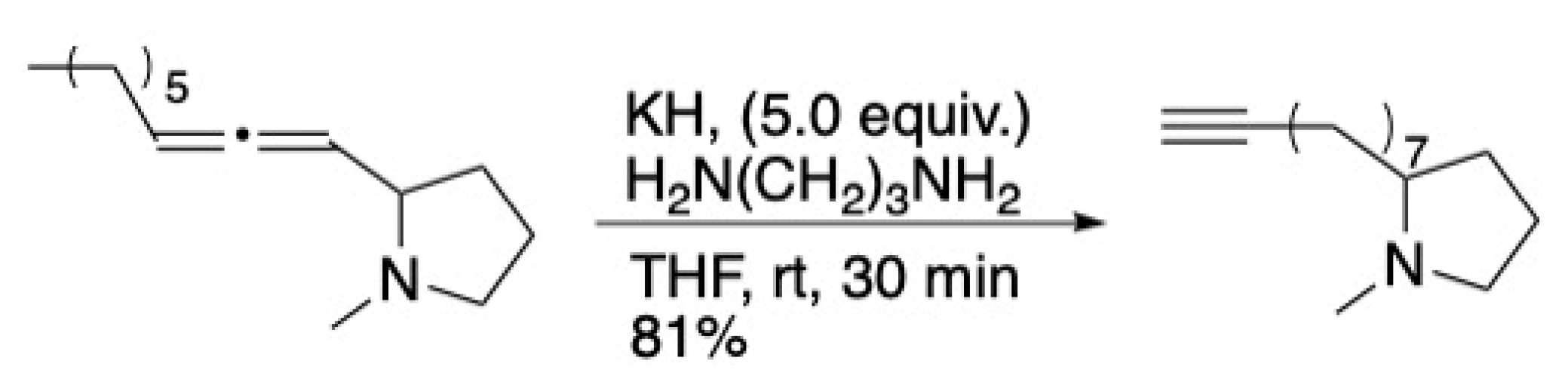
3. The Alkyne Zipper Reaction in Synthesis
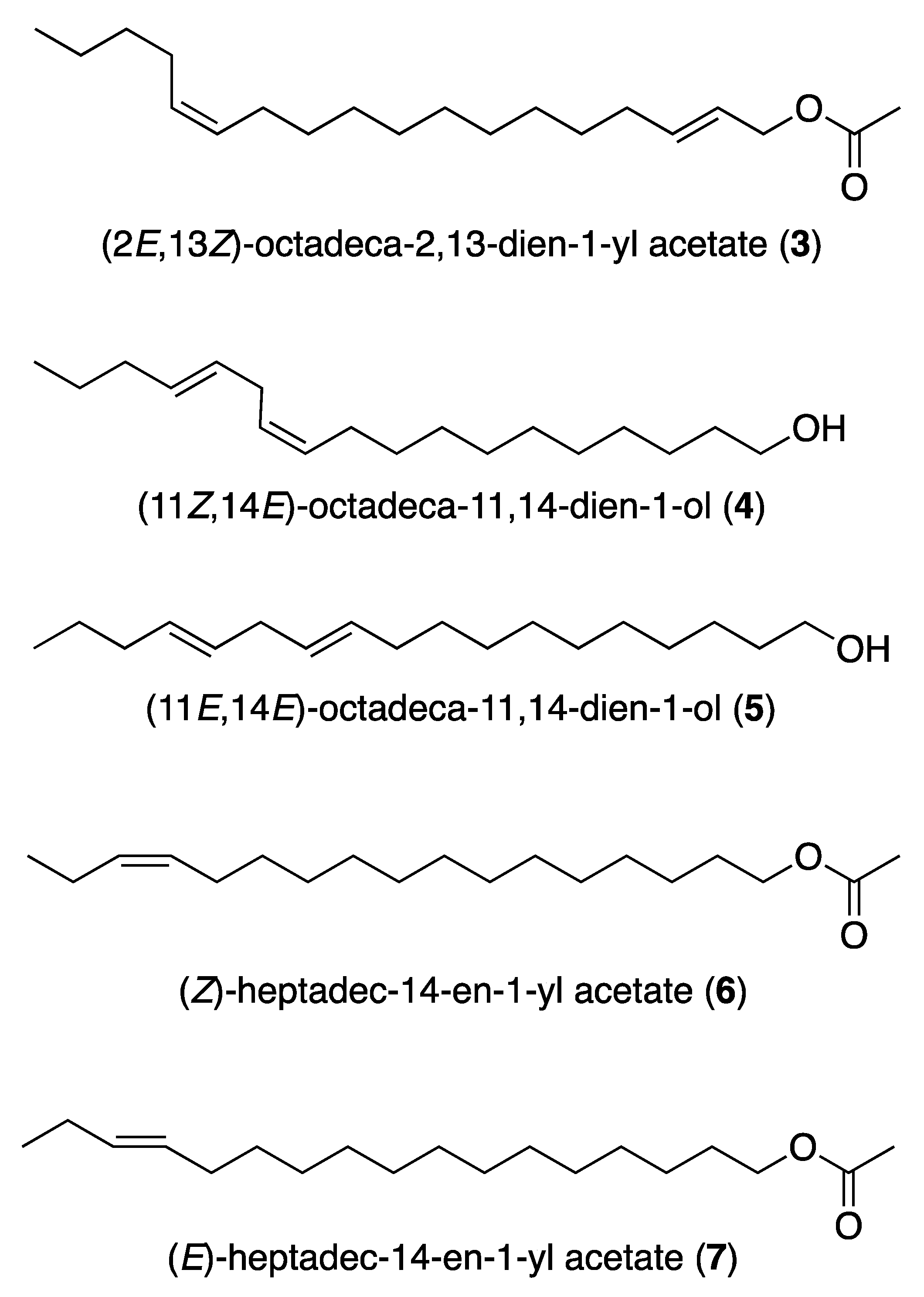
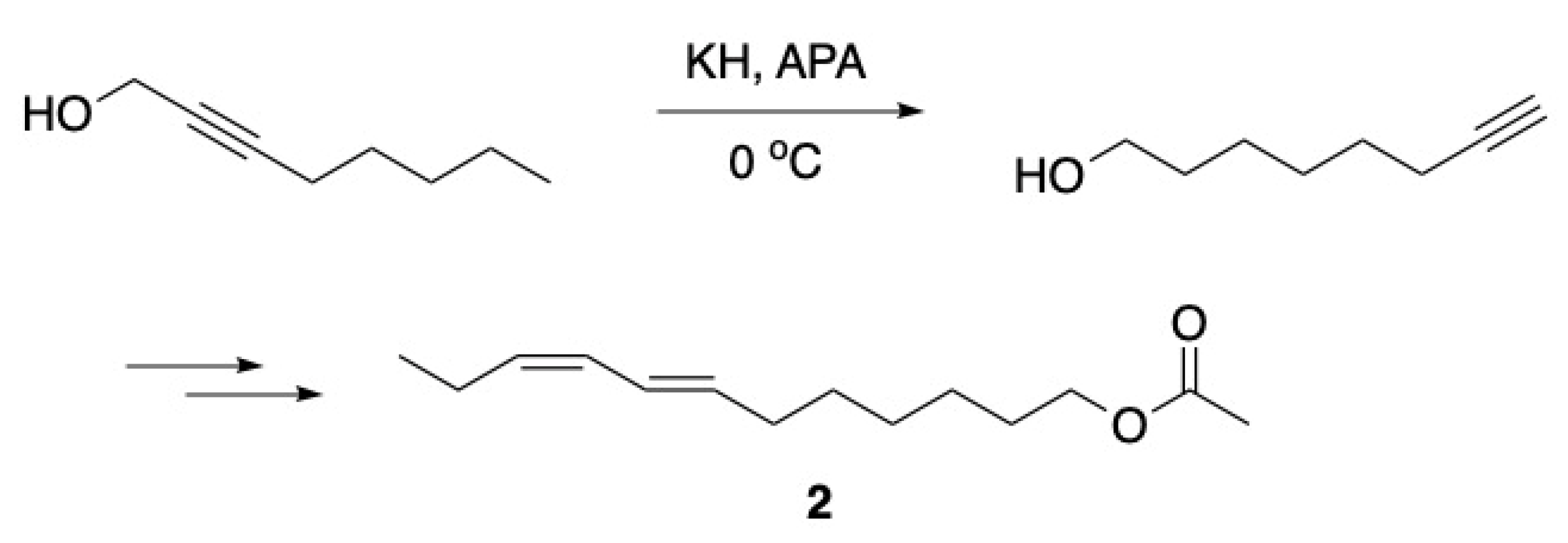
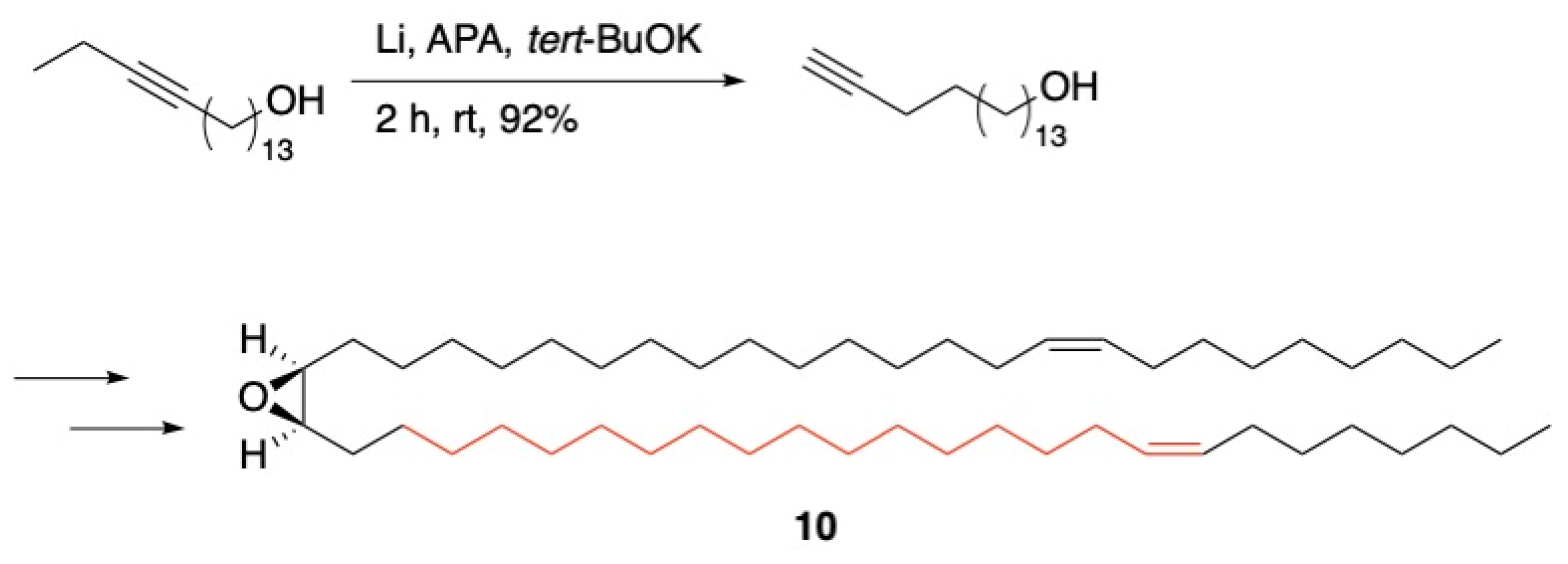
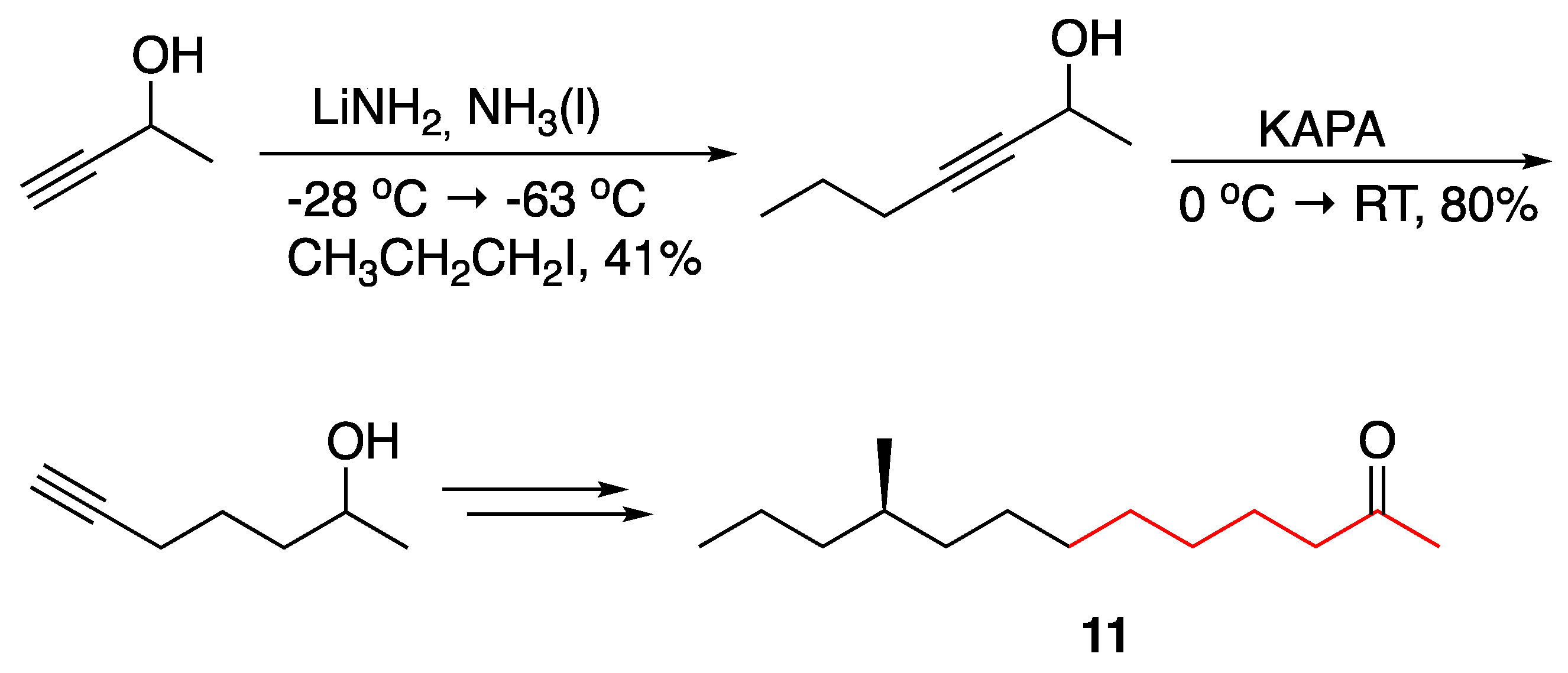
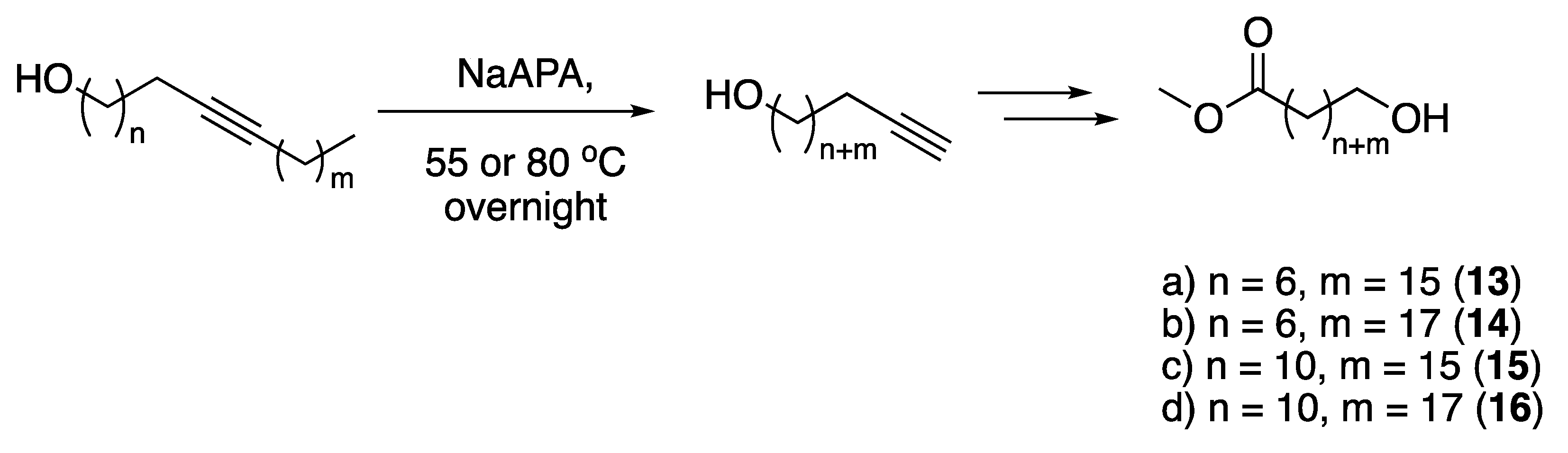
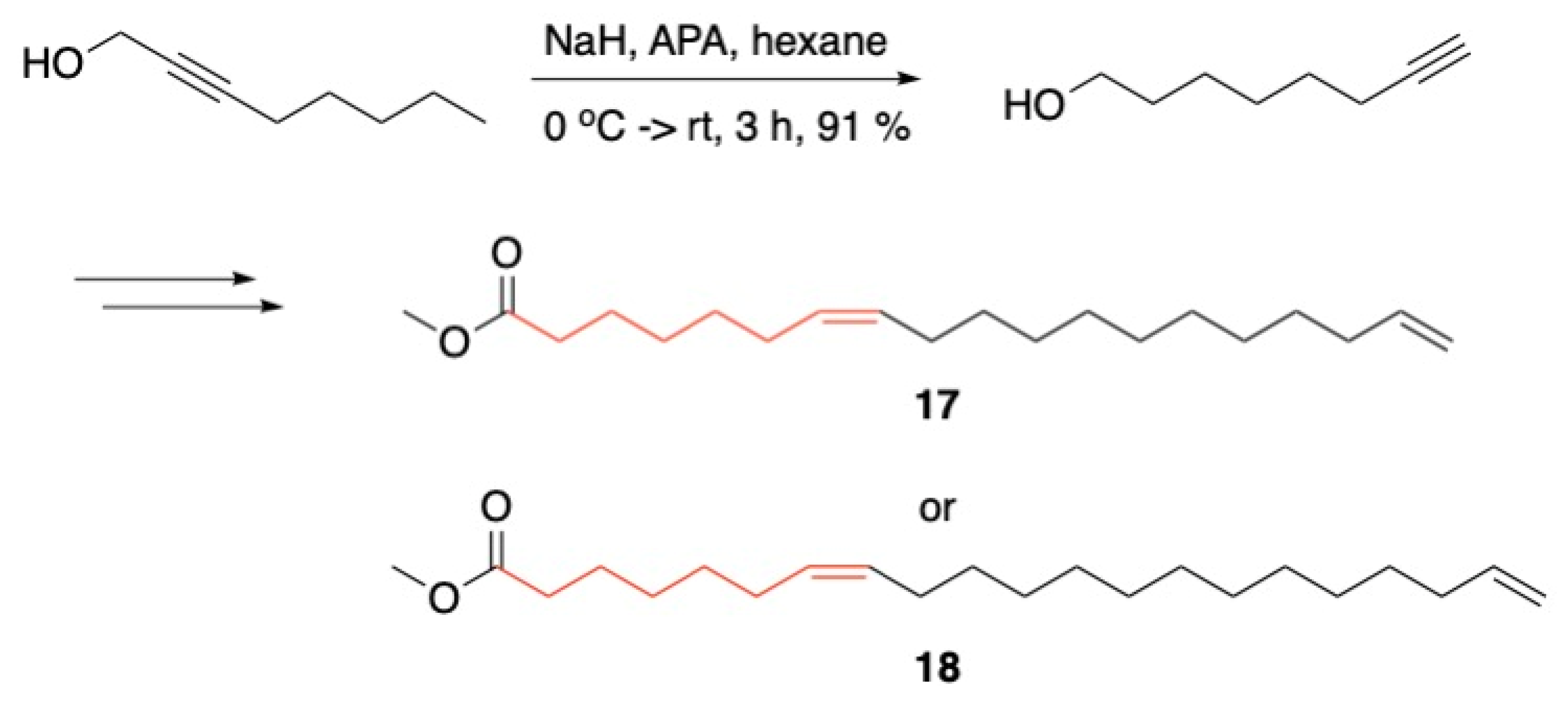
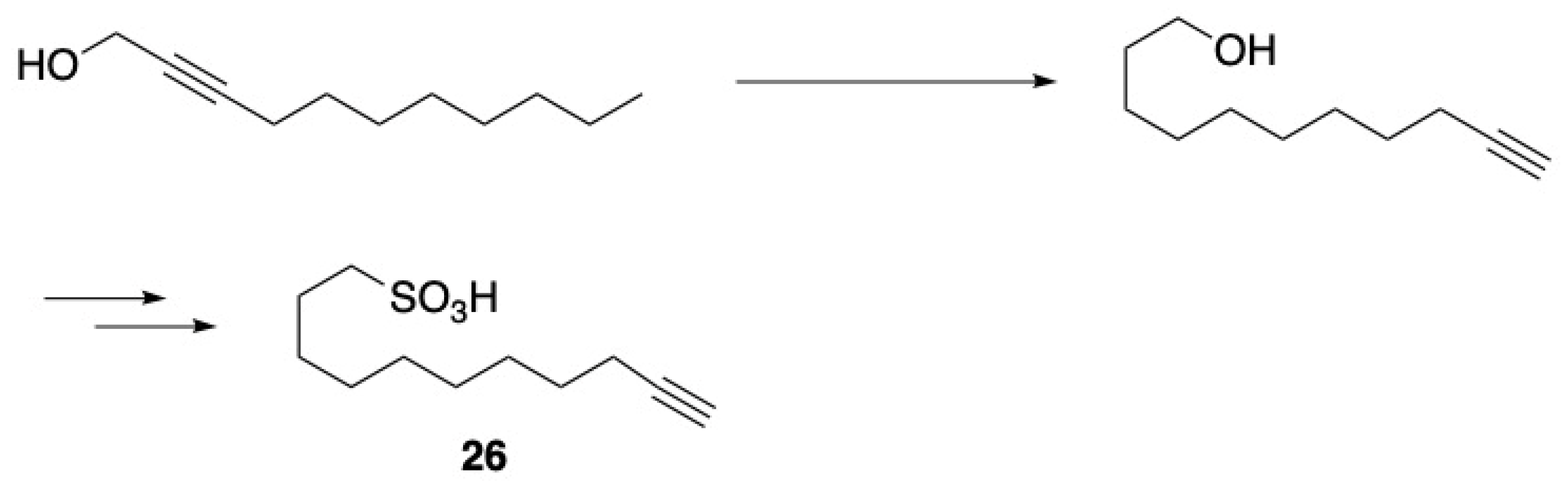
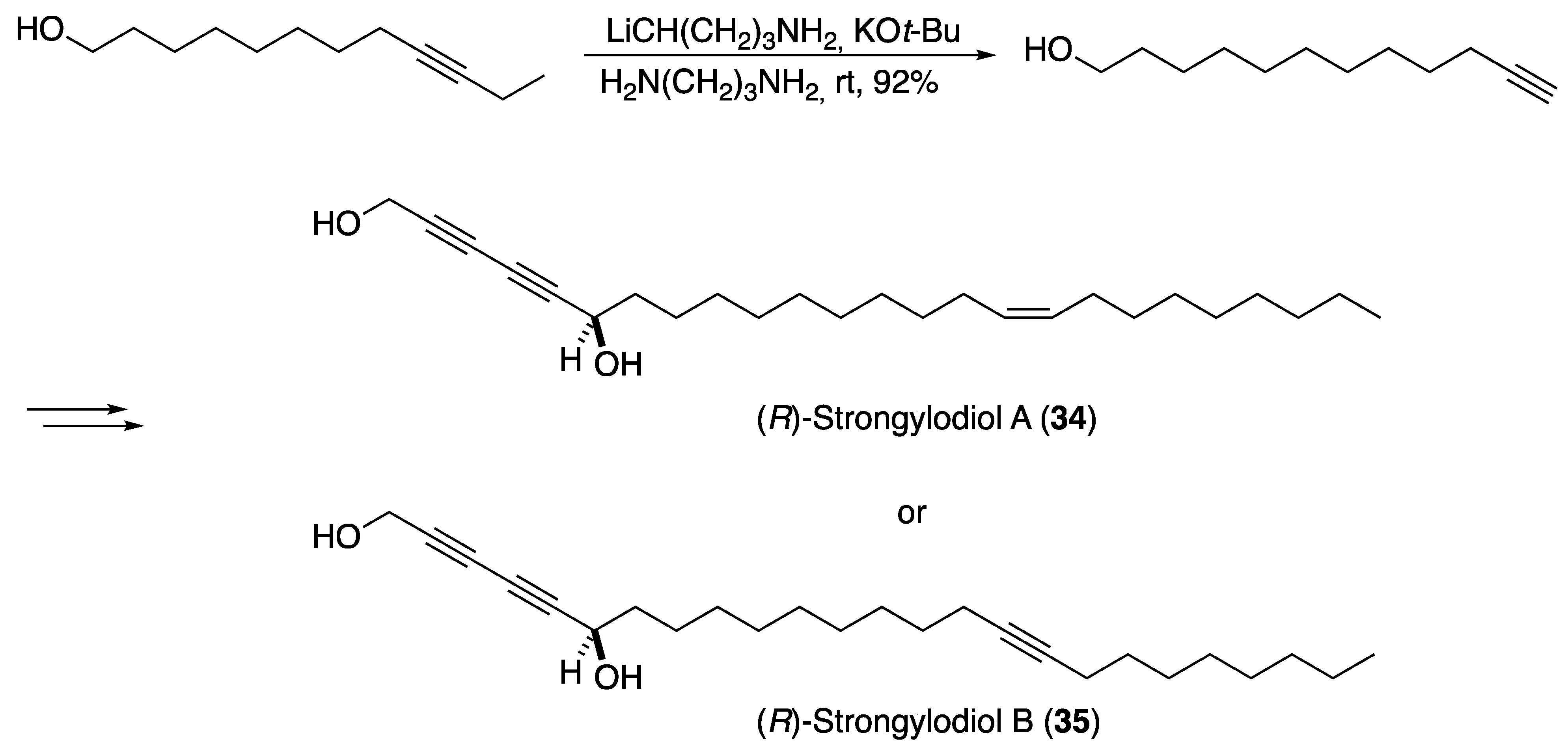
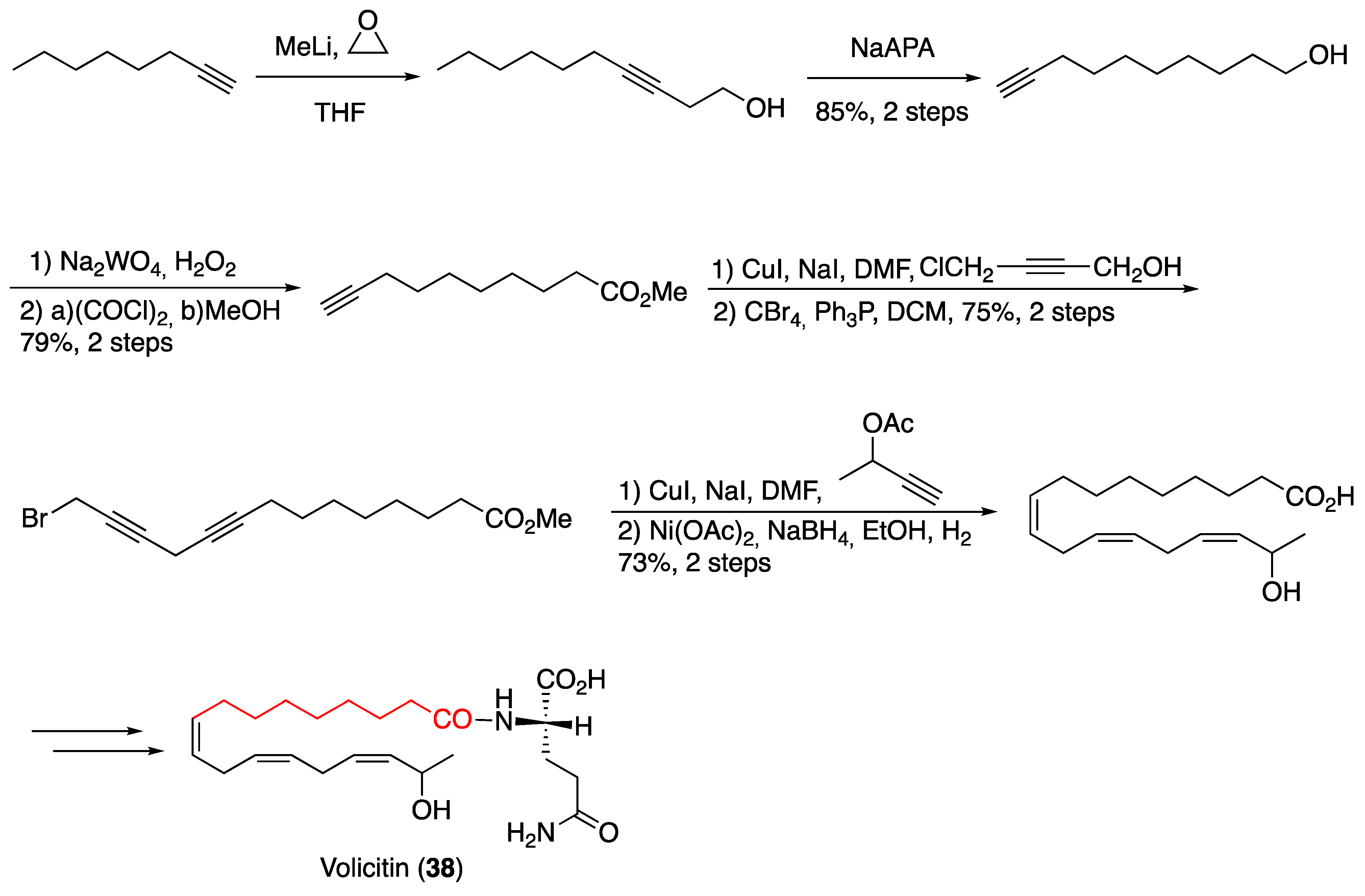
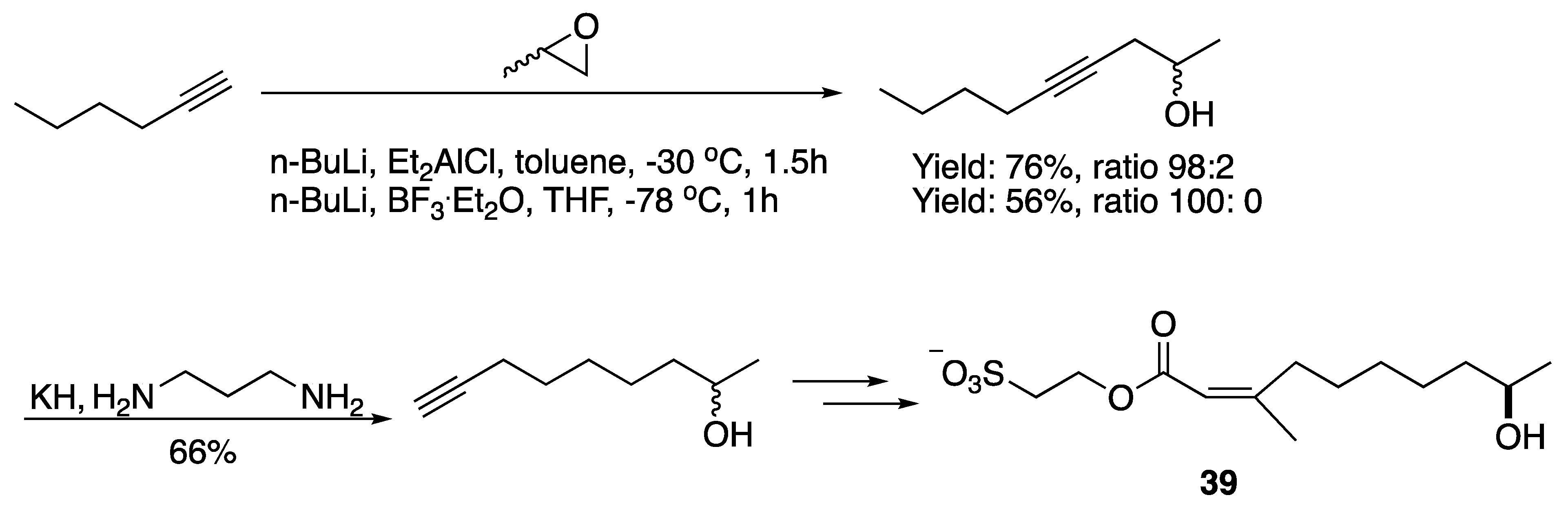
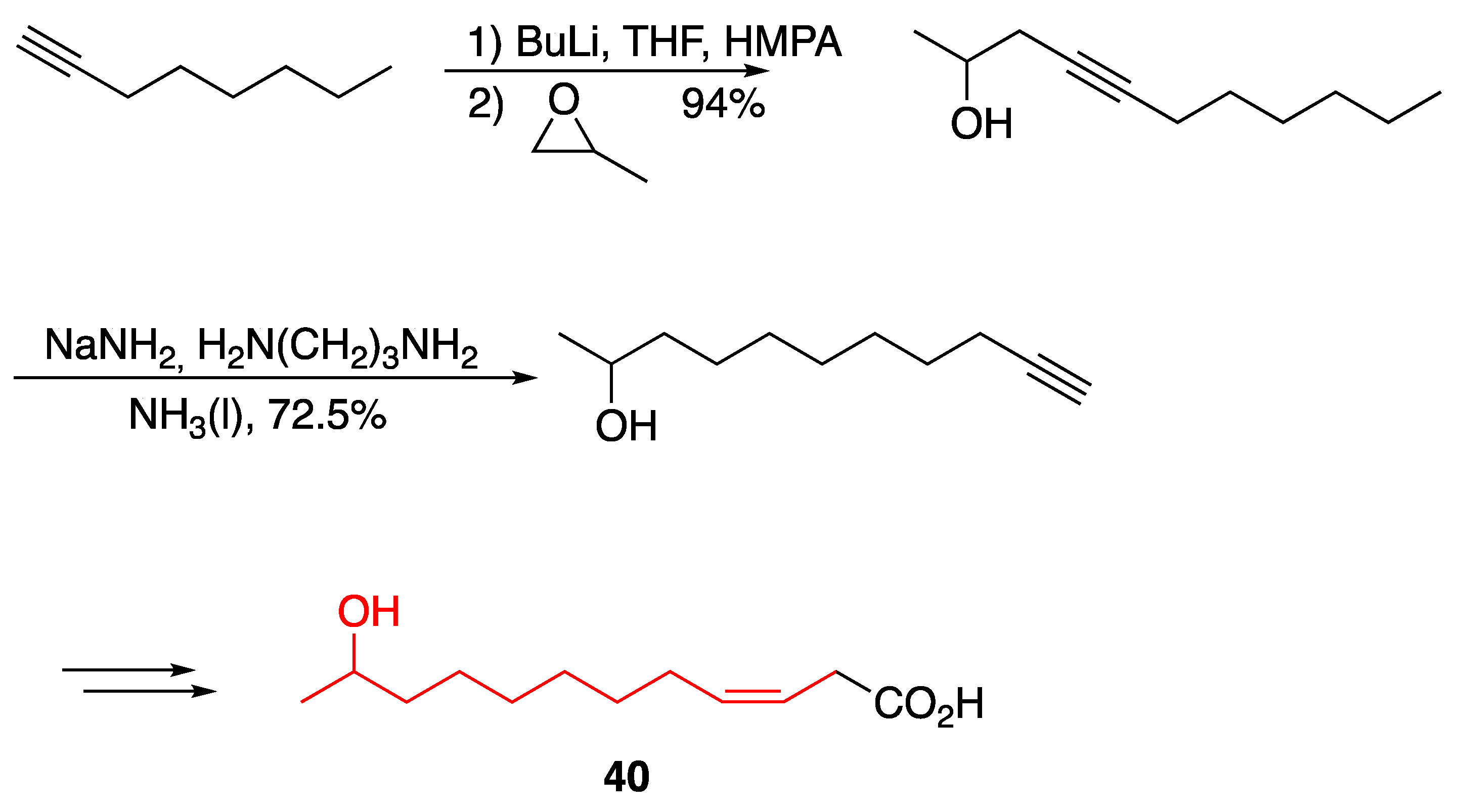
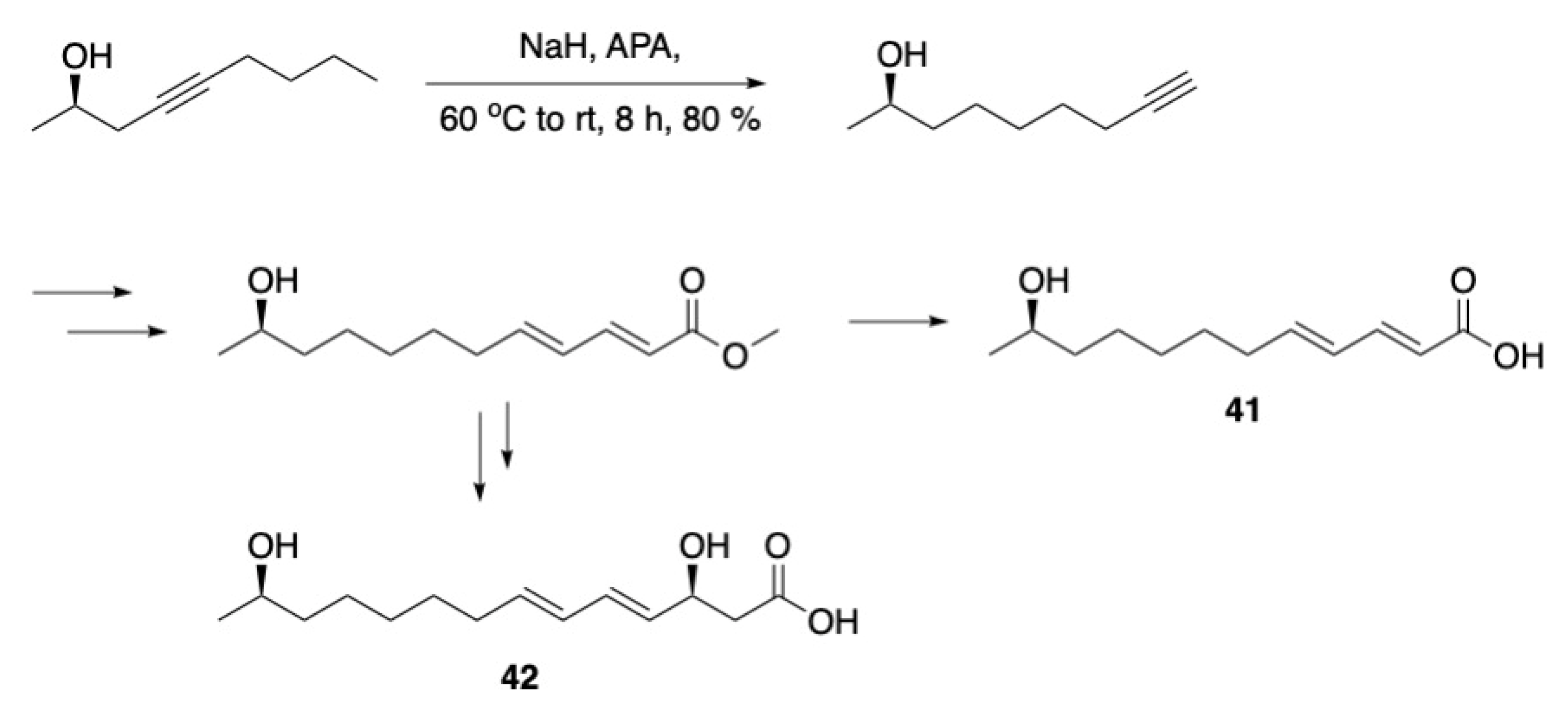
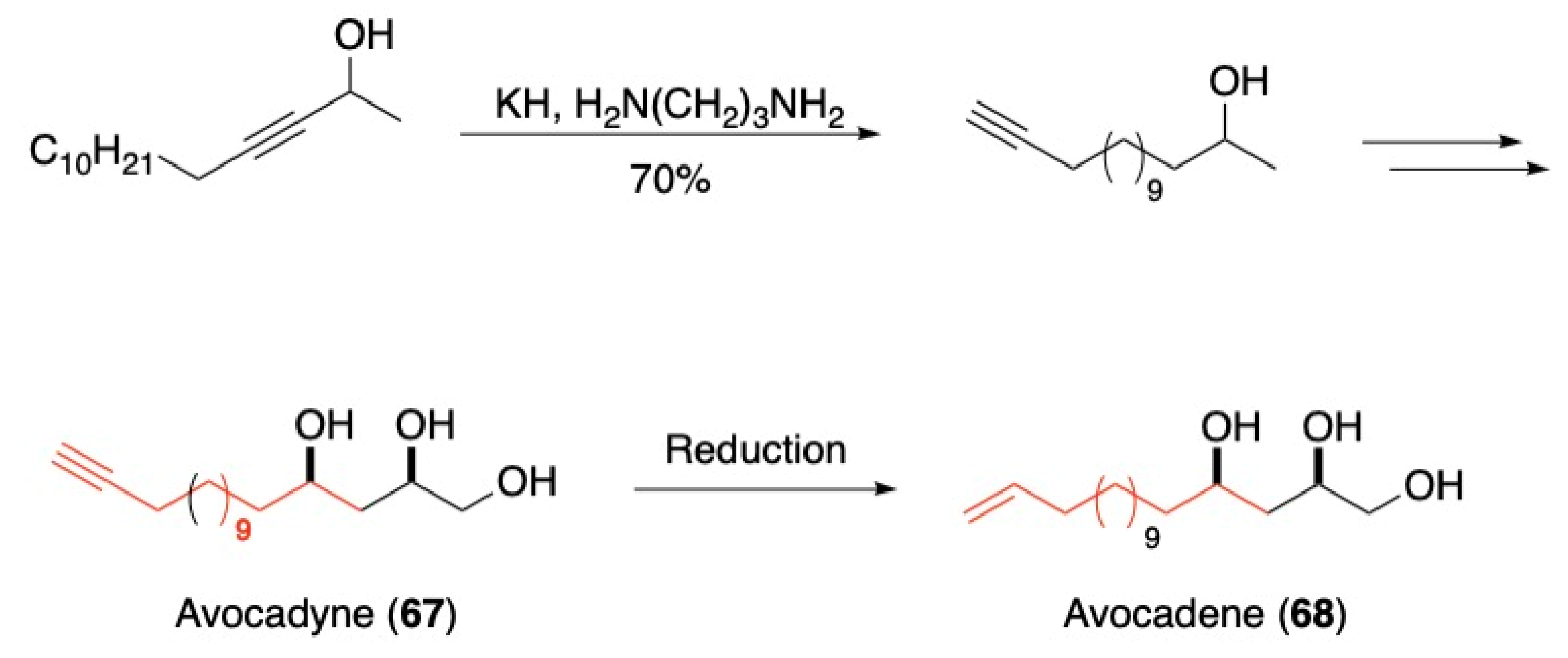
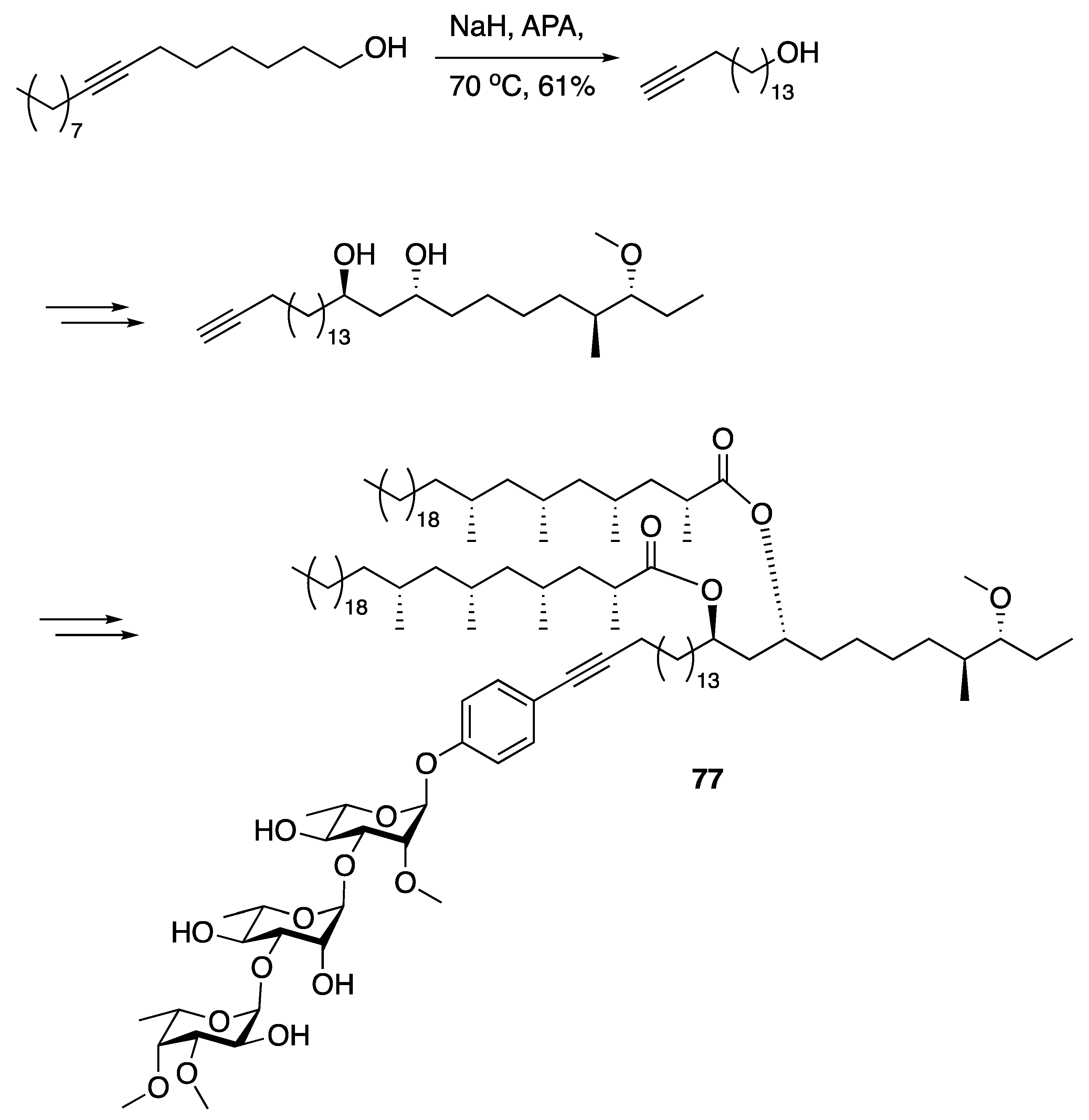
3.1. Pheromones and Long Chained Alcohols
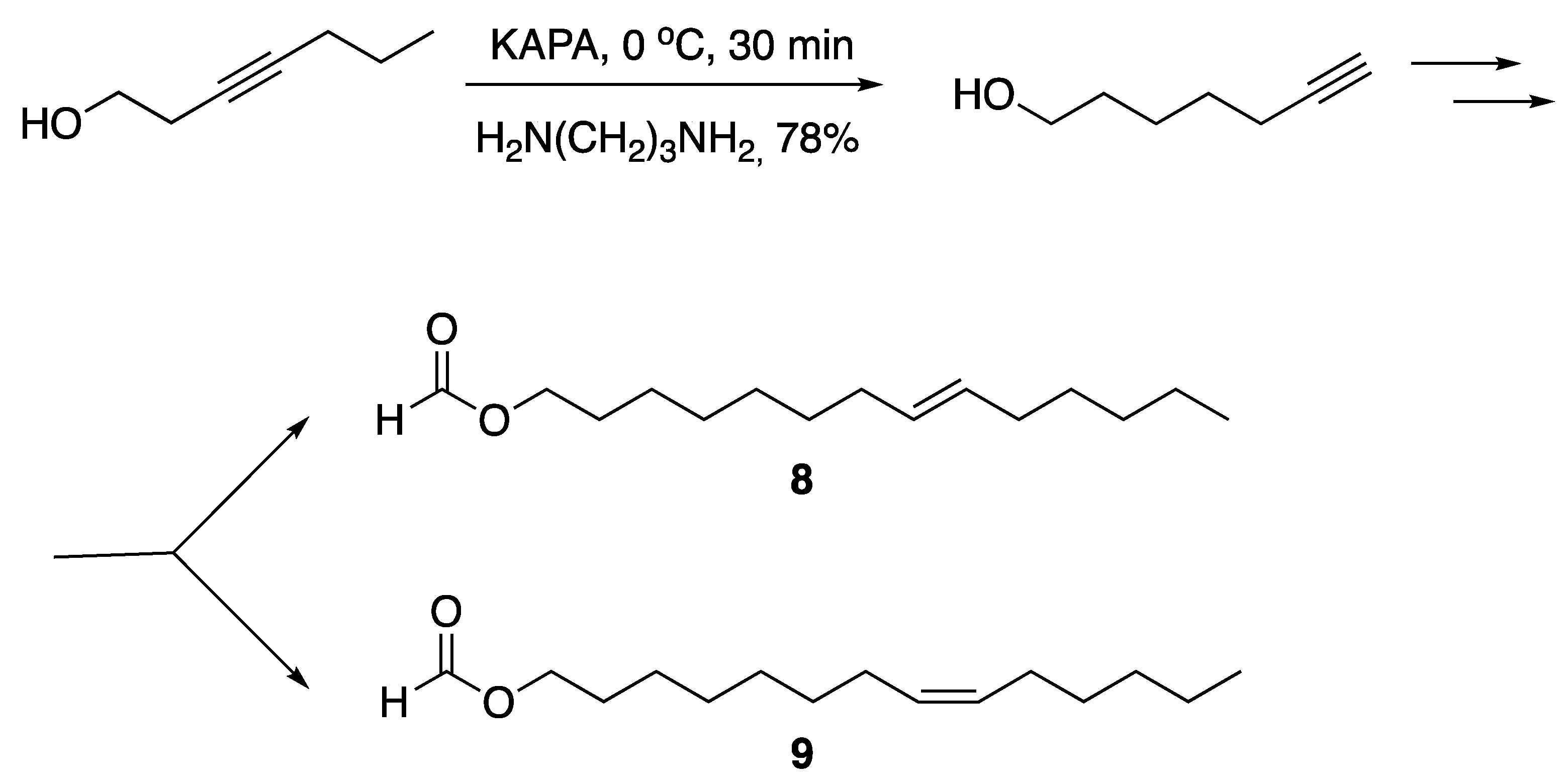
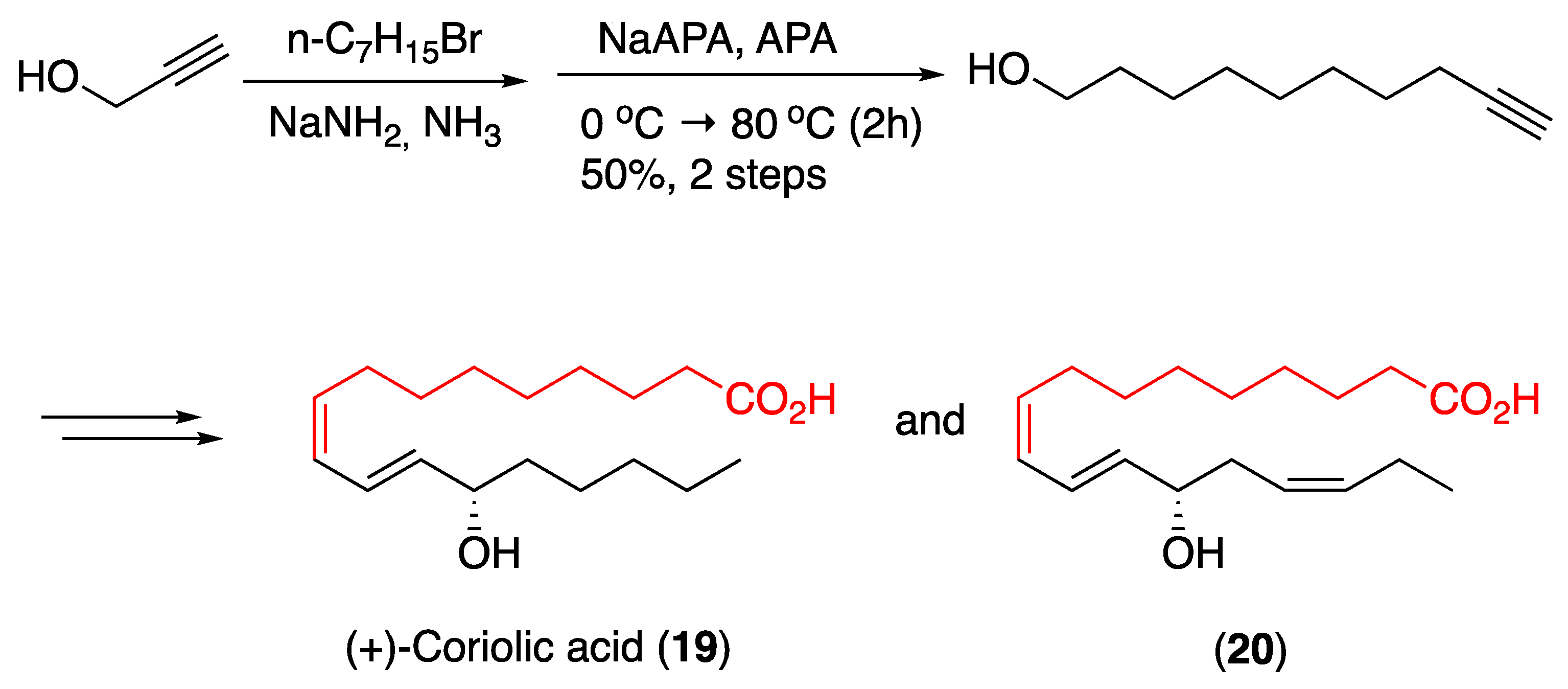
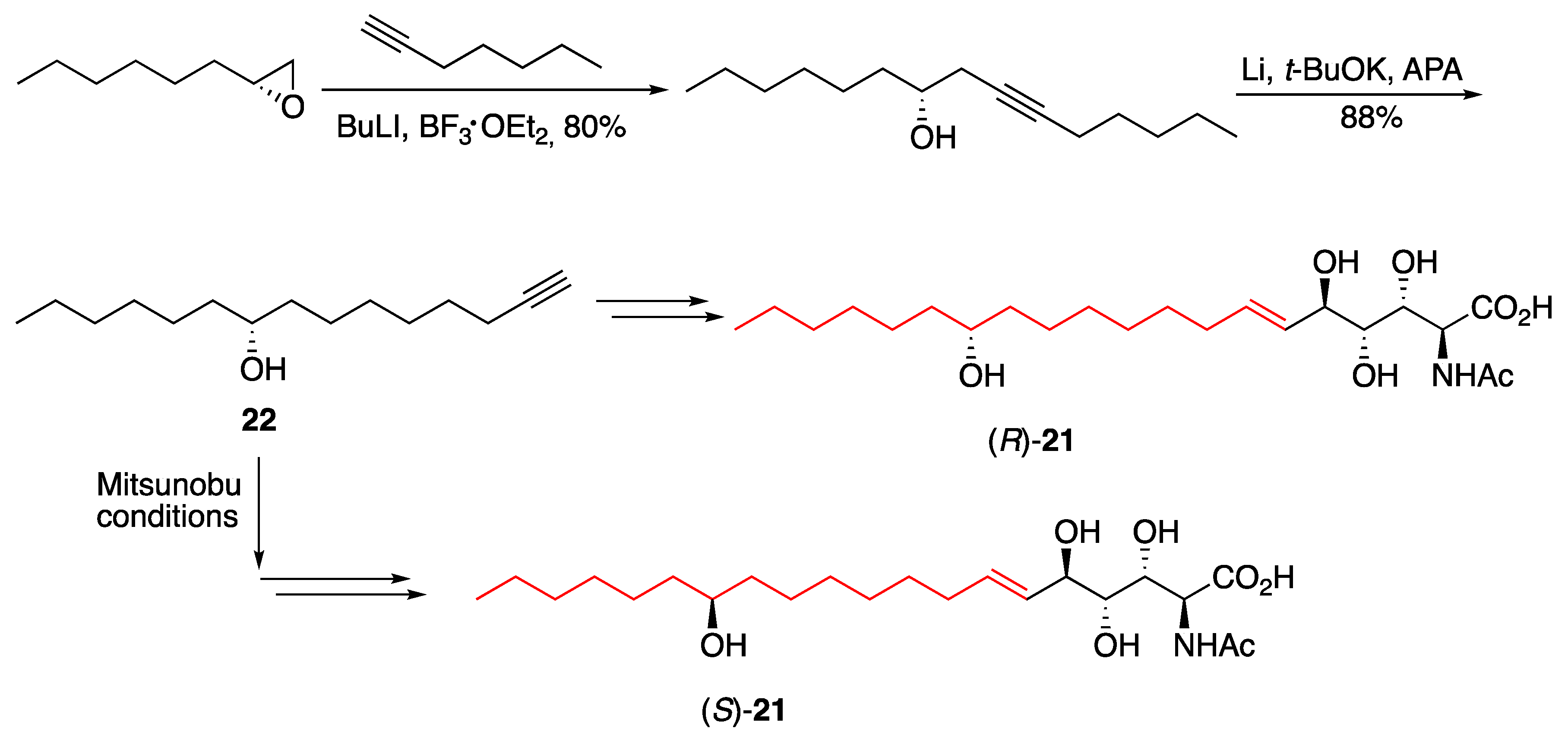
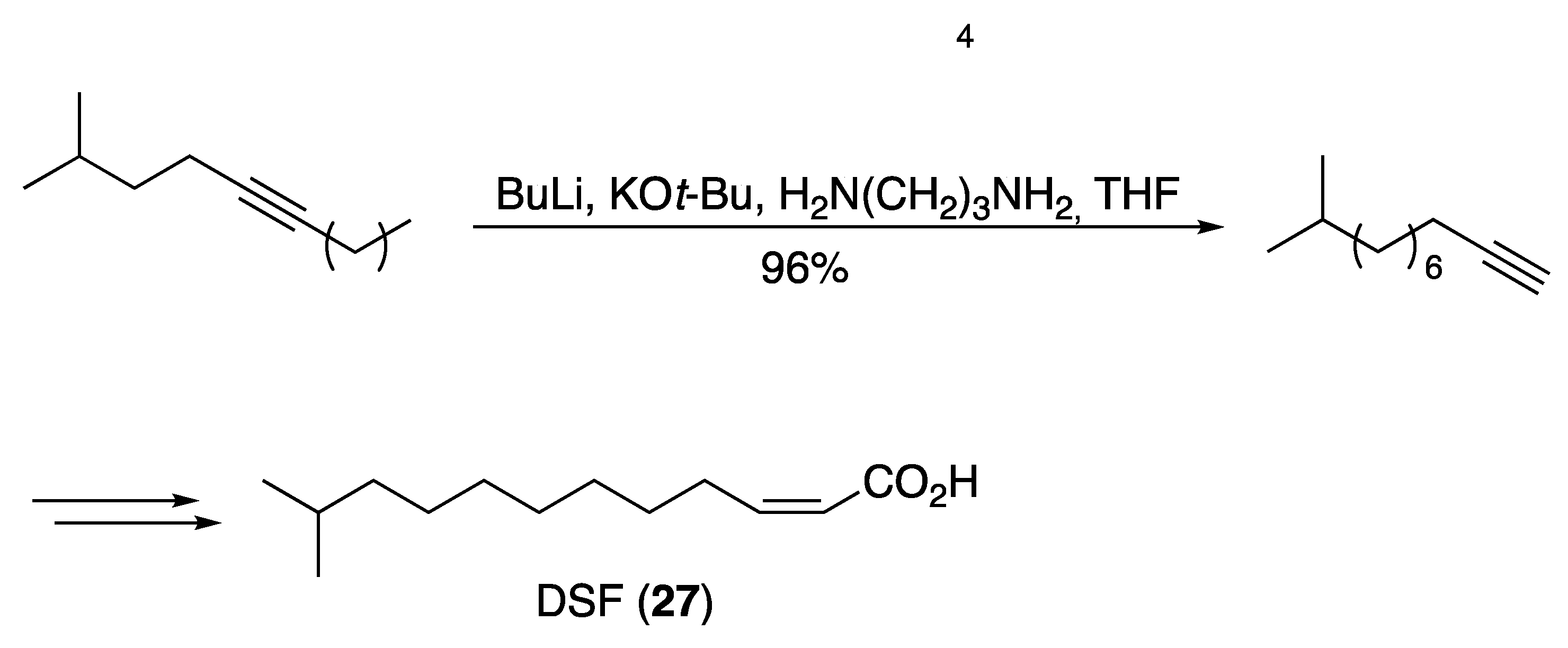
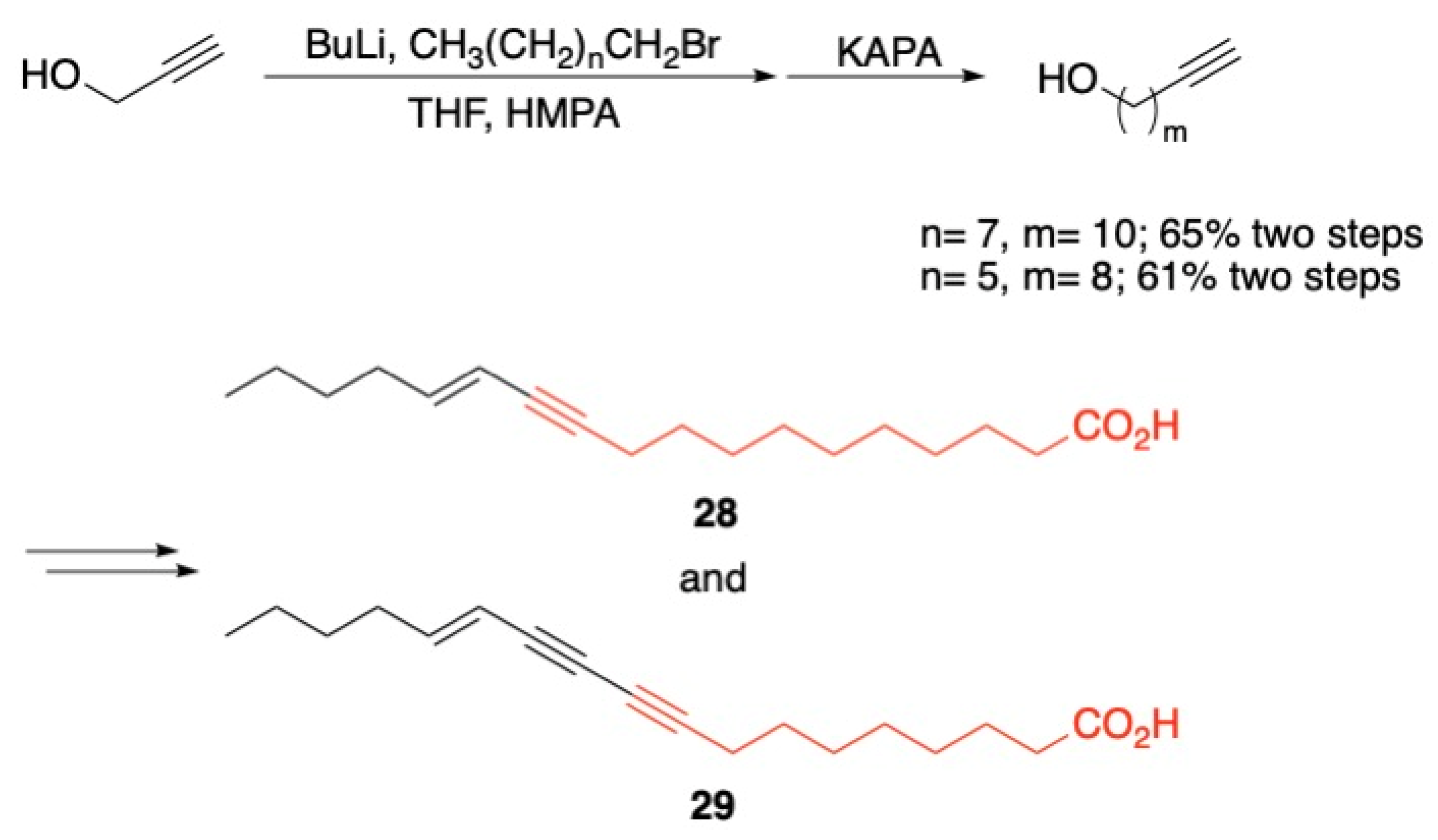
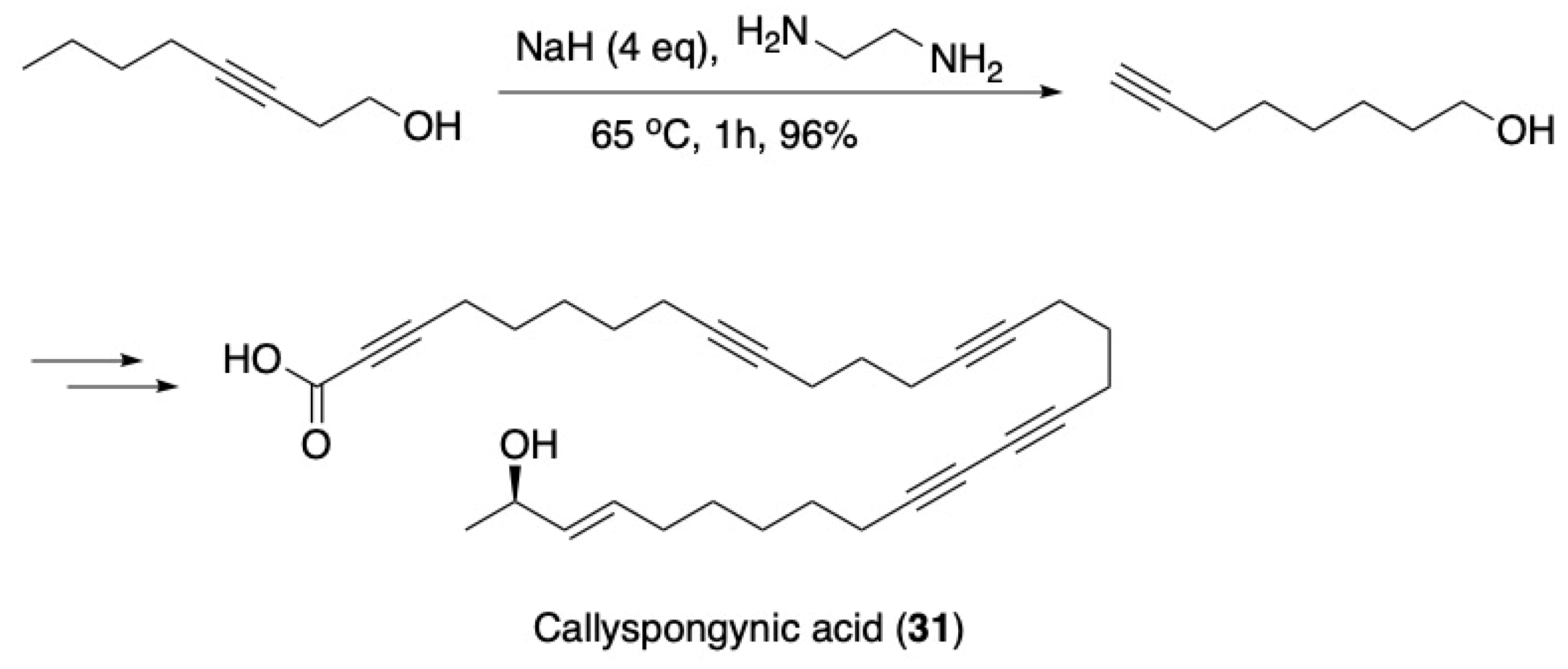
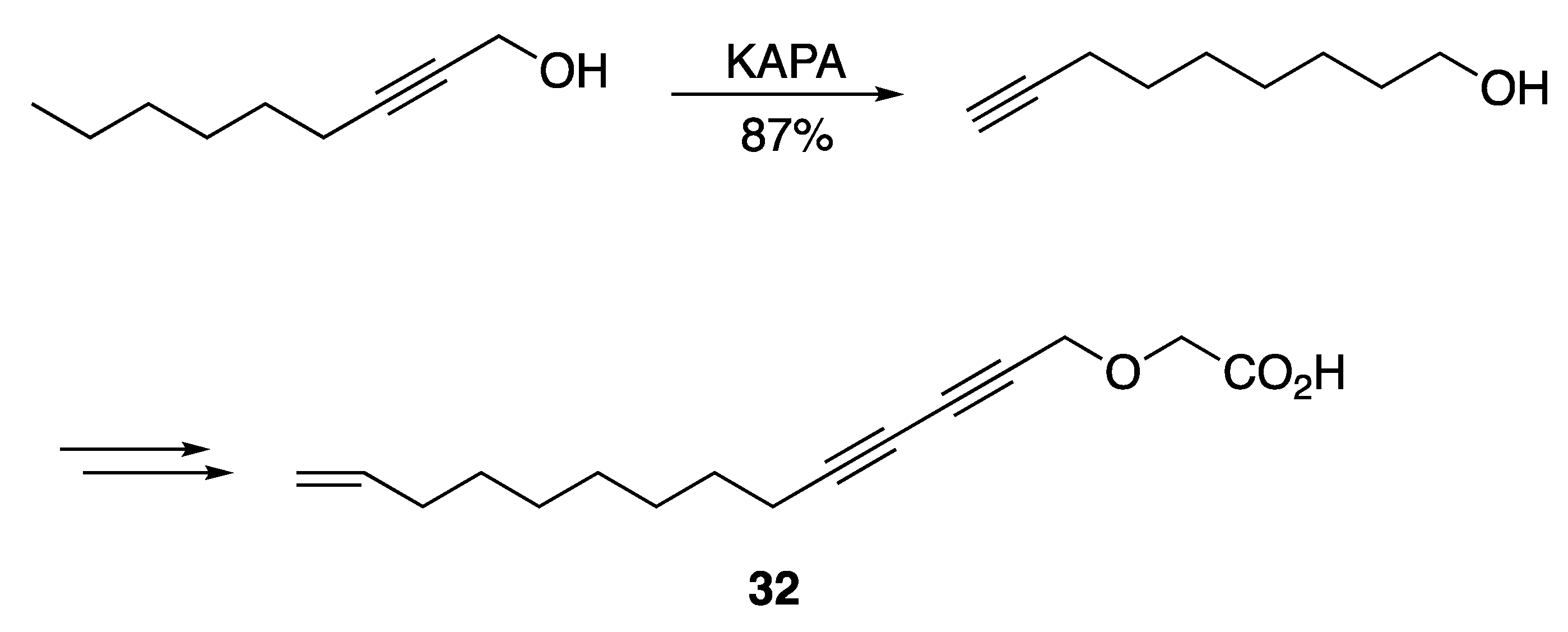
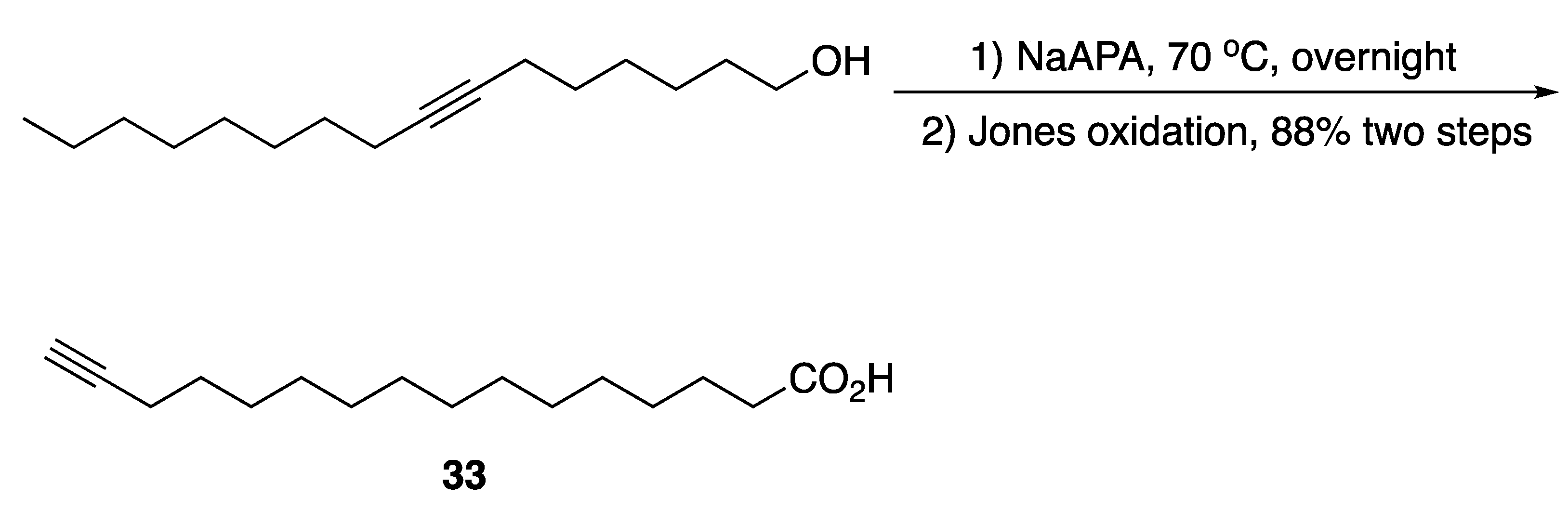
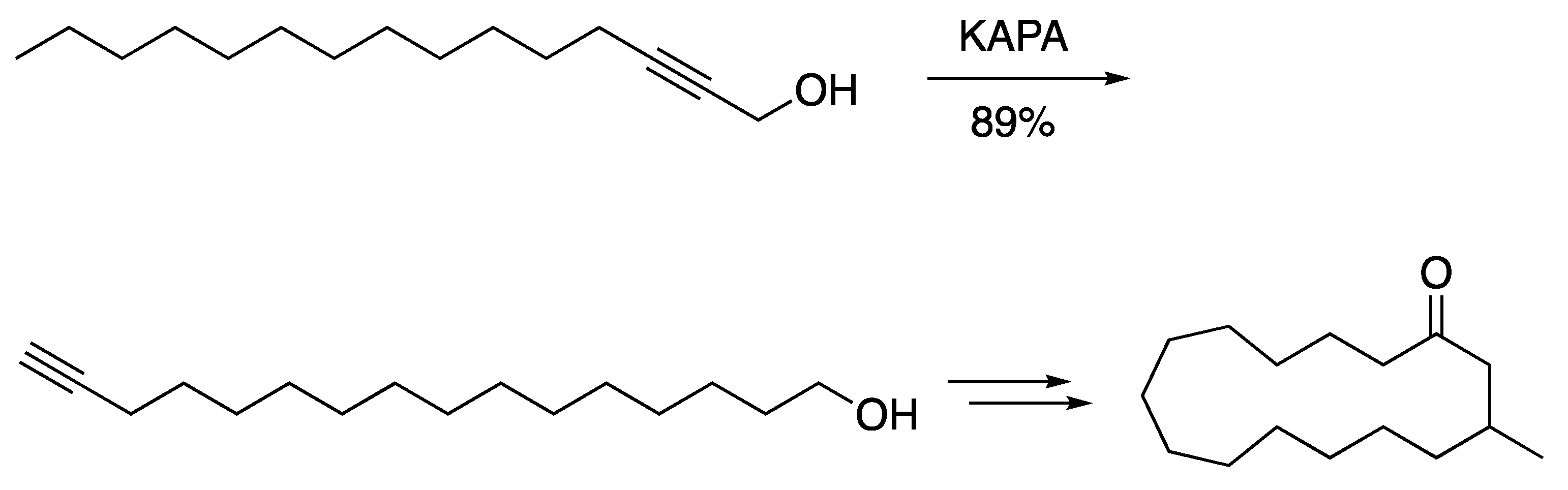
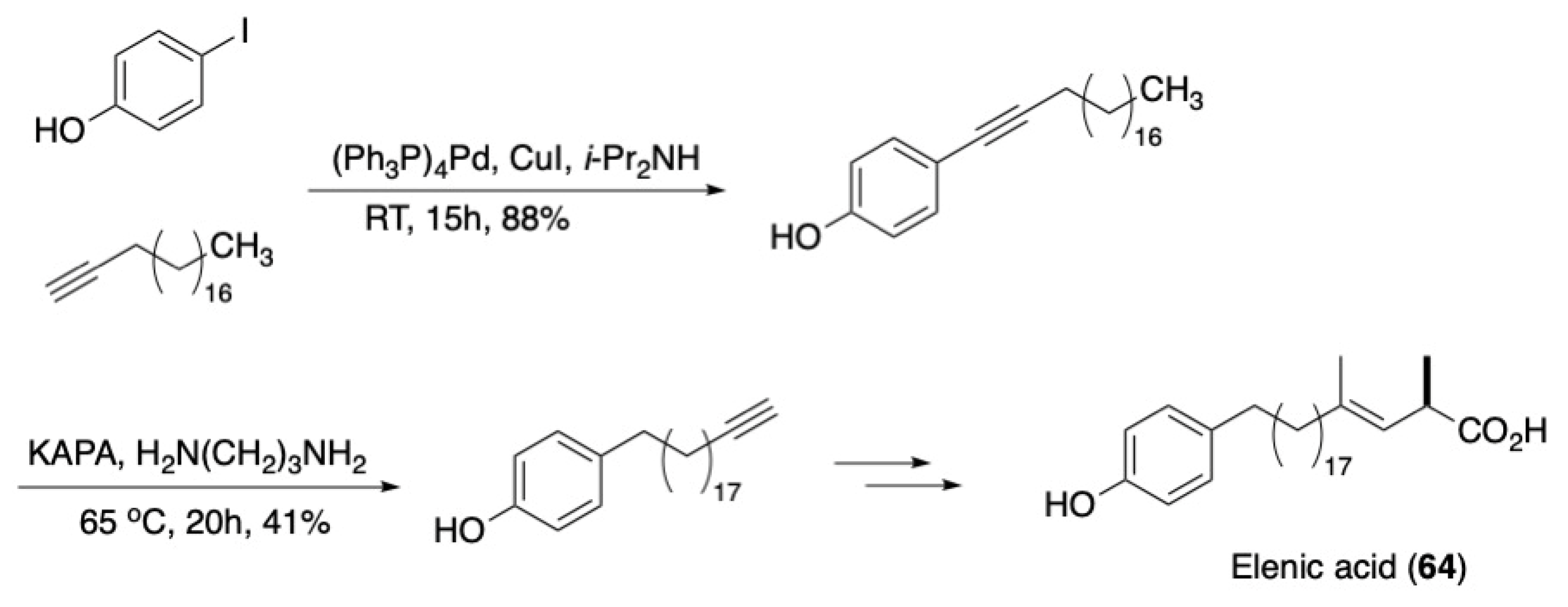
3.2. Fatty Acids and Derived Structures
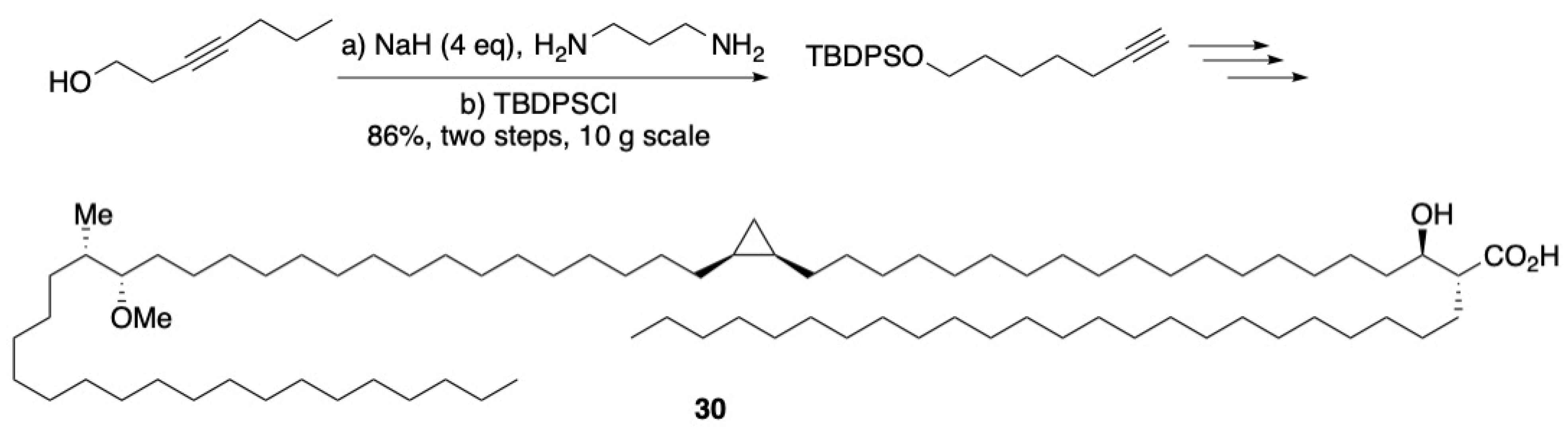
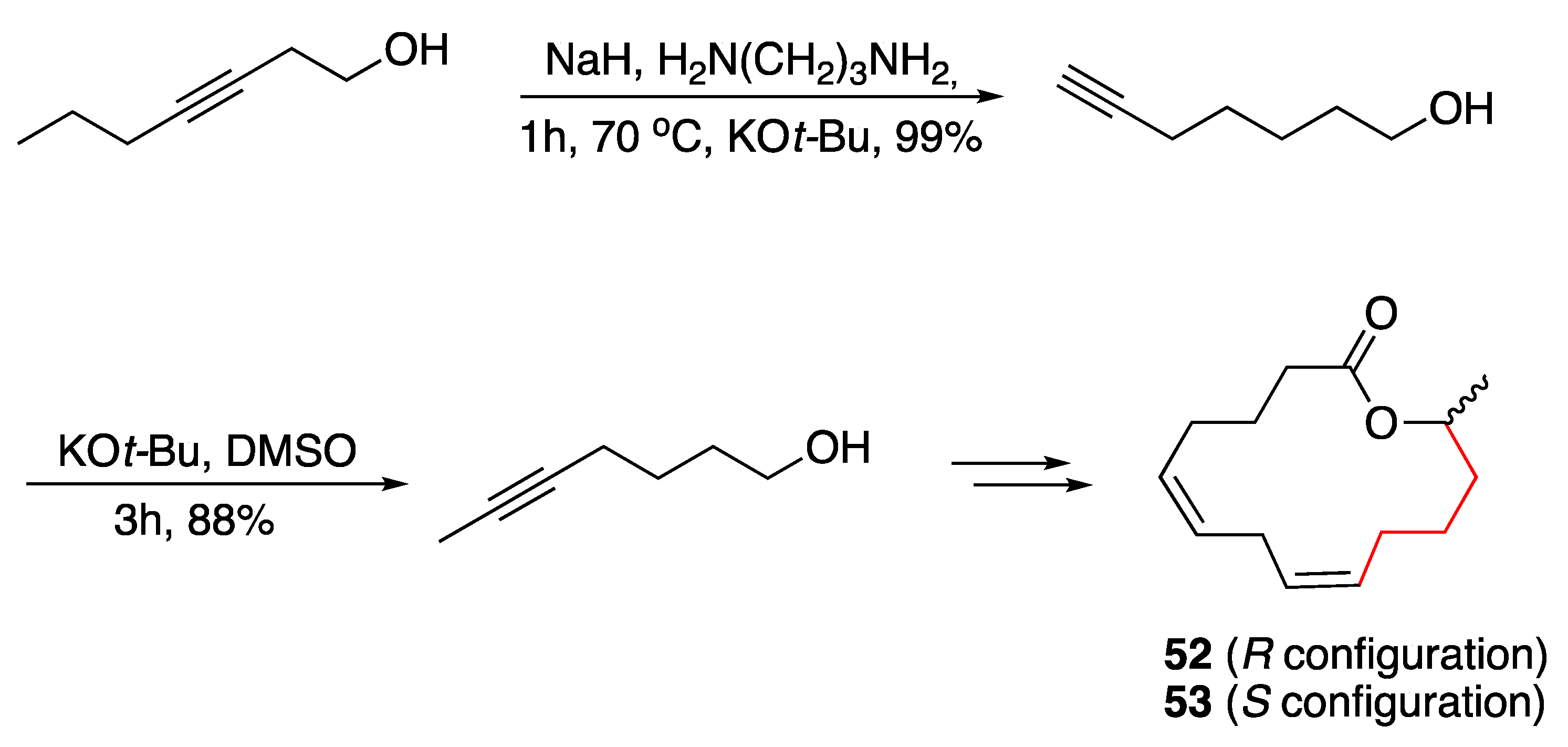
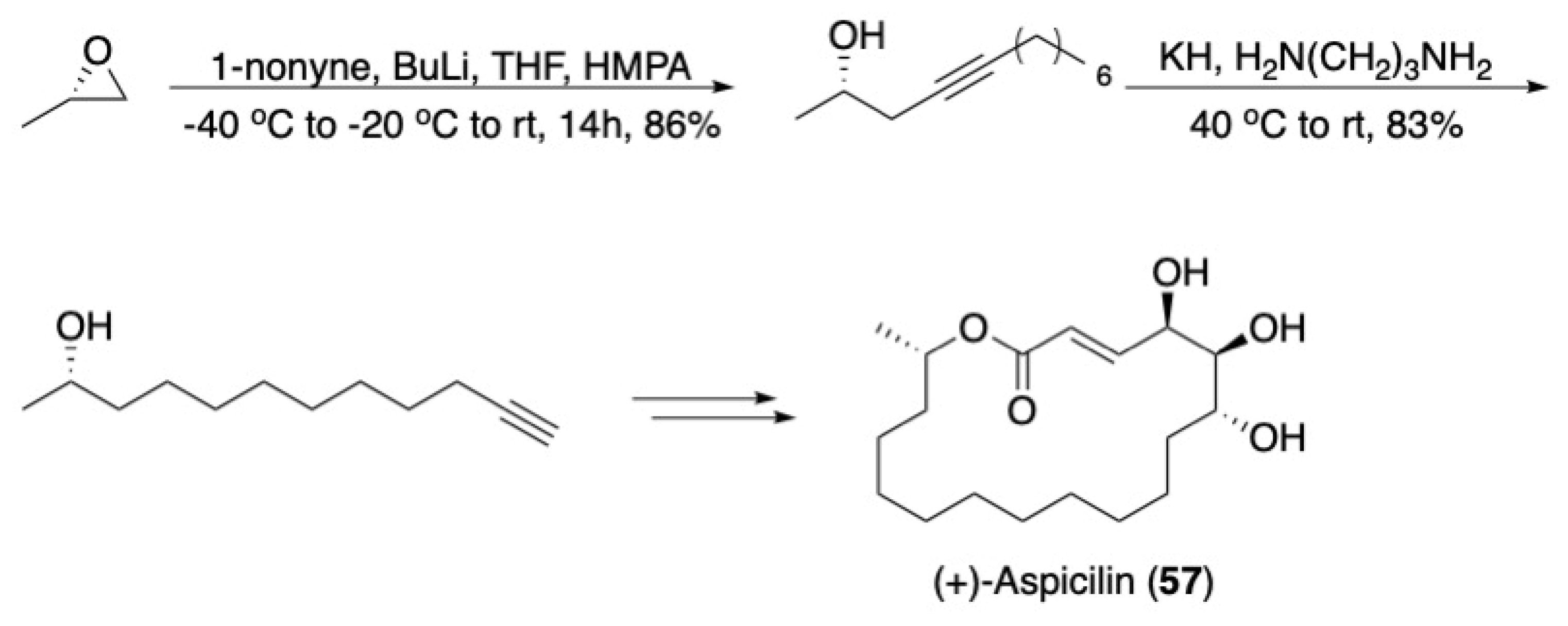
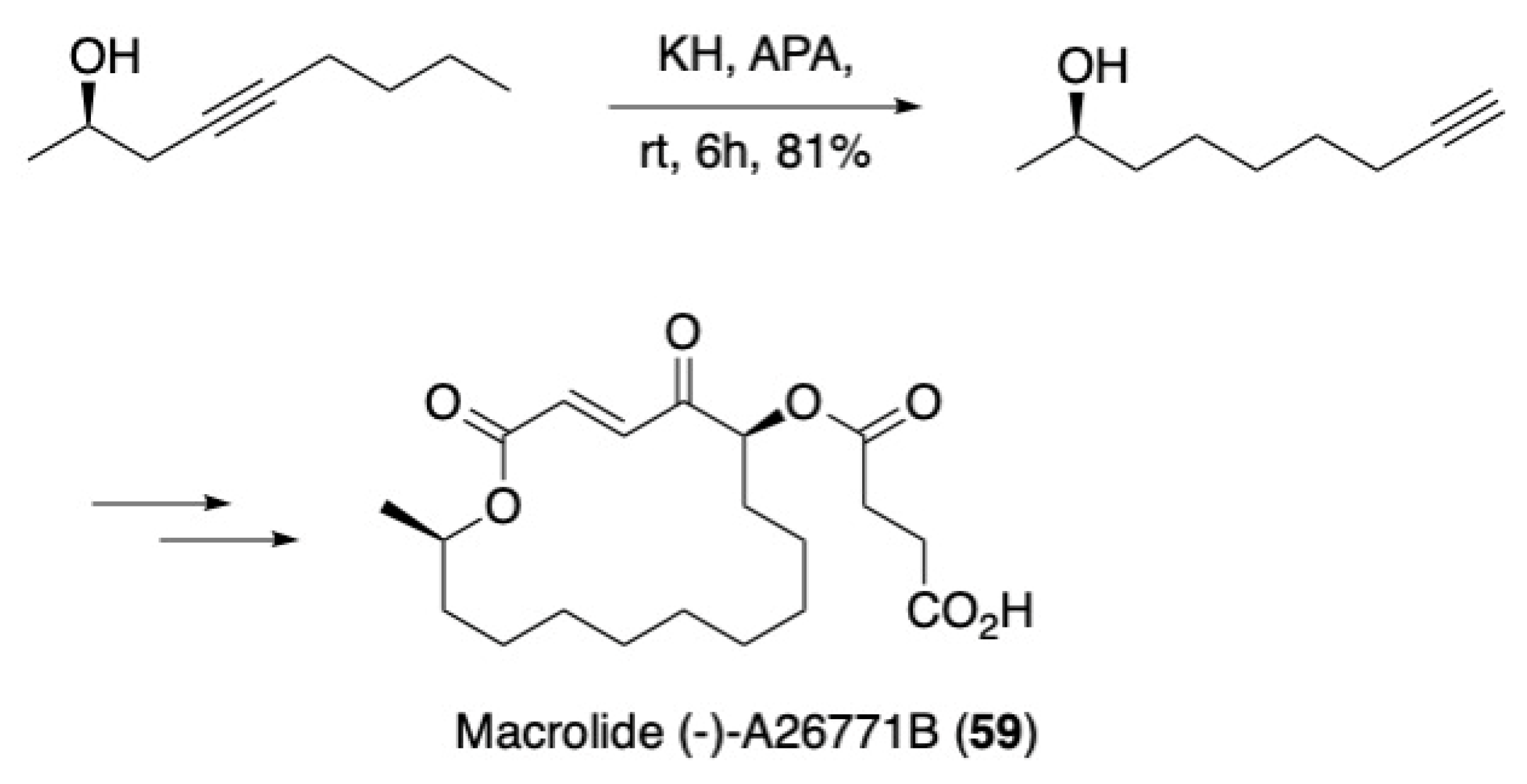
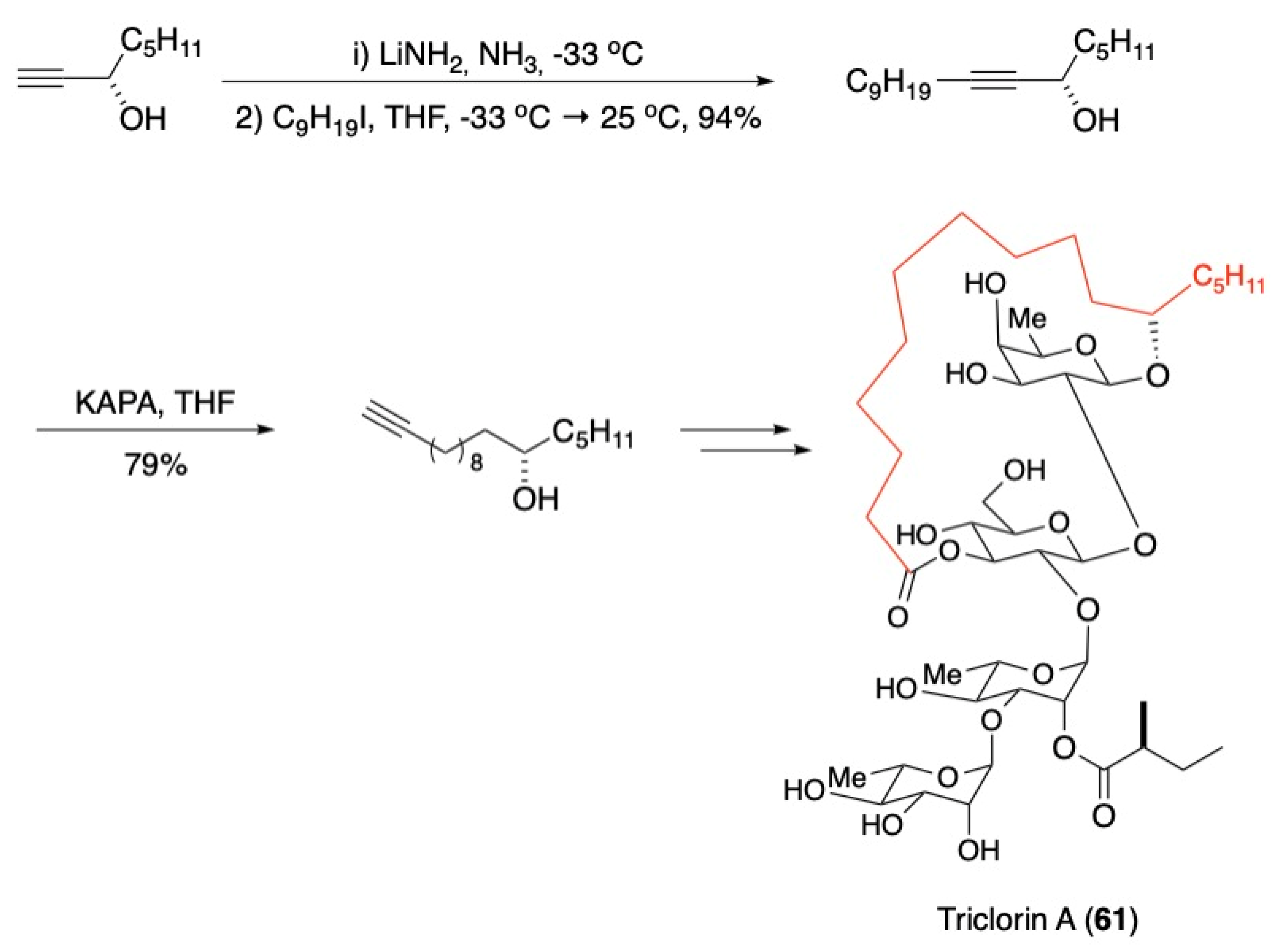
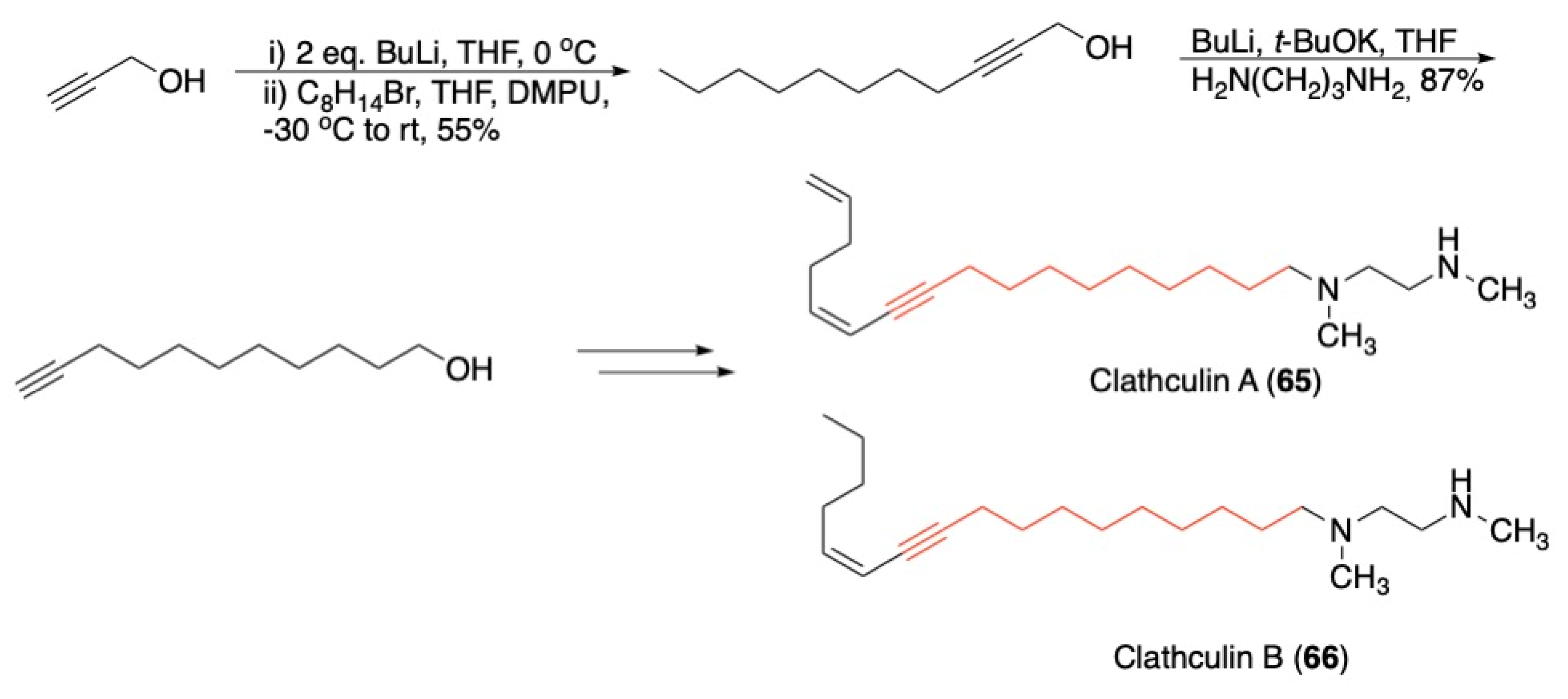
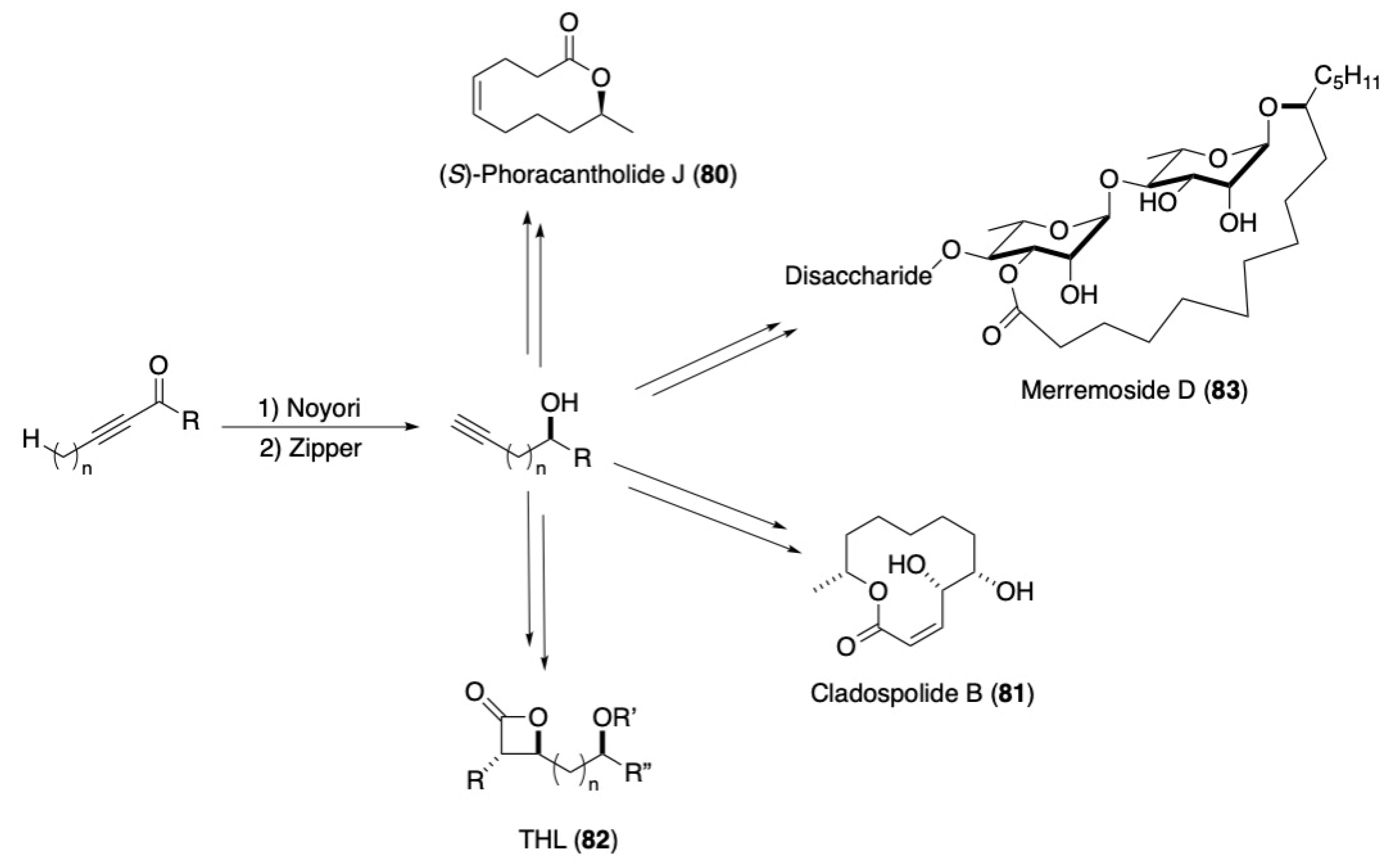
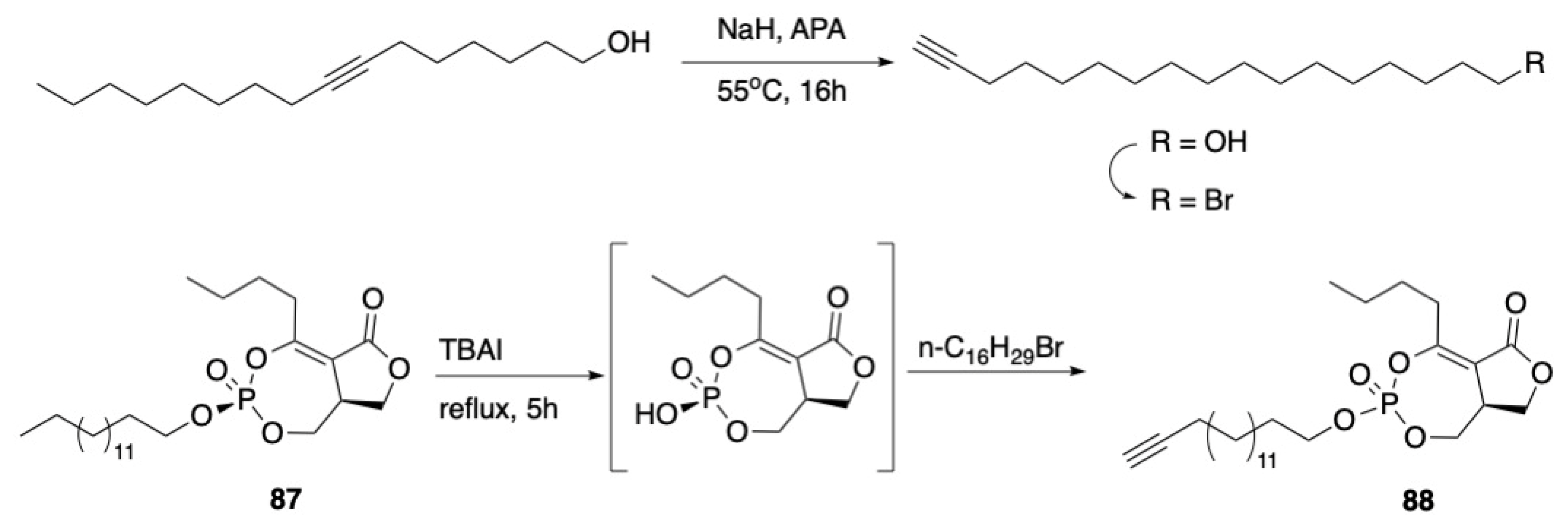
3.3. Macrocycles
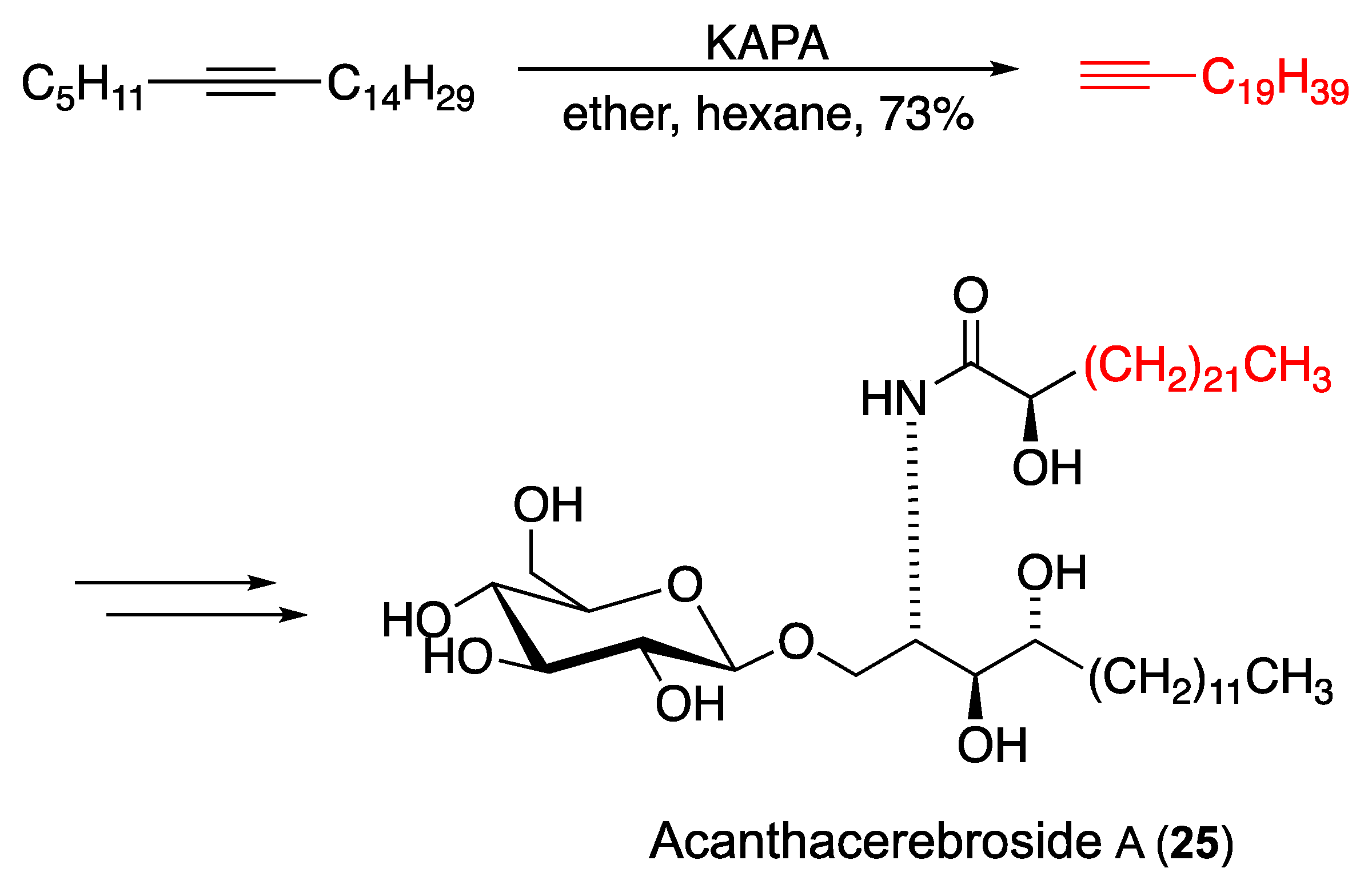
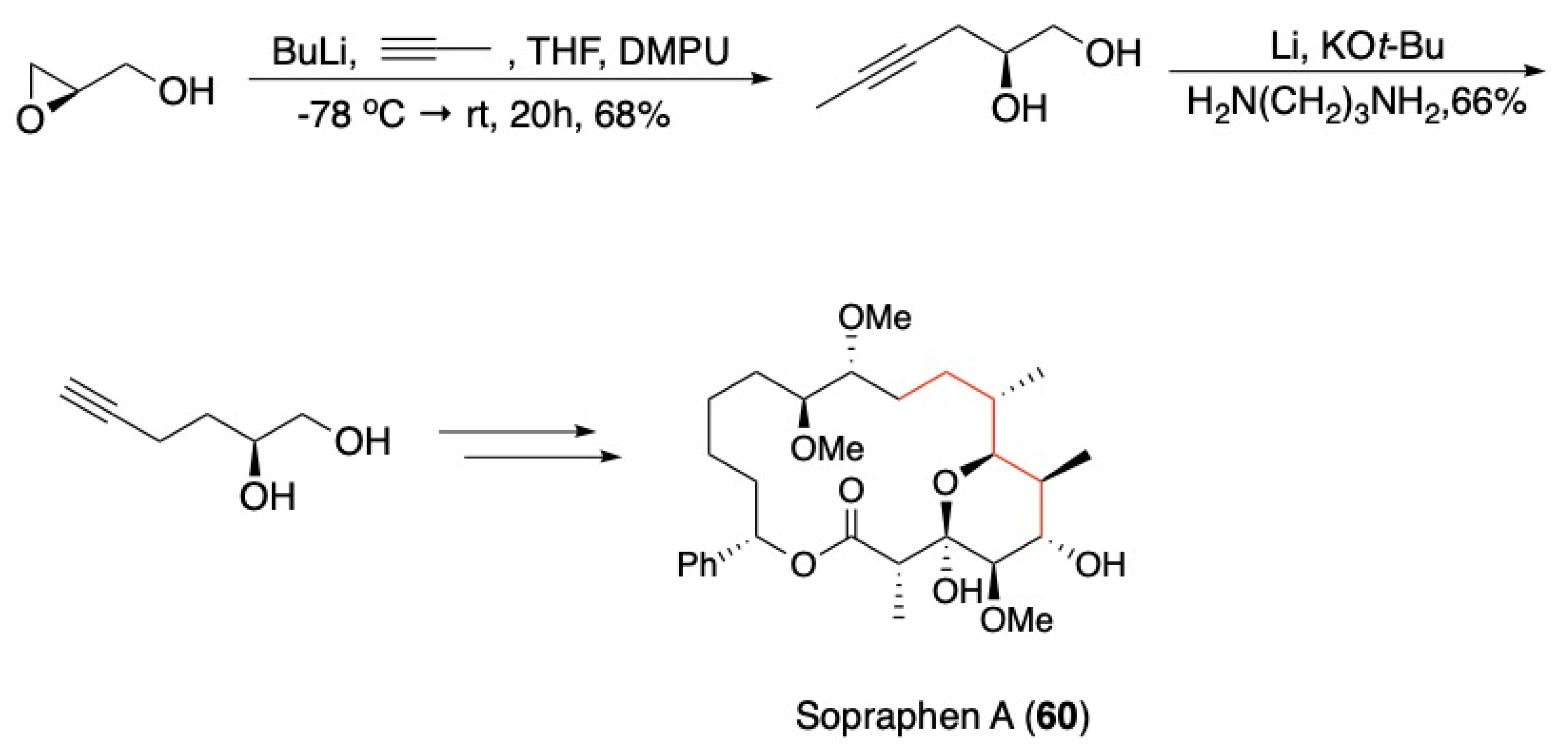
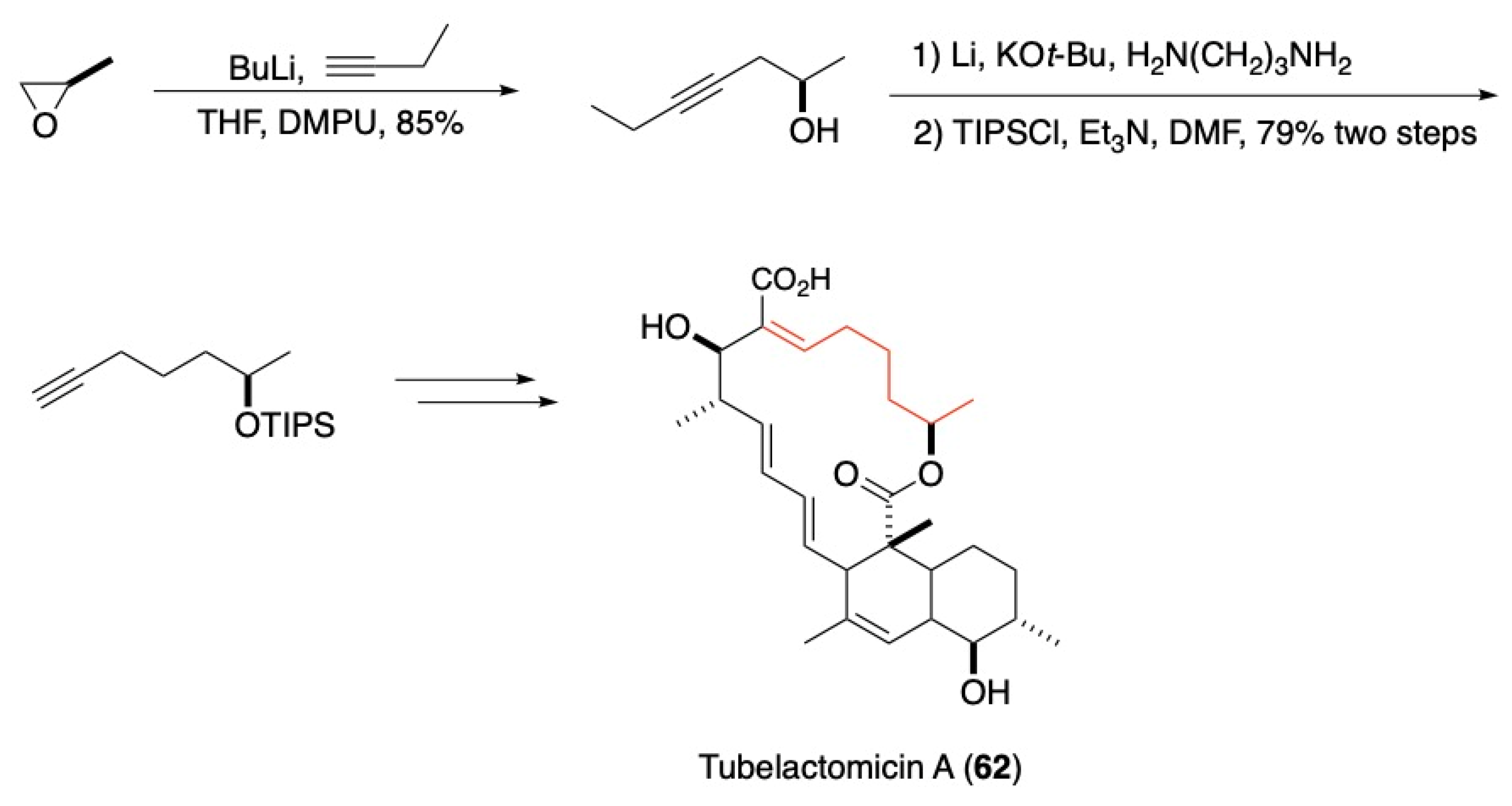
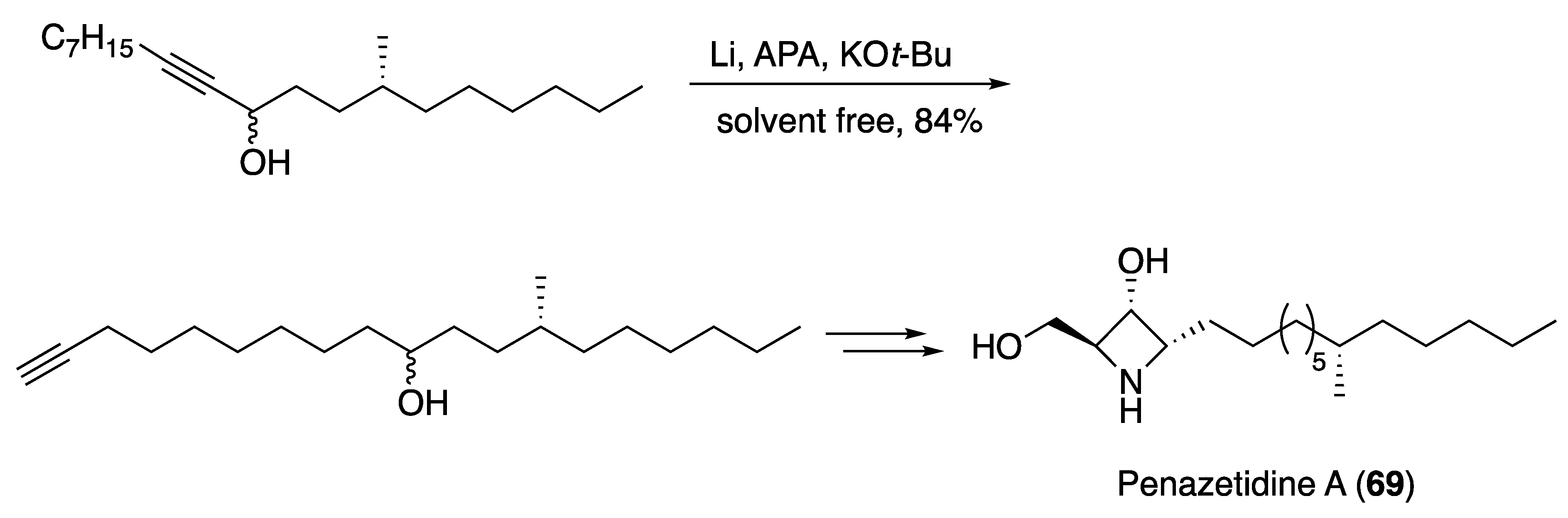
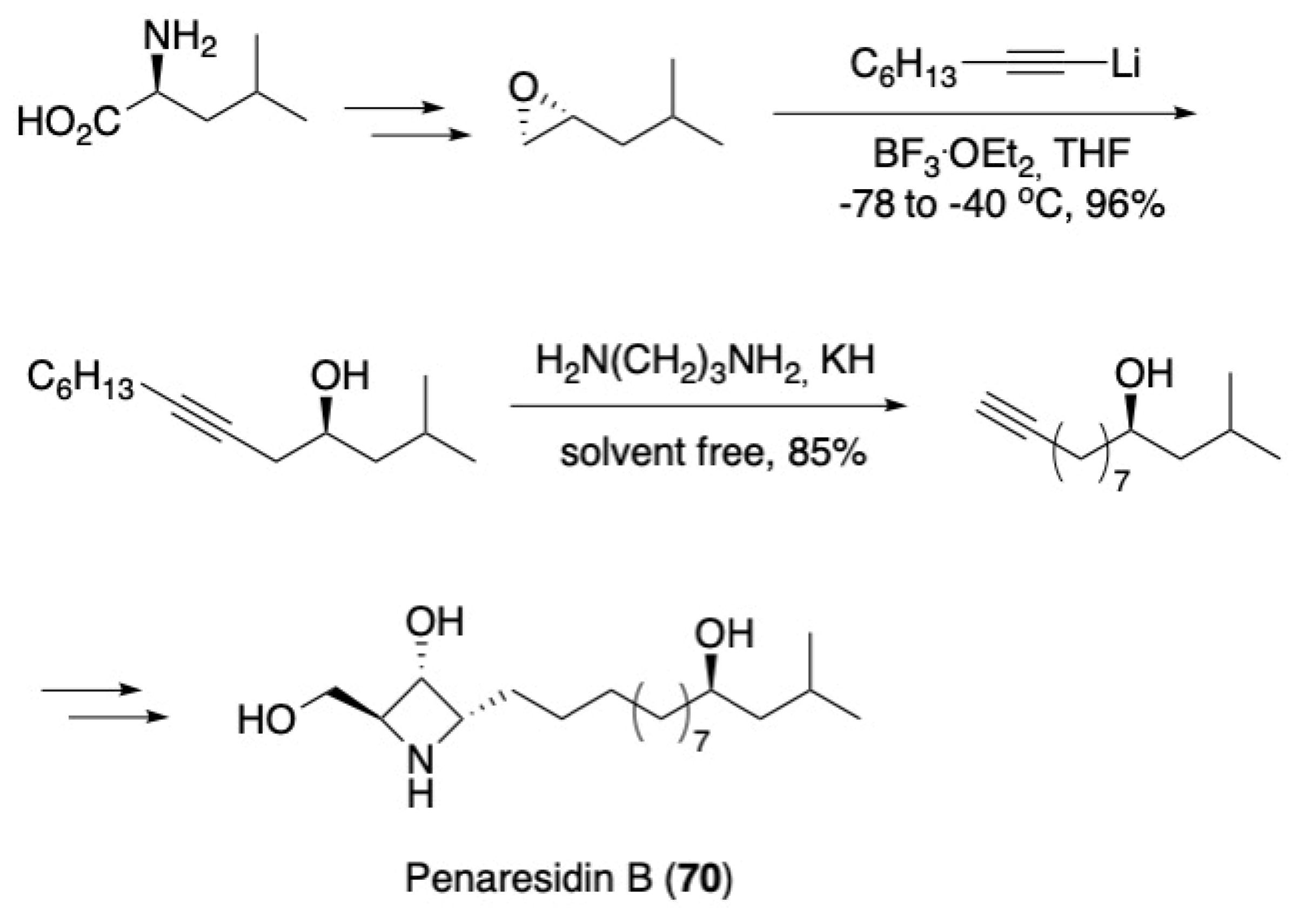
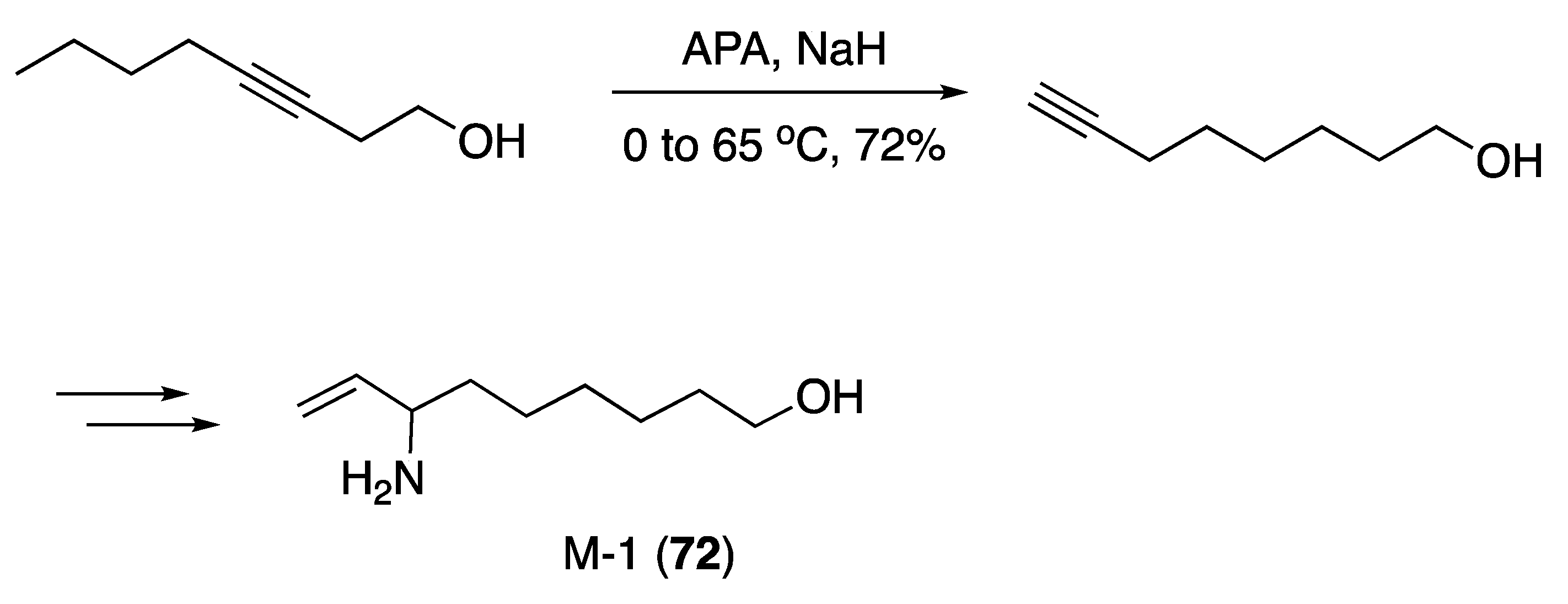
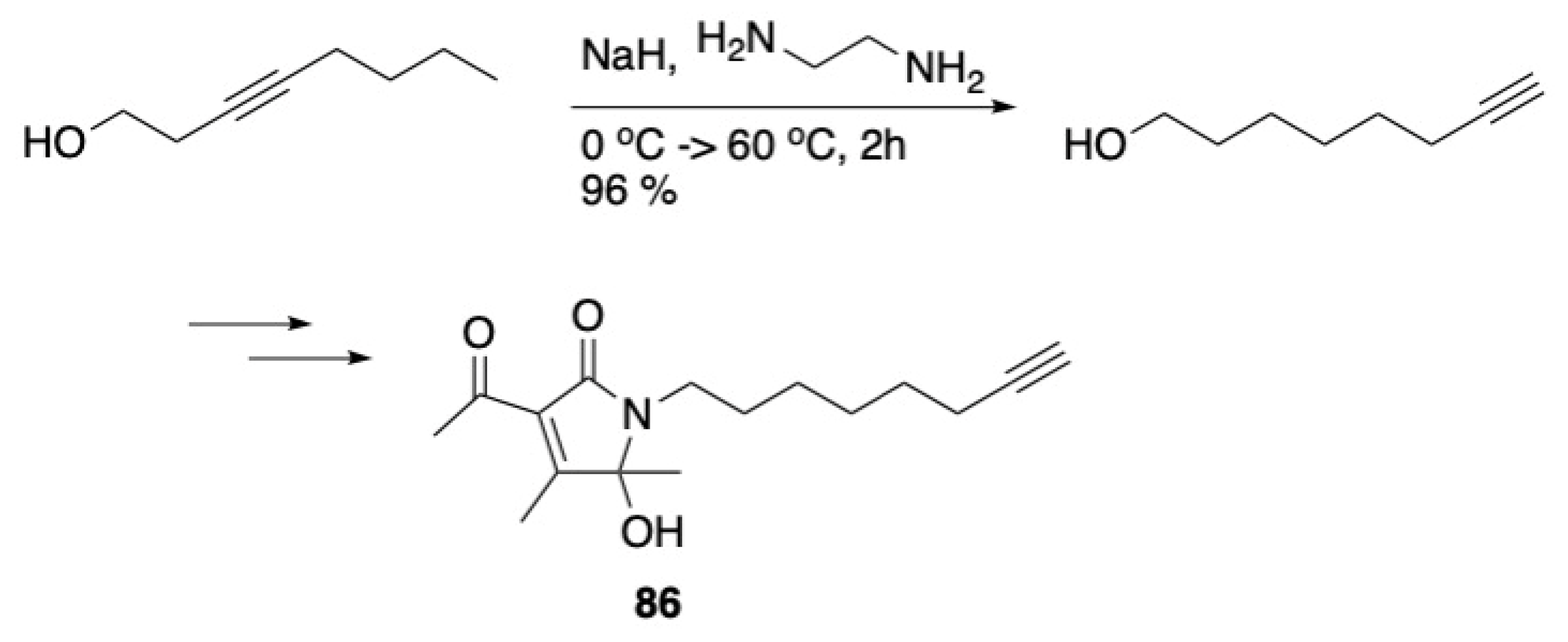
3.4. Other Natural Product Classes
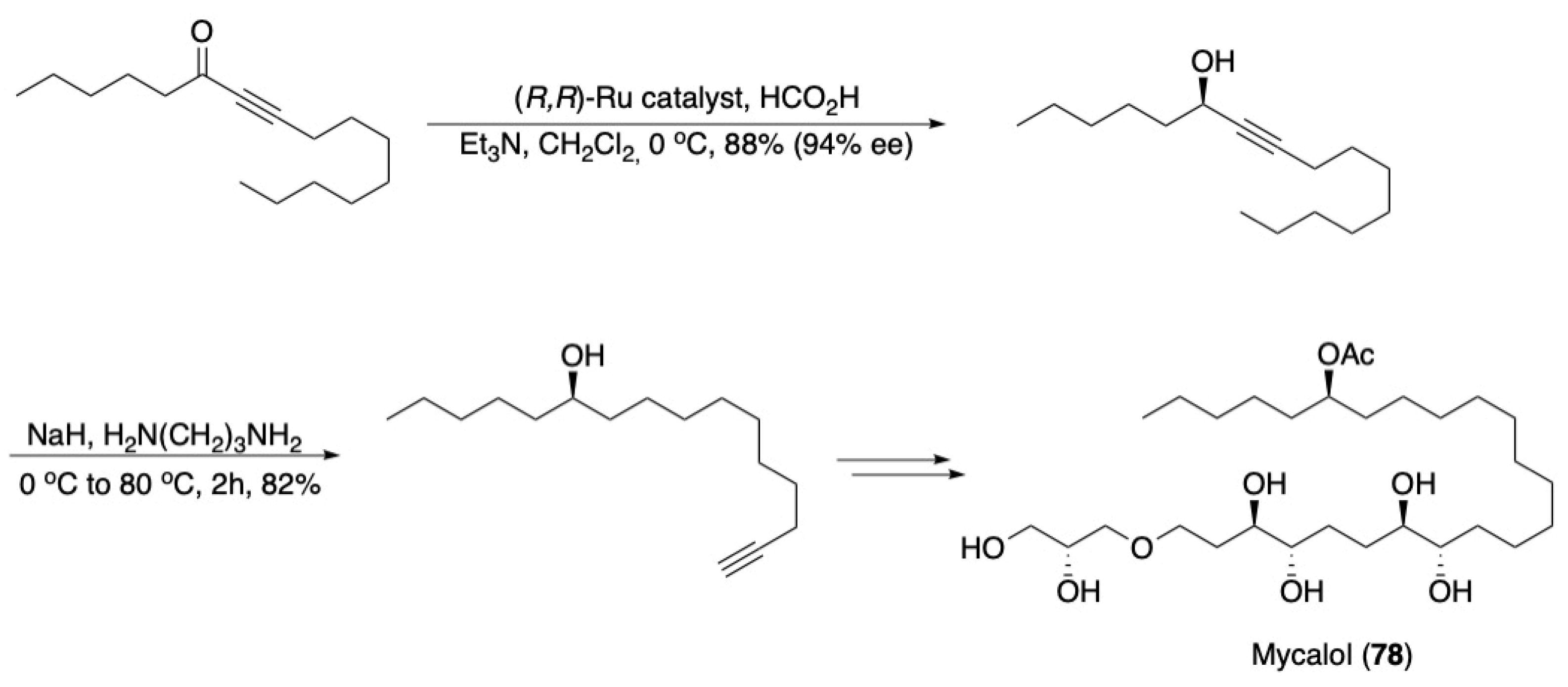
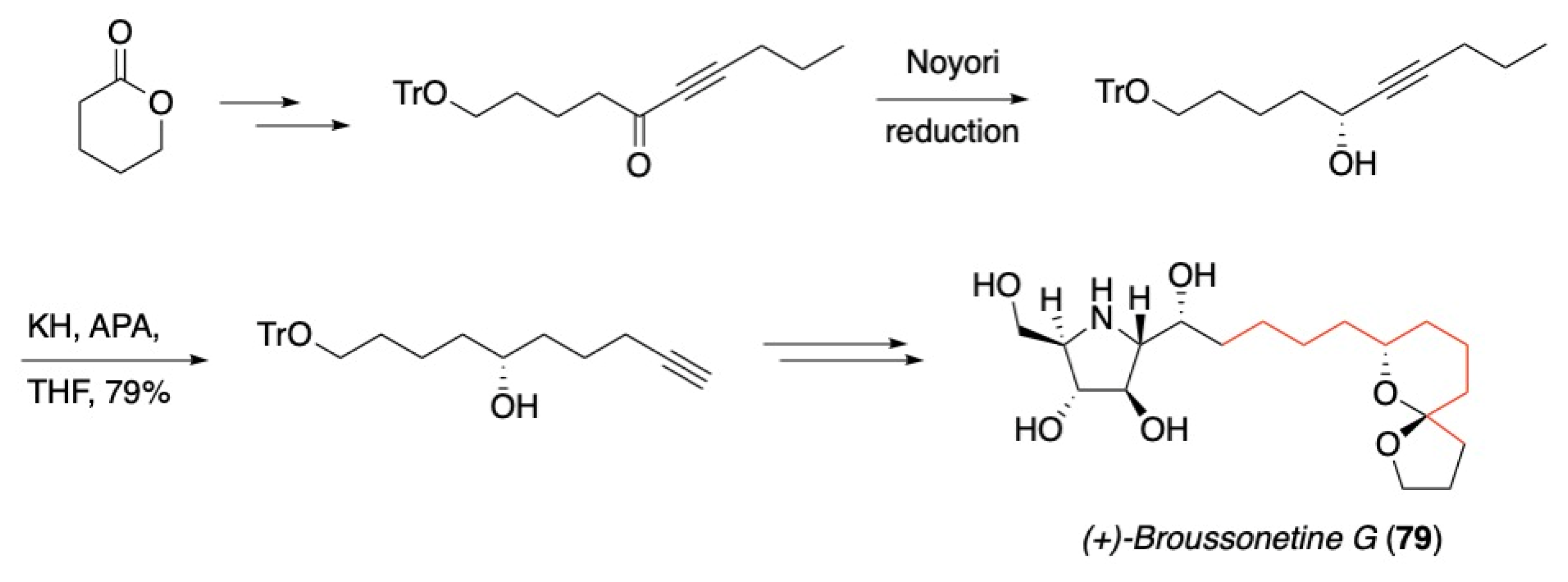
3.5. Noyori Reduction/Acetylene Zipper Combination
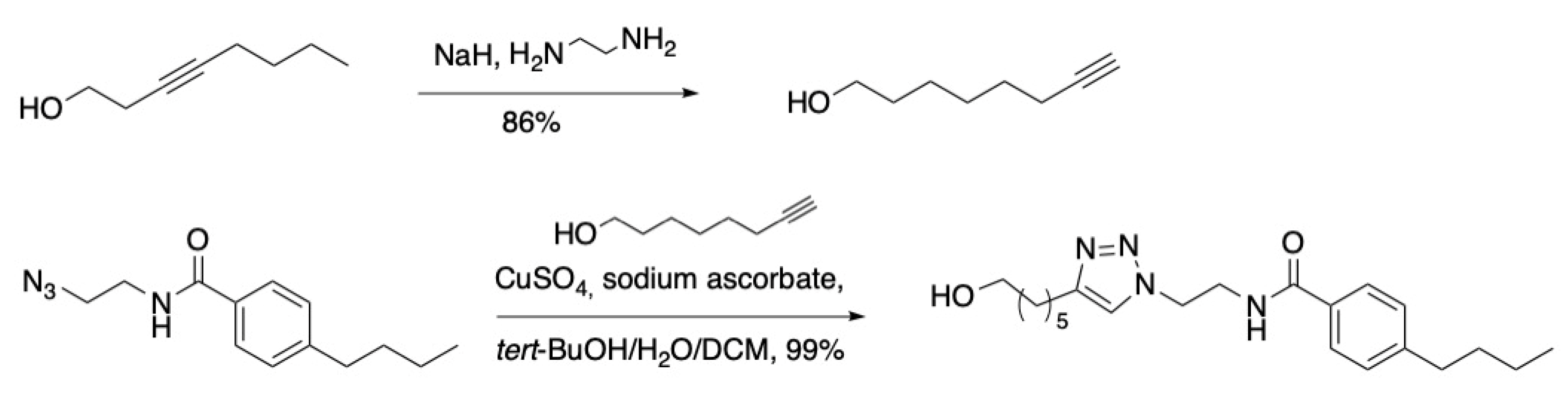
3.6. Probes via Copper Catalyzed Azide–Alkyne Cycloaddition (Click Chemistry)
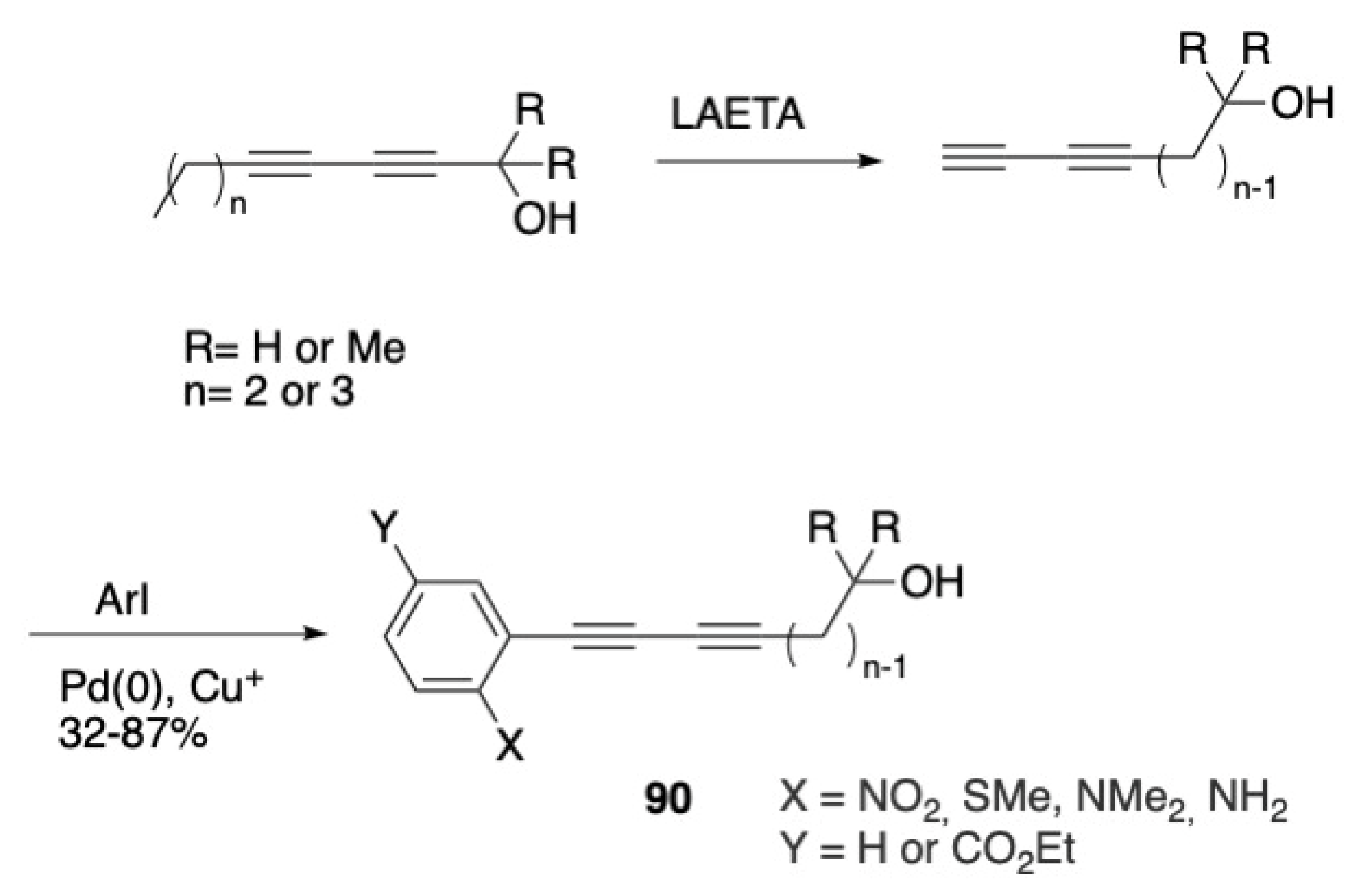
3.7. Synthesis of Arylalkadiynols via an Acetylene Zipper-Sonogashira Reaction Sequence
3.8. Polyurethane Materials
4. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
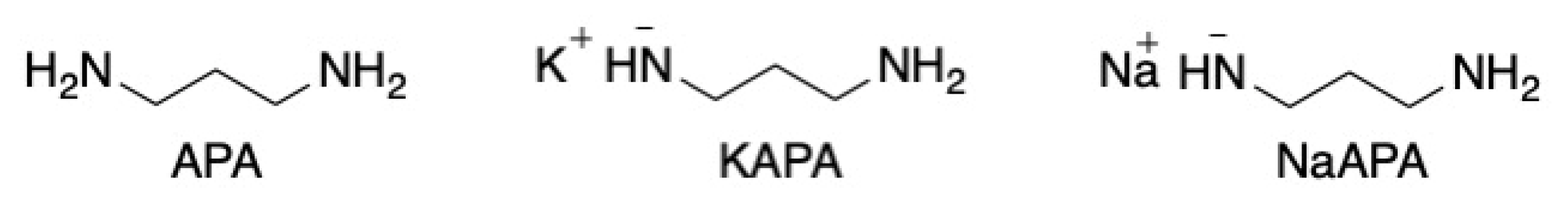
Abbreviations
| APA | 1,3-Diaminopropane |
| CTAB | Cetyltrimethylammonium bromide |
| CuAAC | Copper catalyzed azide–alkyne cycloaddition |
| DCM | Dichloromethane |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DSF | Diffusible signal factor |
| HMPA | Hexamethylphosphoramide |
| KAPA/PAPA | Potassium 3-aminopropylamide |
| KOt-Bu | Potassium tert-butoxide |
| LAETA | Lithium 2-aminoethylamide |
| MOM | Methoxymethyl |
| NaAPA | Sodium 3-aminopropylamide |
| PU | Polyurethane |
| rt | Room temperature |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| TAMRA-N3 | Tetramethylrhodamine azide |
| TBAI | Tetrabutylammonium iodide |
| TBDPS | tert-Butyldiphenylsilyl |
| THF | Tetrahydrofuran |
References
- Brown, C.A.; Yamashita, A. Saline hydrides and superbases in organic reactions. IX. Acetylene zipper. Exceptionally facile contrathermodynamic multipositional isomeriazation of alkynes with potassium 3-aminopropylamide. J. Am. Chem. Soc. 1975, 97, 891. [Google Scholar] [CrossRef]
- Brown, C.A. Saline hydrides and superbases in organic reactions. VII. Potassium hydride, highly active new hydride reagent. Reactivity, applications, and techniques in organic and organometallic reactions. J. Org. Chem. 1974, 39, 3913–3918. [Google Scholar] [CrossRef]
- Wotiz, J.H.; Billups, W.E.; Christian, D.T. The Sodium Amide Catalyzed Rearrangement of Some Acetylenes in Ethylenediamine1a. J. Org. Chem. 1966, 31, 2069–2073. [Google Scholar] [CrossRef]
- Smadja, W. Chimie Organique—L’emploi du tertibutylate de sodium dans l’alcool tertiobutylique anhydre comme agent disomerisation des hydrocarbures acetyleniques. Comptes Rendus Hebd. Des Séances De L’académie Des Sci. 1963, 256, 2426–2428. [Google Scholar]
- Smadja, W. Carbanionic isomerization of linear hydrocarbons-acetylenes-allenes-conjugated dienes. Ann. Chim. 1965, 10, 105–144. [Google Scholar]
- Smadja, W. Obtention de dienes conjuges par isomerisation basique dehydrocarbures acetyleniques et alleniques. Isomerisation basique de malanges d’heptadienes conjuges. Comptes Rendus Hebd. Des Séances De L’académie Des Sci. 1964, 258, 5461–5464. [Google Scholar]
- Wotiz, J.H.; Barelski, P.M.; Koster, D.F. Mechanism of the base-catalyzed prototropic propargylic rearrangement in vicinal diamines. J. Org. Chem. 1973, 38, 489–493. [Google Scholar] [CrossRef]
- Avocetien, K.; Li, Y.; O’Doherty, G.A. The Alkyne Zipper Reaction in Asymmetric Synthesis. In Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations; Barry, M., Trost, C.-J.L., Eds.; Wiley and Sons: Weinheim, Germany, 2014; pp. 365–394. [Google Scholar] [CrossRef]
- Becker, J.Y. Isomerization of mono- and diacetylenic hydrocarbons. Tetrahedron 1976, 32, 3041–3043. [Google Scholar] [CrossRef]
- Brown, C.A.; Yamashita, A. Exceptionally easy isomerization of acetylenic alcohols with potassium 3-aminopropylamide. A new, high yield synthesis of functionally differentiated αω-difunctional structures. J. Chem. Soc. Chem. Commun. 1976, 23, 959–960. [Google Scholar] [CrossRef]
- Lindhoudt, J.C.; van Mourik, G.L.; Pabon, H.J.J. Multipositional isomerisation of functionally substituted alkynes catalysed by potassium 3-aminopropylamide. Tetrahedron Lett. 1976, 17, 2565–2568. [Google Scholar] [CrossRef]
- Macaulay, S.R. The rearrangement of isomeric linear decyn-1-ols by reaction with the sodium salt of 1,3-diaminopropane. Can. J. Chem. 1980, 58, 2567–2572. [Google Scholar] [CrossRef]
- Abrams, S.R.; Shaw, A.C. Triple-Bond Isomerizations: 2-to 9-Decyn-1-ol. Org. Synth. 2003, 66, 127. [Google Scholar] [CrossRef]
- Abrams, S.R. A general synthesis of long chain ω- and (ω-1)-hydroxy fatty acids. Chem. Phys. Lipids 1981, 28, 379–384. [Google Scholar] [CrossRef]
- Abrams, S.R. Alkyne isomerization reagents: Mixed alkali metal amides. Can. J. Chem. 1984, 62, 1333–1334. [Google Scholar] [CrossRef]
- Abrams, S.R.; Nucciarone, D.D.; Steck, W.F. Some alkali metal alkyl amides as alkyne isomerization reagents: Selective isomerization of one triple bond of a diynol. Can. J. Chem. 1983, 61, 1073–1076. [Google Scholar] [CrossRef]
- Takaki, K.S. Potassium 3-Aminopropylamide. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Hommes, H.; Brandsma, L. A modified procedure for the isomerization of alkynes and acetylenic alcohols to terminal acetylenes:(Preliminary communication). Recl. Des Trav. Chim. Des Pays Bas 1977, 96, 160. [Google Scholar] [CrossRef]
- Kimmel, T.; Becker, D. New procedures for preparation of potassium 3-aminopropylamide. J. Org. Chem. 1984, 49, 2494–2496. [Google Scholar] [CrossRef]
- Midland, M.M.; Halterman, R.L.; Brown, C.A.; Yamaichi, A. The isomerization of optically-active propargyl alcohols to terminal acetylenes. Tetrahedron Lett. 1981, 22, 4171–4172. [Google Scholar] [CrossRef]
- Jacobs, T.L.; Akawie, R.; Cooper, R.G. Rearrangements Involving 1-Pentyne, 2-Pentyne and 1,2-Pentadiene1. J. Am. Chem. Soc. 1951, 73, 1273–1276. [Google Scholar] [CrossRef]
- Cram, D.J.; Willey, F.; Fischer, H.P.; Scott, D.A. Base-Catalyzed Intramolecular 1,3- and 1,5-Proton Transfers. J. Am. Chem. Soc. 1964, 86, 5370–5371. [Google Scholar] [CrossRef]
- Bushby, R.J. Base-catalysed isomerisation of acetylenes. Q. Rev. Chem. Soc. 1970, 24, 585–600. [Google Scholar] [CrossRef]
- Bushby, R.J.; Whitham, G.H. Researches on acetylenic compounds. Part LXVI. Base catalysed interconversions between pent-2-ynoic, penta-2,3-dienoic, and pent-3-ynoic acids—A mechanistic study. J. Chem. Soc. B Phys. Org. 1969, 67–73. [Google Scholar] [CrossRef]
- Bushby, R.J.; Whitham, G.H. Researches on acetylenic compounds. Part LXVII. Base-catalysed isomerisation of hepta-2,4-diynoic, hepta-2,5-diynoic, hepta-4,5-dien-2-ynoic, and hepta-2,3-dien-5-ynoic acids. J. Chem. Soc. B Phys. Org. 1970, 563–569. [Google Scholar] [CrossRef]
- Bowden, K.; Cook, R.S. Reactions in strongly basic solutions. Part VI. Correlation of the rates of rearrangement of weak carbon acids in aqueous dimethyl sulphoxide with an acidity function. Substituent and kinetic isotope effects. J. Chem. Soc. Perkin Trans. 2 1972, 10, 1407–1411. [Google Scholar] [CrossRef]
- Carr, M.D.; Gan, L.H.; Reid, I. Acetylene–allene isomerisations. Part I. Base catalysis by potassium t-butoxide in t-butyl alcohol. J. Chem. Soc. Perkin Trans. 2 1973, 5, 668–672. [Google Scholar] [CrossRef]
- Brandsma, L.; Wijers, H.E.; Arens, J.F. Chemistry of acetylenic ethers, 67: Allenyl thioethers from alkynyl thioethers. Recl. Des Trav. Chim. Des Pays-Bas 1963, 82, 1040–1046. [Google Scholar] [CrossRef]
- Cram, D.J. Chapter V—Isomerization by Proton Transfer in Unsaturated Systems. In Organic Chemistry; Cram, D.J., Ed.; Elsevier: Amsterdam, The Netherlands, 1965; Volume 4, pp. 175–210. [Google Scholar]
- Cram, D.J.; Uyeda, R.T. Intramolecular Proton Transfer in a Basecatalyzed Allylic Rearrangement. J. Am. Chem. Soc. 1962, 84, 4358–4359. [Google Scholar] [CrossRef]
- Reggel, L.; Friedman, S.; Wender, I. The Lithium-Ethylenediamine System. II. Isomerization of Olefins and Dehydrogenation of Cyclic Dienes1,2. J. Org. Chem. 1958, 23, 1136–1139. [Google Scholar] [CrossRef]
- Abrams, S.R. Isomerization of acetylenic acids with sodium salt of 1,2-diaminoethane: A one step synthesis of megatomoic acid. Can. J. Chem. 1982, 60, 1238–1243. [Google Scholar] [CrossRef]
- Hass, E.C.; Mezey, P.G.; Abrams, S.R. Theoretical studies on “acetylenic zipper” reaction intermediates. J. Comput. Chem. 1982, 3, 185–190. [Google Scholar] [CrossRef]
- Zhang, W.; Werness, J.B.; Tang, W. Base-Catalyzed Intramolecular Hydroamination of Conjugated Enynes. Org. Lett. 2008, 10, 2023–2026. [Google Scholar] [CrossRef]
- Hubert, A.J.; Anciaux, A.J. Base-Catalysed Prototropic Isomerization IV. Isomerization of Conjugated Diynes on a Basic Catalyst. The Mechanism of the Base-Catalysed Isomerization of Triple-Bonds. Bull. Des Sociétés Chim. Belg. 1968, 77, 513–520. [Google Scholar] [CrossRef]
- Abrams, S.R. Isomerization of alkyl branched alkynoic acids. Can. J. Chem. 1986, 64, 457–463. [Google Scholar] [CrossRef]
- Augustin, K.E.; Schäfer, H.J. Conversion of oleic acid to 17- and 18-substituted stearic acid derivatives by way of the „acetylene zipper”. Liebigs Ann. Der Chem. 1991, 1991, 1037–1040. [Google Scholar] [CrossRef]
- Li, X.; Lv, J.-M.; Hu, D.; Abe, I. Biosynthesis of alkyne-containing natural products. RSC Chem. Biol. 2021, 2, 166–180. [Google Scholar] [CrossRef]
- Shi Shun, A.L.K.; Tykwinski, R.R. Synthesis of Naturally Occurring Polyynes. Angew. Chem. Int. Ed. 2006, 45, 1034–1057. [Google Scholar] [CrossRef]
- Menger, F.; Chen, X.; Brocchini, S.; Hopkins, H.; Hamilton, D. Synthesis and thermotropic properties of macrocyclic lipids related to archaebacterial membranes. J. Am. Chem. Soc. 1993, 115, 6600–6608. [Google Scholar] [CrossRef]
- Barancelli, D.A.; Mantovani, A.C.; Jesse, C.; Nogueira, C.W.; Zeni, G. Synthesis of Natural Polyacetylenes Bearing Furan Rings. J. Nat. Prod. 2009, 72, 857–860. [Google Scholar] [CrossRef]
- Paik, S.; Carmeli, S.; Cullingham, J.; Moore, R.E.; Patterson, G.M.L.; Tius, M.A. Mirabimide E, an Unusual N-Acylpyrrolinone from the Blue-Green Alga Scytonema mirabile: Structure Determination and Synthesis. J. Am. Chem. Soc. 1994, 116, 8116–8125. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Gonzalez-Rodriguez, E.; Kawade, R.K.; Stepanov, A.A.; Vasilevsky, S.F. Alkynes as Synthetic Equivalents of Ketones and Aldehydes: A Hidden Entry into Carbonyl Chemistry. Molecules 2019, 24, 1036. [Google Scholar] [CrossRef] [Green Version]
- Godt, A.; Duda, S.; Ünsal, Ö.; Thiel, J.; Härter, A.; Roos, M.; Tschierske, C.; Diele, S. An Efficient Synthesis of Liquid Crystalline Gigantocycles Combining Banana-Shaped and Rod-Like Mesogenic Units. Chem. A Eur. J. 2002, 8, 5094–5106. [Google Scholar] [CrossRef]
- Diederich, F.; Stang, P.J.; Tykwinski, R.R. Acetylene Chemistry: Chemistry, Biology and Material Science; John Wiley & Sons: Weinheim, Germany, 2006. [Google Scholar]
- Nicolaou, K.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef]
- Wang, D.; Gao, S. Sonogashira coupling in natural product synthesis. Org. Chem. Front. 2014, 1, 556–566. [Google Scholar] [CrossRef]
- Fürstner, A. Alkyne metathesis on the rise. Angew. Chem. Int. Ed. 2013, 52, 2794–2819. [Google Scholar] [CrossRef]
- Fürstner, A.; Davies, P.W. Alkyne metathesis. Chem. Commun. 2005, 23, 2307–2320. [Google Scholar] [CrossRef]
- Diver, S.T.; Giessert, A.J. Enyne Metathesis (Enyne Bond Reorganization). Chem. Rev. 2004, 104, 1317–1382. [Google Scholar] [CrossRef]
- Mori, M. Synthesis of Natural Products and Related Compounds using Enyne Metathesis. Adv. Synth. Catal. 2007, 349, 121–135. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. ‘Click’chemistry in polymer and materials science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Negishi, E.-I.; Abramovitch, A. A highly efficient chemo, regio, and stereoselective synthesis of (7E, 9Z)-dodecadien-1-yl acetate, a sex pheromone of lobesia botrana, via a functionalized organoborate. Tetrahedron Lett. 1977, 18, 411–414. [Google Scholar] [CrossRef]
- Schwarz, M.; Klun, J.A.; Leonhardt, B.A.; Johnson, D.T. (E,Z)-2,13-octadecadien-1-ol acetate. A new pheromone structure for sesiid moths. Tetrahedron Lett. 1983, 24, 1007–1010. [Google Scholar] [CrossRef]
- Ho, H.; Tao, Y.; Tsai, R.; Wu, Y.; Tseng, H.; Chow, Y. Isolation, identification, and synthesis of sex pheromone components of female tea cluster caterpillar, Andraca bipunctata Walker (Lepidoptera: Bombycidae) in Taiwan. J. Chem. Ecol. 1996, 22, 271–285. [Google Scholar] [CrossRef]
- Camps, F.; Fabriàs, G.; Gasol, V.; Guerrero, A.; Hernández, R.; Montoya, R. Analogs of sex pheromone of processionary moth,Thaumetopoea pityocampa: Synthesis and biological activity. J. Chem. Ecol. 1988, 14, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Watanabe, H.; Fujiwhara, M.; Kuwahara, S. Pheromone synthesis, CXXII. (E)- and (Z)-tetradecenyl formate, potent sex pheromone mimics against the yellow peach moth. Liebigs Ann. Der Chem. 1990, 1990, 1257–1259. [Google Scholar] [CrossRef]
- Mori, K.; Argade, N.P. Pheromone Synthesis, CLXIII. Synthesis of (9Z,25S,26R,43Z)-25,26-Epoxy-9,43-henpentacontadiene and Its Antipode, Components of the Nymph Recognition Pheromone Produced by Nymphs of the Cockroach Nauphoeta cinerea. Liebigs Ann. Der Chem. 1994, 1994, 695–700. [Google Scholar] [CrossRef]
- Senda, S.; Mori, K. Synthesis of (R)-(—)-10-Methyl-2-tridecanone, the Pheromone of the Southern Corn Rootworm. Agric. Biol. Chem. 1983, 47, 795–798. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.J.; Mays, S.G.; Baillie, M.T.; Howard, R.B.; Culver, D.G.; Saindane, M.; Pruett, S.T.; Holt, J.J.; Menaldino, D.S.; Evers, T.J.; et al. Discovery of a Fluorinated Enigmol Analog with Enhanced in Vivo Pharmacokinetic and Anti-Tumor Properties. ACS Med. Chem. Lett. 2016, 7, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Yadav, J.S.; Deshpande, P.K.; Sharma, G.V.M. Stereoselective synthesis of (S)-13-hydroxy octadeca-(9Z, 11E)-di- and (9Z, 11E, 15Z)-trienoic acids: Selfdefensive substances against rice blast disease. Tetrahedron 1992, 48, 4465–4474. [Google Scholar] [CrossRef]
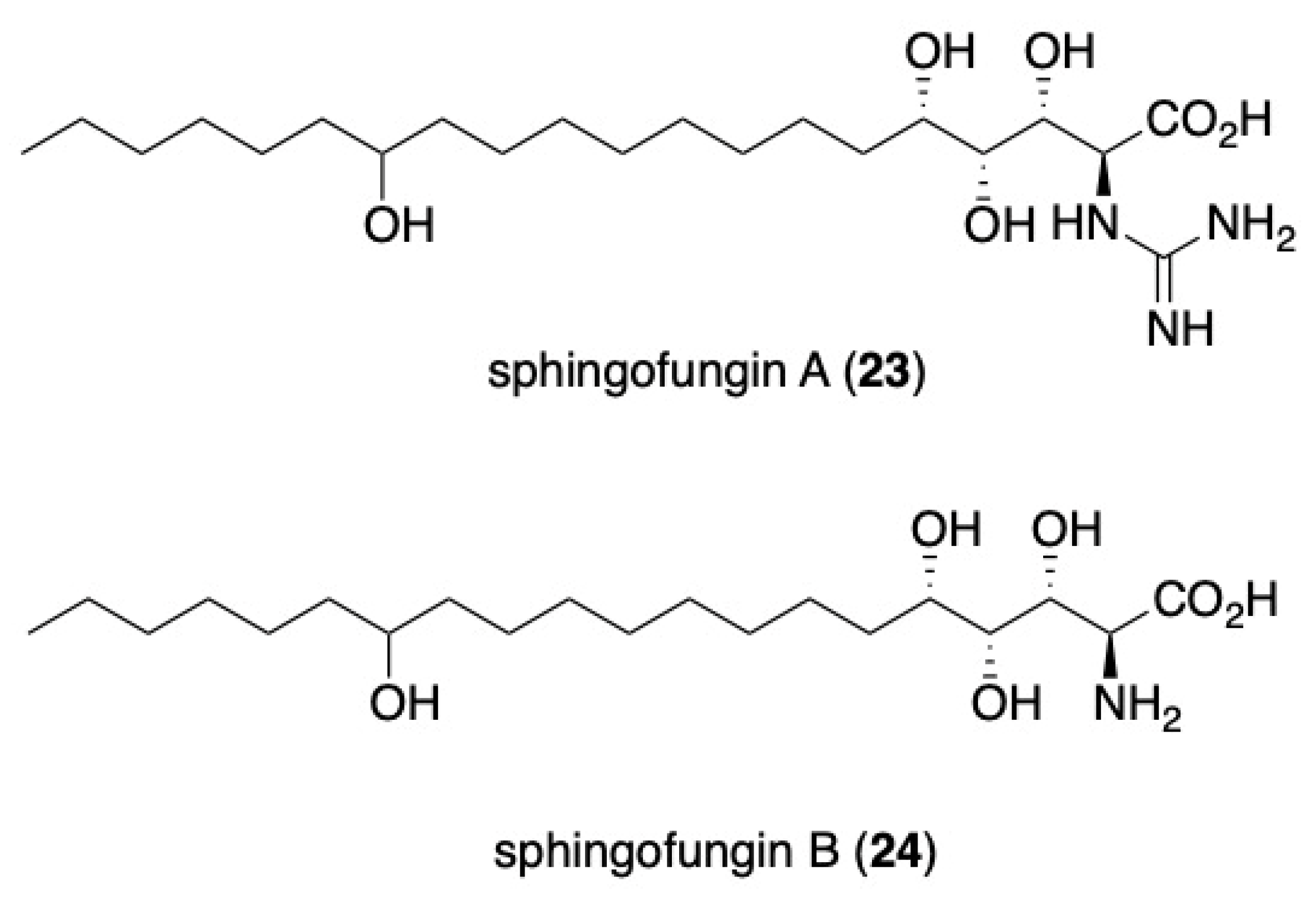
- Mori, K.; Otaka, K. Synthesis of sphingofungin D and its stereoisomer at C-14. Tetrahedron Lett. 1994, 35, 9207–9210. [Google Scholar] [CrossRef]
- Otaka, K.; Mori, K. Synthesis of Sphingofungin D and Its Three Diastereomers. Eur. J. Org. Chem. 1999, 1999, 1795–1802. [Google Scholar] [CrossRef]
- Chida, N.; Ikemoto, H.; Noguchi, A.; Amano, S.; Ogawa, S. Total Synthesis and Absolute Configuration of Sphingofungin D (N-Acetyl Asperfungin). Nat. Prod. Lett. 1995, 6, 295–302. [Google Scholar] [CrossRef]
- Sugiyama, S.; Honda, M.; Komori, T. Biologically active glycosides from asteroidea, XV. Asymmetric synthesis of phytosphingosine and phytosphingosine anhydro base: Assignment of the absolute stereochemistry. Liebigs Ann. Der Chem. 1988, 1988, 619–625. [Google Scholar] [CrossRef]
- Sugiyama, S.; Honda, M.; Komori, T. Biologically active glycosides from Asteroidea, XXIII. Synthesis of acanthacerebroside A. Liebigs Ann. Der Chem. 1990, 1990, 1063–1068. [Google Scholar] [CrossRef]
- Macaulay, S.R. Isomerization of internal triple bonds of alkyn-1-ols with sodium hydride in 1, 3-diaminopropane. J. Org. Chem. 1980, 45, 734–735. [Google Scholar] [CrossRef]
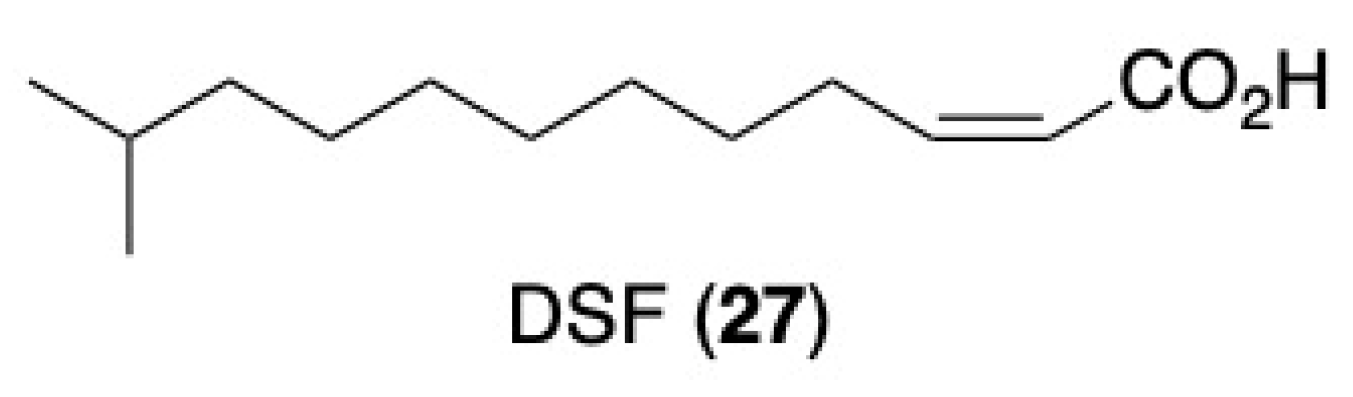
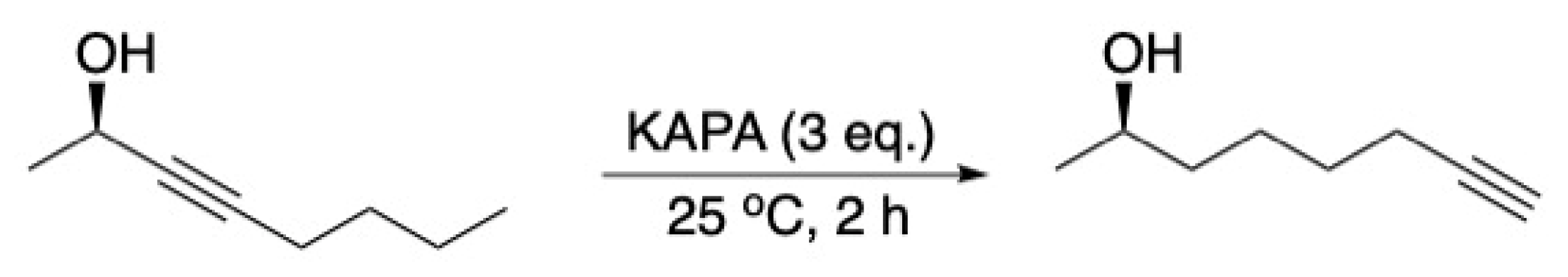
- Kumar, V.P.; Gupta, M.K.; Horgan, C.; O’Sullivan, T.P. Synthesis of the quorum sensing molecule Diffusible Signal Factor using the alkyne zipper reaction. Tetrahedron Lett. 2018, 59, 2193–2195. [Google Scholar] [CrossRef]
- Alves, D.; Nogueira, C.W.; Zeni, G. Synthesis of polyacetylenic acids isolated from Nanodea muscosa. Tetrahedron Lett. 2005, 46, 8761–8764. [Google Scholar] [CrossRef]
- Zeni, G.; Panatieri, R.B.; Lissner, E.; Menezes, P.H.; Braga, A.L.; Stefani, H.A. Synthesis of Polyacetylenic Acids Isolated from Heisteria acuminata. Org. Lett. 2001, 3, 819–821. [Google Scholar] [CrossRef]
- Tahiri, N.; Fodran, P.; Jayaraman, D.; Buter, J.; Witte, M.D.; Ocampo, T.A.; Moody, D.B.; Van Rhijn, I.; Minnaard, A.J. Total Synthesis of a Mycolic Acid from Mycobacterium tuberculosis. Angew. Chem. Int. Ed. 2020, 59, 7555–7560. [Google Scholar] [CrossRef] [Green Version]
- Nickel, S.; Serwa, R.A.; Kaschani, F.; Ninck, S.; Zweerink, S.; Tate, E.W.; Kaiser, M. Chemoproteomic Evaluation of the Polyacetylene Callyspongynic Acid. Chem. A Eur. J. 2015, 21, 10721–10728. [Google Scholar] [CrossRef]
- Stefani, H.A.; Costa, I.M.; Zeni, G. Synthesis of polyacetylenic montiporic acids A and B. Tetrahedron Lett. 1999, 40, 9215–9217. [Google Scholar] [CrossRef]
- Hannoush, R.N.; Arenas-Ramirez, N. Imaging the Lipidome: ω-Alkynyl Fatty Acids for Detection and Cellular Visualization of Lipid-Modified Proteins. ACS Chem. Biol. 2009, 4, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, J.E.D.; Courtney, T.D.L.; Lee, V.; Baldwin, J.E. Asymmetric synthesis of cytotoxic sponge metabolites R-strongylodiols A and B. Tetrahedron Lett. 2004, 45, 5645–5647. [Google Scholar] [CrossRef]
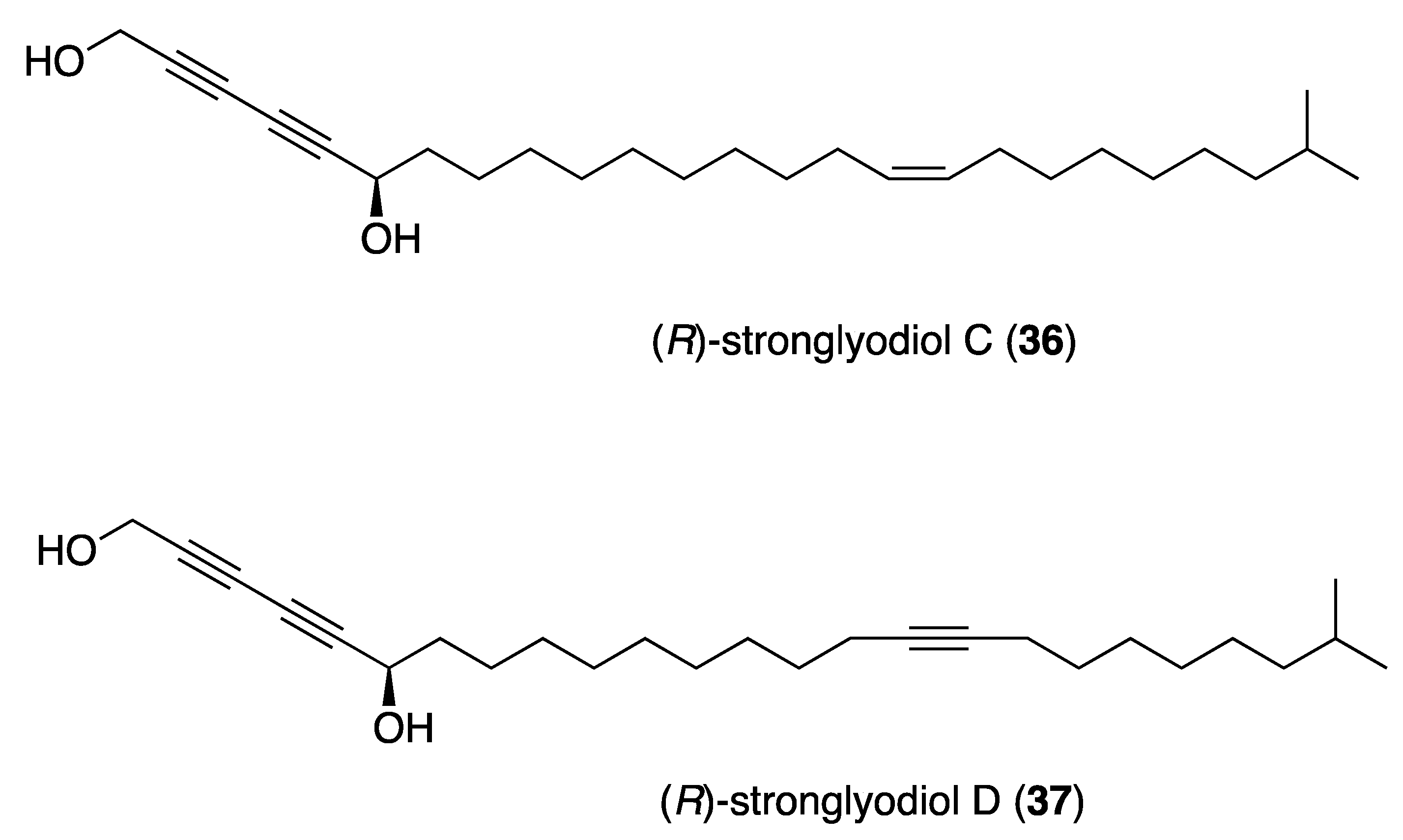
- Liu, F.; Zhong, J.; Li, S.; Li, M.; Wu, L.; Wang, Q.; Mao, J.; Liu, S.; Zheng, B.; Wang, M.; et al. Total Syntheses of (R)-Strongylodiols C and D. J. Nat. Prod. 2016, 79, 244–247. [Google Scholar] [CrossRef]
- Hansen, T.V.; Stenstrom, Y. A Facile Formal Synthesis of Volicitin. Synth. Commun. 2000, 30, 2549–2557. [Google Scholar] [CrossRef]
- Alborn, H.; Turlings, T.; Jones, T.; Stenhagen, G.; Loughrin, J.; Tumlinson, J. An elicitor of plant volatiles from beet armyworm oral secretion. Science 1997, 276, 945–949. [Google Scholar] [CrossRef]
- Dobbs, A.P.; Venturelli, A.; Butler, L.A.; Parker, R.J. First Total Synthesis of the Irciniasulfonic Acids. Synlett 2005, 2005, 652–654. [Google Scholar] [CrossRef]
- Oehlschlager, A.; Czyzewska, E.; Aksela, R.; Pierce, H., Jr. Improved syntheses of hydroxy acid precursors of macrolide pheromones of cucujid grain beetles. Can. J. Chem. 1986, 64, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Chinnababu, B.; Reddy, S.P.; Reddy, D.K.; Rao, D.C.; Venkateswarlu, Y. Stereoselective Concise Total Synthesis of Leodomycin C and D. Synthesis 2012, 44, 311–315. [Google Scholar] [CrossRef]
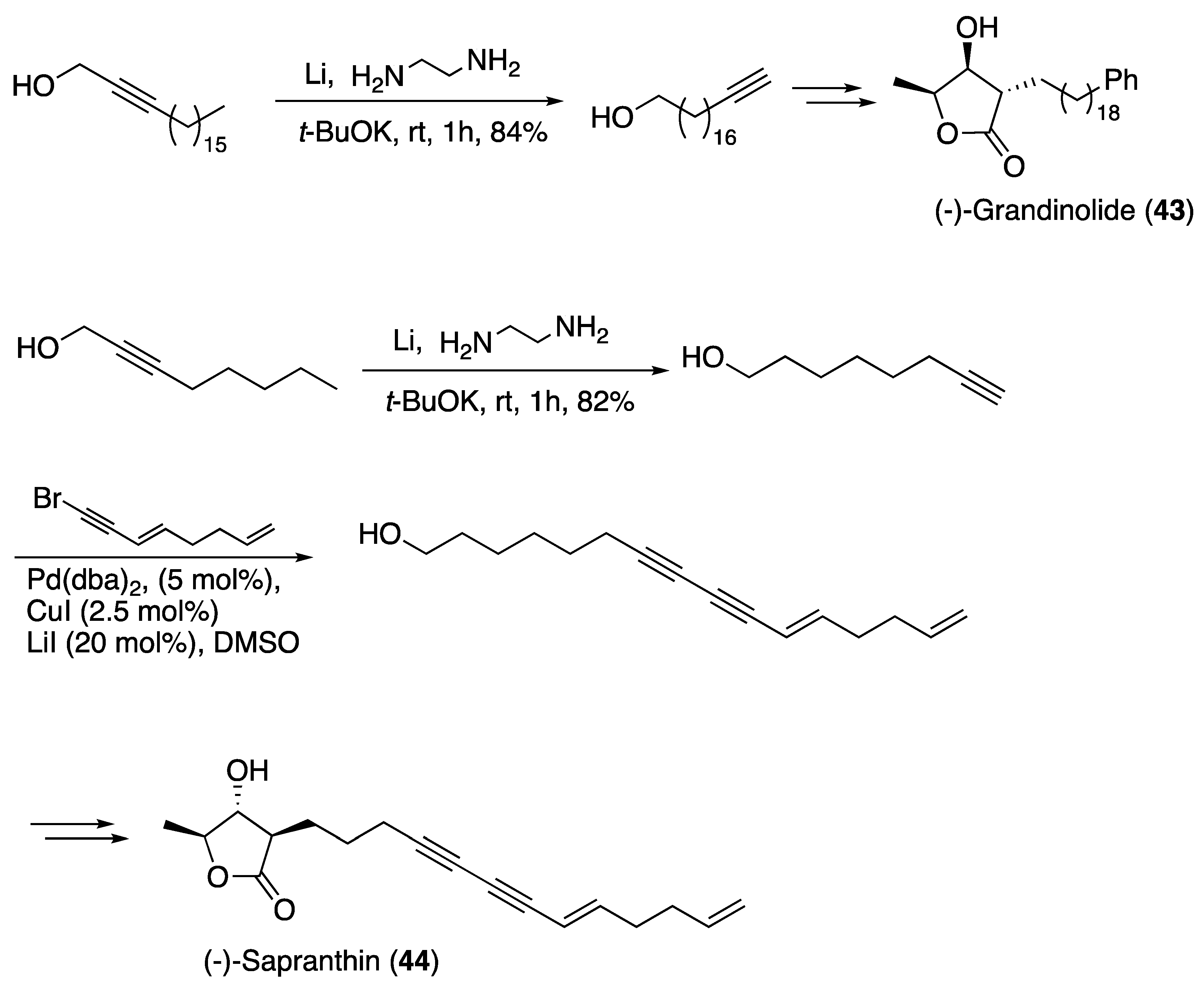
- Harcken, C.; Brückner, R.; Rank, E. Total Syntheses of (−)-Grandinolide and (−)-Sapranthin by the Sharpless Asymmetric Dihydroxylation of Methyl trans-3-Pentenoate: Elucidation of the Stereostructure of (−)-Sapranthin. Chem. A Eur. J. 1998, 4, 2342–2352. [Google Scholar] [CrossRef]
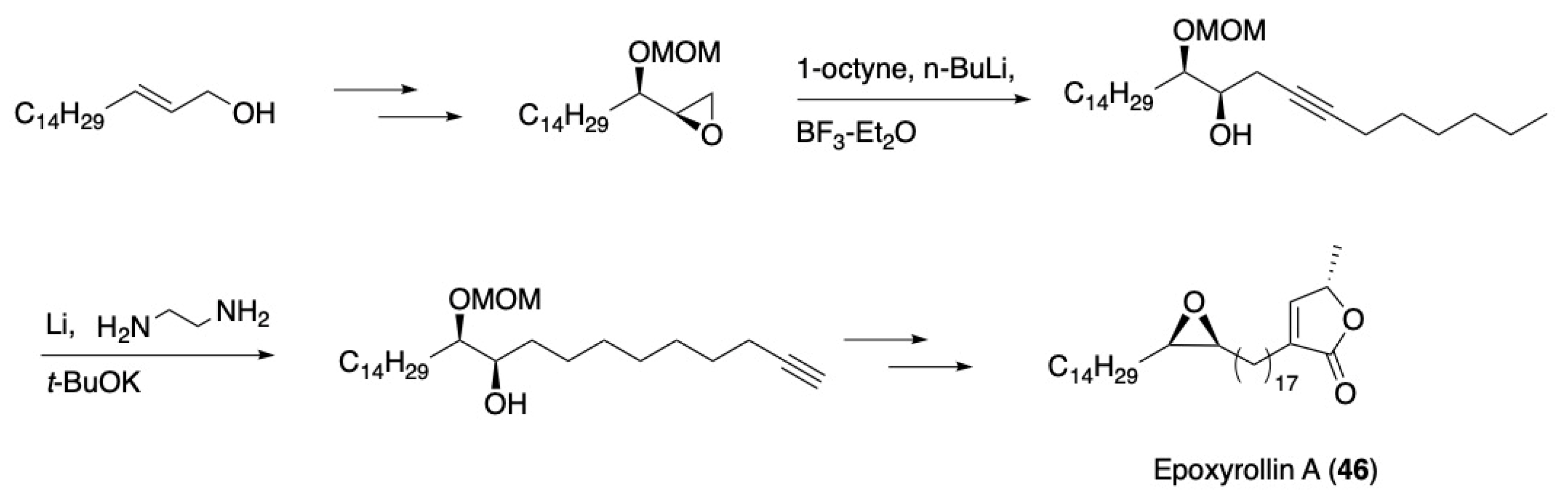
- Konno, H.; Makabe, H.; Tanaka, A.; Oritani, T. Synthesis of two possible diastereoisomers of the epoxy lactone proposed for epoxyrollin A. Biosci. Biotechnol. Biochem. 1995, 59, 2355–2357. [Google Scholar] [CrossRef]
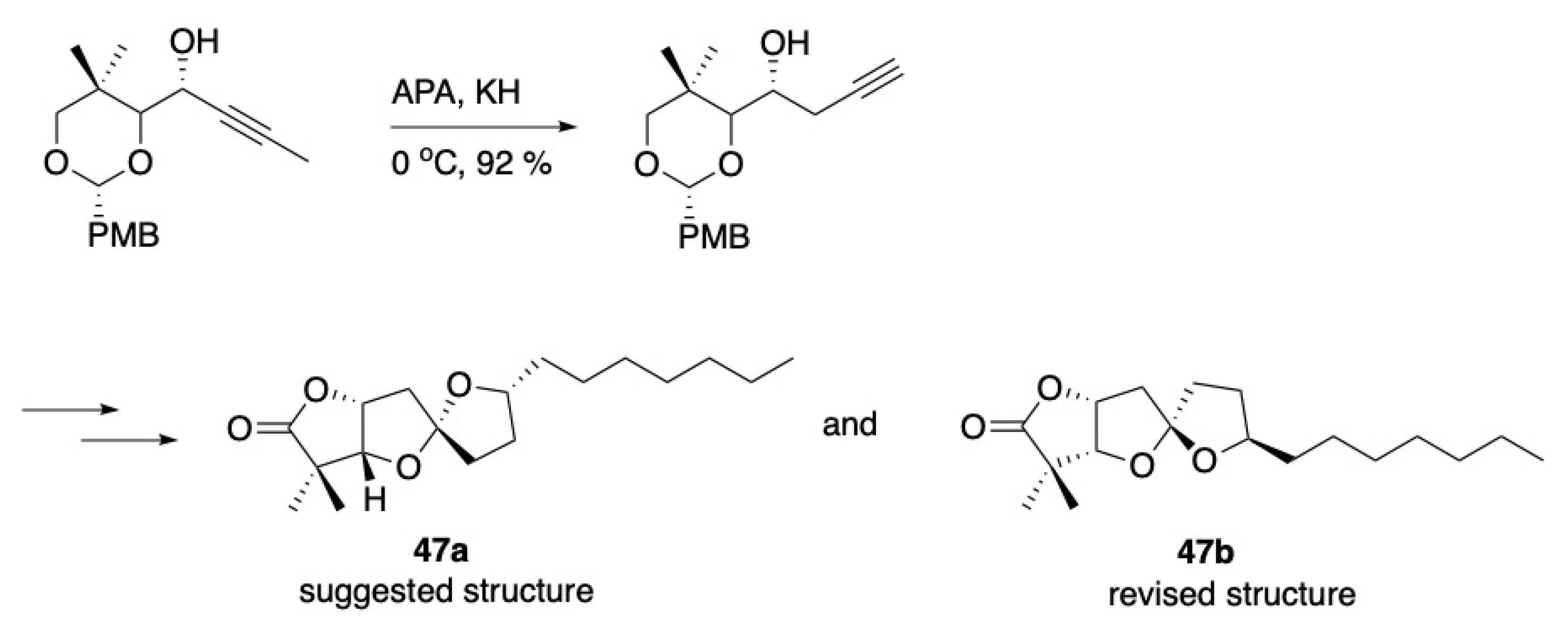
- Tlais, S.F.; Dudley, G.B. A gold-catalyzed alkyne-diol cycloisomerization for the synthesis of oxygenated 5,5-spiroketals. Beilstein J. Org. Chem. 2011, 7, 570–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tlais, S.F.; Dudley, G.B. Stereocontrol of 5,5-Spiroketals in the Synthesis of Cephalosporolide H Epimers. Org. Lett. 2010, 12, 4698–4701. [Google Scholar] [CrossRef] [PubMed]
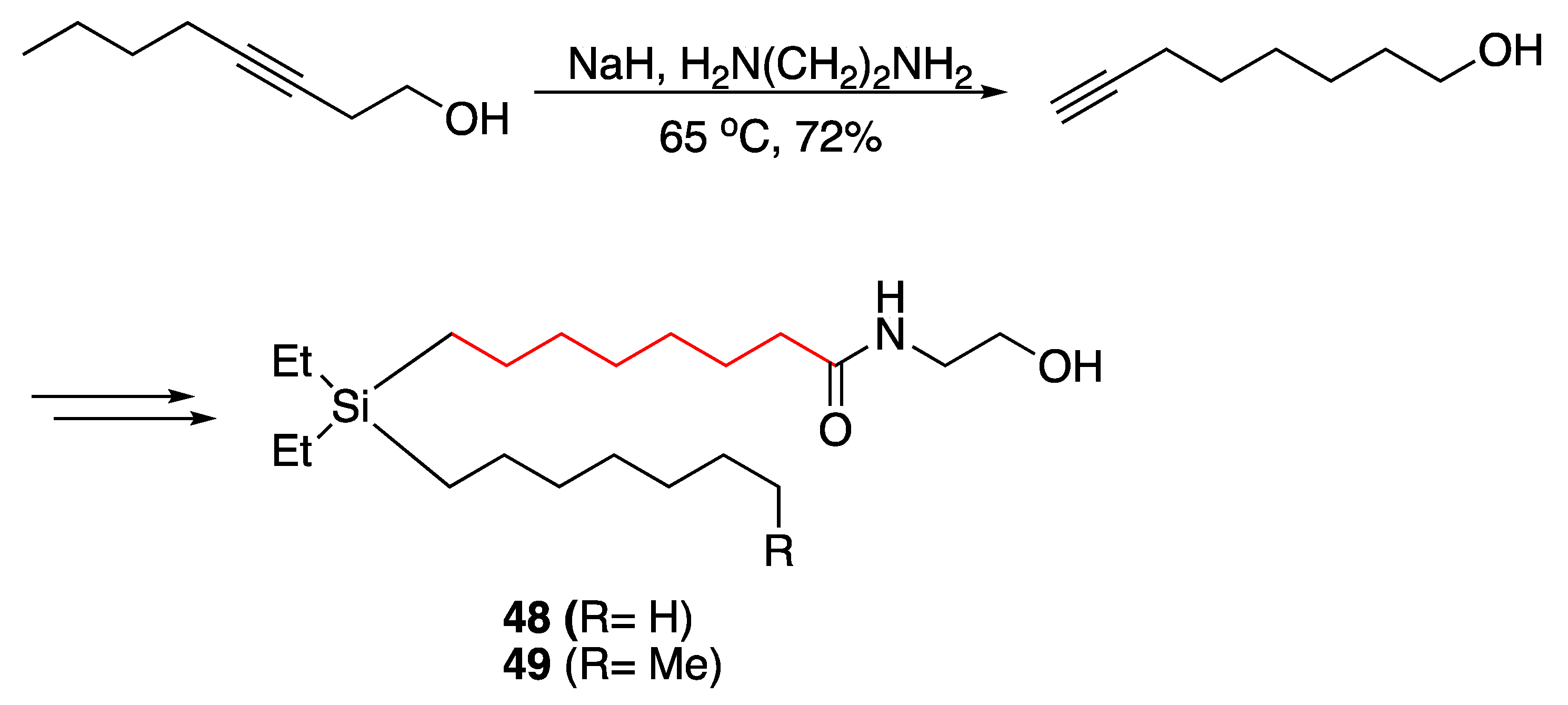
- Kajita, D.; Nakamura, M.; Matsumoto, Y.; Ishikawa, M.; Hashimoto, Y.; Fujii, S. Design and synthesis of silicon-containing fatty acid amide derivatives as novel peroxisome proliferator-activated receptor (PPAR) agonists. Bioorganic Med. Chem. Lett. 2015, 25, 3350–3354. [Google Scholar] [CrossRef]
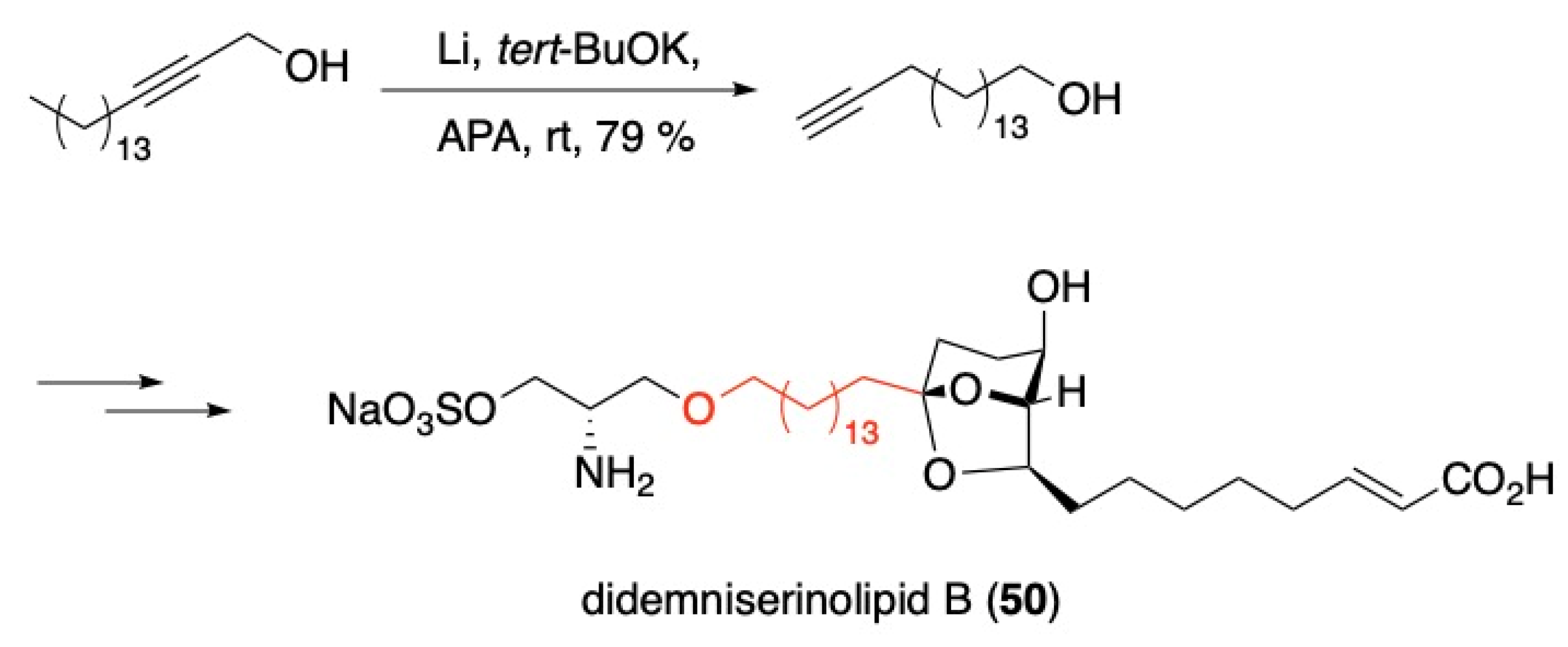
- Ramana, C.V.; Induvadana, B. A formal synthesis of (+)-didemniserinolipid B employing a Pd-mediated 6-endo selective alkynediol cycloisomerization. Tetrahedron Lett. 2009, 50, 271–273. [Google Scholar] [CrossRef]
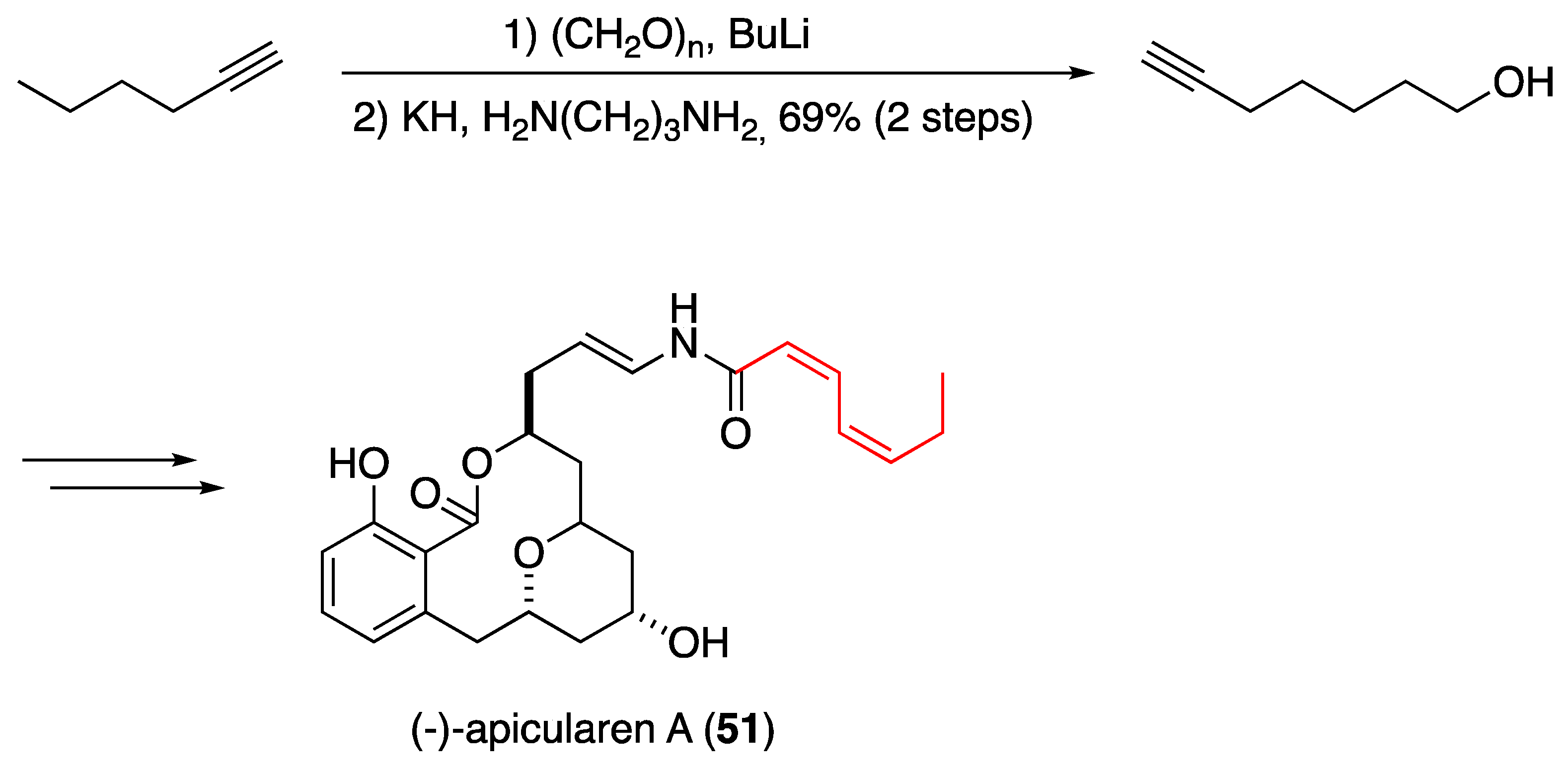
- Li, M.; O’Doherty, G.A. De Novo Formal Synthesis of (−)-Apicularen A via an Iterative Asymmetric Hydration Sequence. Org. Lett. 2006, 8, 6087. [Google Scholar] [CrossRef] [Green Version]
- Hötling, S.; Haberlag, B.; Tamm, M.; Collatz, J.; Mack, P.; Steidle, J.L.M.; Vences, M.; Schulz, S. Identification and Synthesis of Macrolide Pheromones of the Grain Beetle Oryzaephilus Surinamensis and the Frog Spinomantis Aglavei. Chem. – A Eur. J. 2014, 20, 3183–3191. [Google Scholar] [CrossRef]
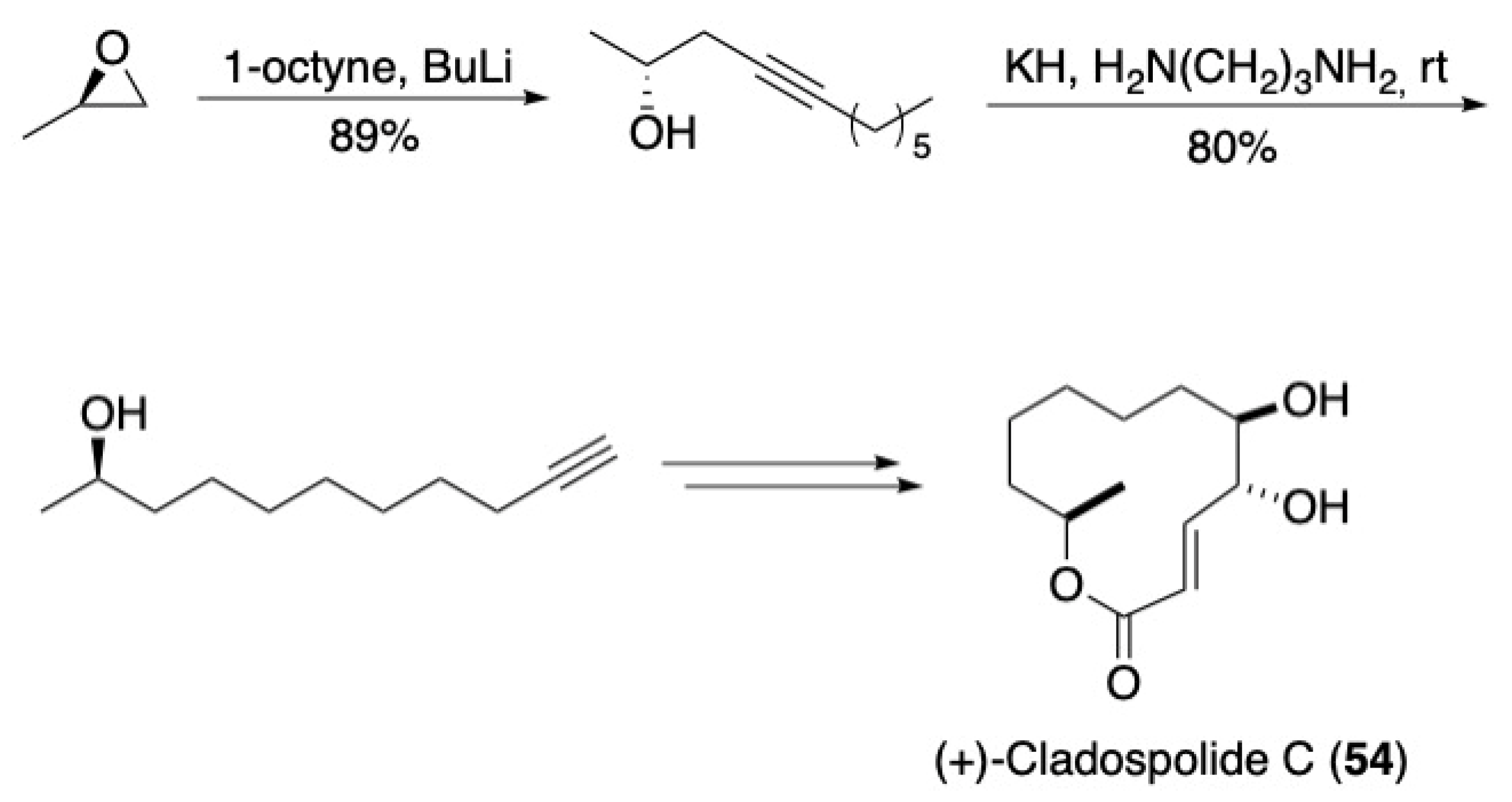
- Raji Reddy, C.; Narsimha Rao, N. An efficient total synthesis of (+)-Cladospolide C. Tetrahedron Lett. 2009, 50, 2478–2480. [Google Scholar] [CrossRef]
- Reddy, C.R.; Suman, D.; Rao, N.N. Alkyne-Mediated Approach for Total Syntheses of Cladospolides A, B, C and iso-Cladospolide B. Eur. J. Org. Chem. 2013, 2013, 3786–3796. [Google Scholar] [CrossRef]
- Reddy, C.R.; Rao, N.N.; Sujitha, P.; Kumar, C.G. Protecting Group-Free Syntheses of (4S,5S,11R)- and (4S,5S,11S)-iso-Cladospolide B and Their Biological Evaluation. Synthesis 2012, 44, 1663–1666. [Google Scholar] [CrossRef]
- Reddy, C.R.; Rao, N.N.; Sujitha, P.; Kumar, C.G. Total Synthesis of (+)-Aspicilin by an Alkyne-Based Approach and Its Biological Evaluation. Eur. J. Org. Chem. 2012, 2012, 1819. [Google Scholar] [CrossRef]
- Quinkert, G.; Heim, N.; Bats, J.W.; Oschkinat, H.; Kessler, H. Die Struktur des Flechten-Makrolids (+)-Aspicilin. Angew. Chem. 1985, 97, 985–986. [Google Scholar] [CrossRef]
- Quinkert, G.; Heim, N.; Bats, J.W.; Oschkinat, H.; Kessler, H. The Structure of the Lichen Macrolide (+)-Aspicilin. Angew. Chem. Int. Ed. Engl. 1985, 24, 987–988. [Google Scholar] [CrossRef]
- Utimoto, K.; Tanaka, M.; Kitai, M.; Nozaki, H. Cyclization of ω-trimethylsilylethynylalkanoyl chlorides. Application of the preparation of large ring ynones and dl- and (R)-muscone. Tetrahedron Lett. 1978, 19, 2301–2304. [Google Scholar] [CrossRef]
- Raji Reddy, C.; Suman, D.; Narsimha Rao, N. Alkyne-Assisted Approach to the Formal Synthesis of Antibiotic Macrolide (-)-A26771B. Synlett 2012, 2012, 272–274. [Google Scholar] [CrossRef]
- Trost, B.M.; Sieber, J.D.; Qian, W.; Dhawan, R.; Ball, Z.T. Asymmetric Total Synthesis of Soraphen A: A Flexible Alkyne Strategy. Angew. Chem. Int. Ed. 2009, 48, 5478–5481. [Google Scholar] [CrossRef]
- Larson, D.P.; Heathcock, C.H. Total synthesis of tricolorin A. J. Org. Chem. 1997, 62, 8406–8418. [Google Scholar] [CrossRef]
- Sommer, H.; Fürstner, A. Hydroxyl-Assisted Carbonylation of Alkenyltin Derivatives: Development and Application to a Formal Synthesis of Tubelactomicin A. Org. Lett. 2016, 18, 3210–3213. [Google Scholar] [CrossRef] [Green Version]
- Hoye, R.C.; Baigorria, A.S.; Danielson, M.E.; Pragman, A.A.; Rajapakse, H.A. Synthesis of Elenic Acid, an Inhibitor of Topoisomerase II. J. Org. Chem. 1999, 64, 2450–2453. [Google Scholar] [CrossRef]
- Hoye, R.C.; Anderson, G.L.; Brown, S.G.; Schultz, E.E. Total Synthesis of Clathculins A and B. J. Org. Chem. 2010, 75, 7400–7403. [Google Scholar] [CrossRef]
- Cunha, V.L.S.; Liu, X.; Lowary, T.L.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Avocadyne, Avocadene, and Avocadane Stereoisomers. J. Org. Chem. 2019, 84, 15718–15725. [Google Scholar] [CrossRef] [PubMed]
- Yajima, A.; Takikawa, H.; Mori, K. Synthesis of Sphingosine Relatives, XVIII!. Synthesis of Penazetidine A, an Alkaloid Inhibitor of Protein Kinase C Isolated from the Marine Sponge Penares sollasi. Liebigs Ann. 1996, 1996, 1083–1089. [Google Scholar] [CrossRef]
- Yoda, H.; Oguchi, T.; Takabe, K. Novel asymmetric synthesis of penaresidin B as a potent actomyosin ATPase activator. Tetrahedron Lett. 1997, 38, 3283–3284. [Google Scholar] [CrossRef]
- Yoda, H.; Uemura, T.; Takabe, K. Novel and practical asymmetric synthesis of an azetidine alkaloid, penaresidin B. Tetrahedron Lett. 2003, 44, 977–979. [Google Scholar] [CrossRef]
- Fujiwara, T.; Hashimoto, K.; Umeda, M.; Murayama, S.; Ohno, Y.; Liu, B.; Nambu, H.; Yakura, T. Divergent total synthesis of penaresidin B and its straight side chain analogue. Tetrahedron 2018, 74, 4578–4591. [Google Scholar] [CrossRef]
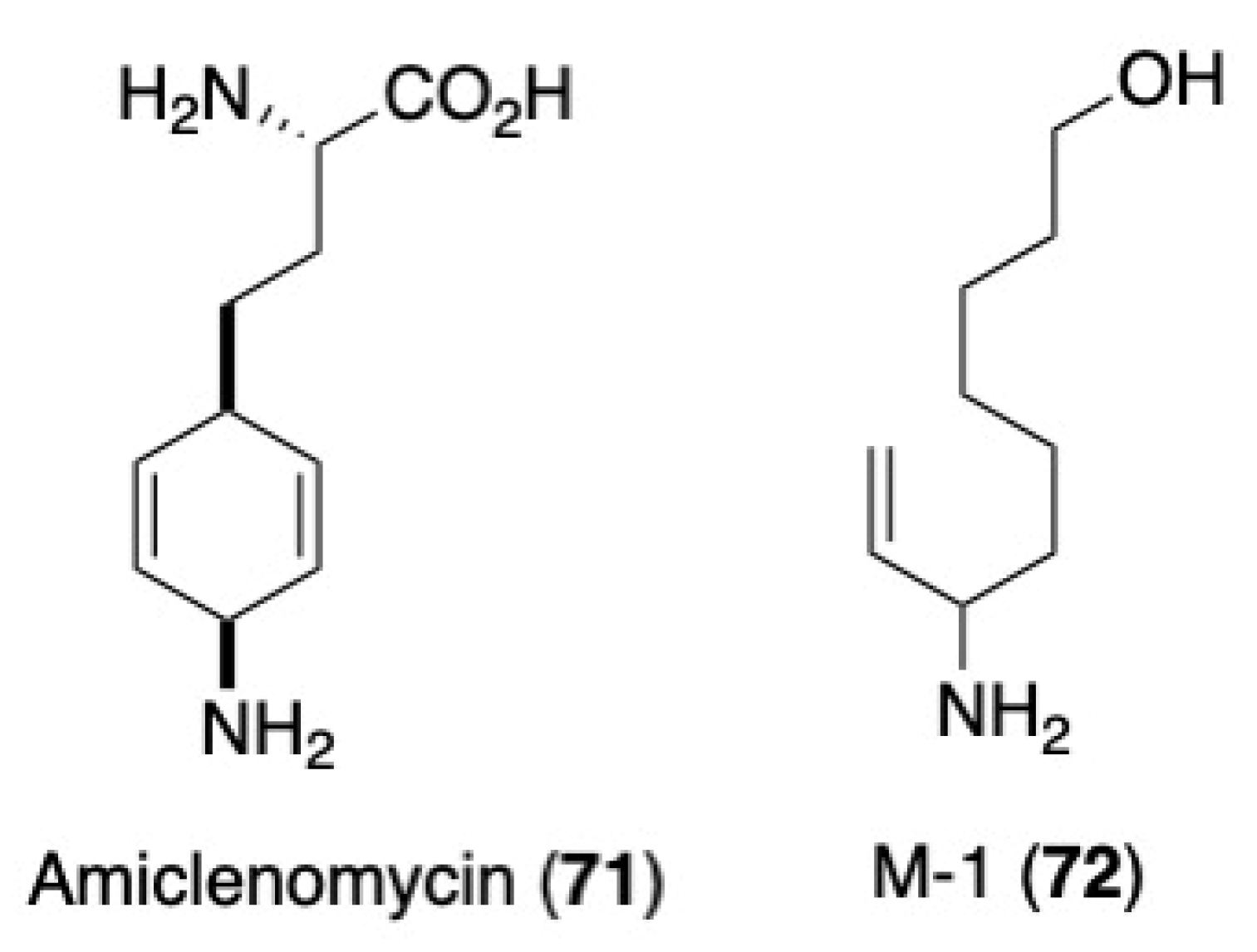
- Shi, C.; Aldrich, C.C. Design and Synthesis of Potential Mechanism-Based Inhibitors of the Aminotransferase BioA Involved in Biotin Biosynthesis. J. Org. Chem. 2012, 77, 6051–6058. [Google Scholar] [CrossRef] [Green Version]
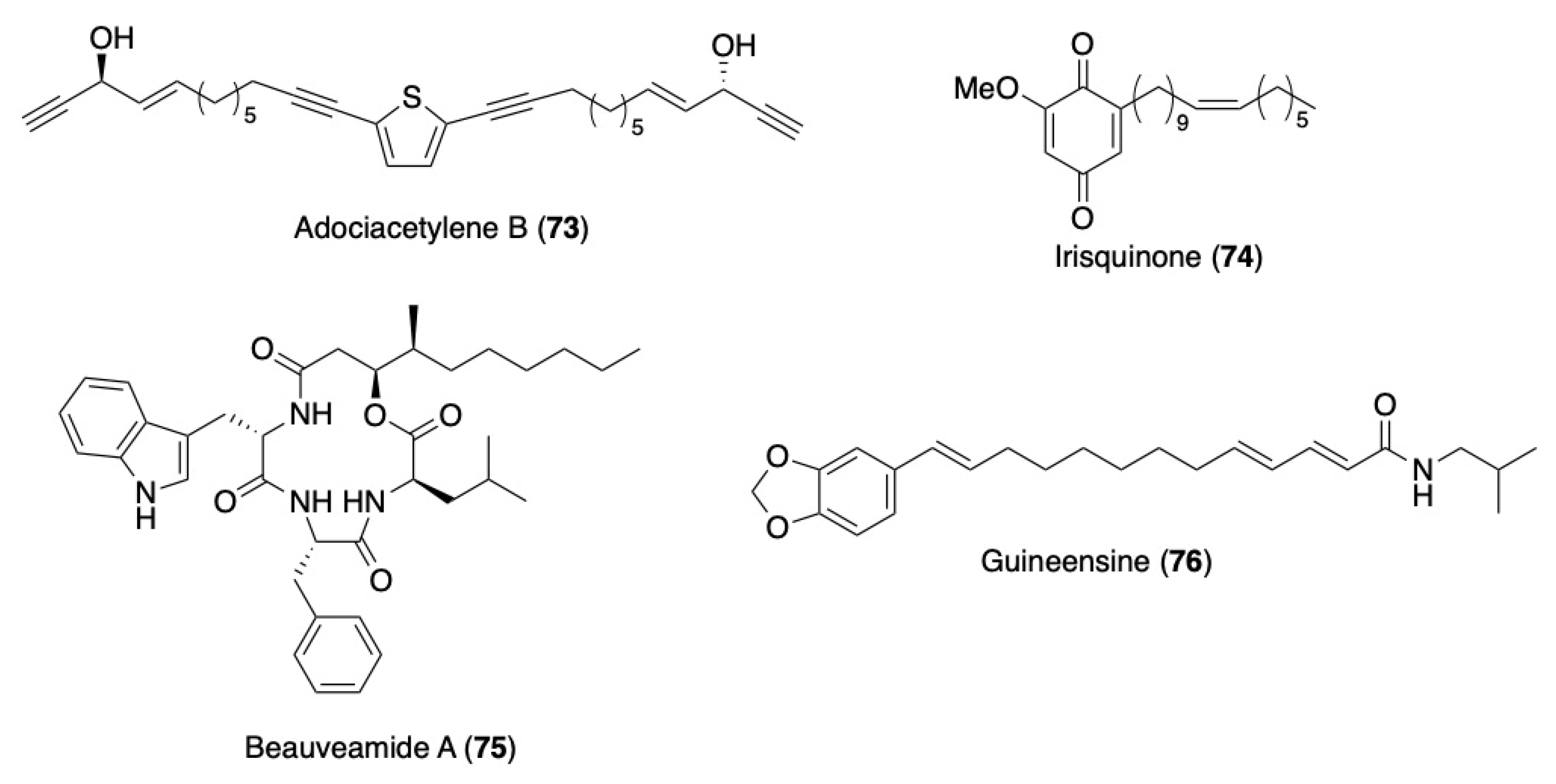
- Gung, B.W.; Dickson, H.; Shockley, S. A concise synthesis of (+)- and (−)-adociacetylene B. Tetrahedron Lett. 2001, 42, 4761–4763. [Google Scholar] [CrossRef]
- Hadfield, J.A.; McGown, A.T.; Butler, J. A High-Yielding Synthesis of the Naturally Occurring Antitumour Agent Irisquinone. Molecules 2000, 5, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Auddy, S.S.; Chatterjee, A.; Sen, P.; Goswami, R.K. Late-Stage Functionalization: Total Synthesis of Beauveamide A and Its Congeners and Their Anticancer Activities. Org. Lett. 2022, 24, 7113–7117. [Google Scholar] [CrossRef]
- Bartholomäus, R.; Nicolussi, S.; Baumann, A.; Rau, M.; Simão, A.C.; Gertsch, J.; Altmann, K.-H. Total Synthesis of the Endocannabinoid Uptake Inhibitor Guineensine and SAR Studies. ChemMedChem 2019, 14, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Barroso, S.; Castelli, R.; Baggelaar, M.P.; Geerdink, D.; ter Horst, B.; Casas-Arce, E.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C.; Minnaard, A.J. Total Synthesis of the Triglycosyl Phenolic Glycolipid PGL-tb1 from Mycobacterium tuberculosis. Angew. Chem. Int. Ed. 2012, 51, 11774–11777. [Google Scholar] [CrossRef]
- Parenty, A.; Campagne, J.-M.; Aroulanda, C.; Lesot, P. Routine Use of Natural Abundance Deuterium NMR in a Polypeptidic Chiral Oriented Solvent for the Determination of the Enantiomeric Composition of Chiral Building Blocks. Org. Lett. 2002, 4, 1663–1666. [Google Scholar] [CrossRef] [PubMed]
- Sharif, E.U.; Wang, H.Y.L.; Akhmedov, N.G.; O’Doherty, G.A. Merremoside D: De novo synthesis of the purported structure, NMR analysis, comparison of spectral data. Org. Lett. 2014, 16, 492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulzer, M.; Tiegs, B.J.; Wang, Y.; Coates, G.W.; O’Doherty, G.A. Total Synthesis of Tetrahydrolipstatin and Stereoisomers via a Highly Regio- and Diastereoselective Carbonylation of Epoxyhomoallylic Alcohols. J. Am. Chem. Soc. 2014, 136, 10814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, Y.; O’Doherty, G.A. The Asymmetric Synthesis of Tetrahydrolipstatin. Asian J. Org. Chem. 2015, 4, 994. [Google Scholar] [CrossRef]
- Xing, Y.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Cladospolide B−D: Structural Reassignment of Cladospolide D via the Synthesis of its Enantiomer. Org. Lett. 2009, 11, 1107. [Google Scholar] [CrossRef]
- Xing, Y.; Penn, J.H.; O’Doherty, G.A. Structure Investigations of (ent)-Cladospolide D by De Novo Synthesis and Kinetic and Thermodynamic Isomerization. Synthesis 2009, 2009, 2847. [Google Scholar] [CrossRef]
- Avocetien, K.F.; Li, J.J.; Liu, X.; Wang, Y.; Xing, Y.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Phoracantholide J. Org. Lett. 2016, 18, 4970–4973. [Google Scholar] [CrossRef]
- Xing, Y.; O’Doherty, G.A. De Novo Asymmetric Approach to Aspergillide-C: Synthesis of 4-epi-seco-Aspergillide-C. ChemistrySelect 2022, 7, e202200266. [Google Scholar] [CrossRef]
- Guo, H.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Daumone via a Palladium-Catalyzed Glycosylation. Org. Lett. 2005, 7, 3921. [Google Scholar] [CrossRef]
- Guo, H.; LaClair, J.; Masler, E.P.; O’Doherty, G.A.; Xing, Y. De novo asymmetric synthesis and biological analysis of the daumone pheromones in Caenorhabditis elegans and in the soybean cyst nematode Heterodera glycines. Tetrahedron 2016, 72, 2280. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.R.; Jithender, E.; Prasad, K.R. Total syntheses of the proposed structure for ieodoglucomides A and B. J. Org. Chem. 2013, 78, 4251. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Bartlett, M.J. Transition-Metal-Catalyzed Synthesis of Aspergillide B: An Alkyne Addition Strategy. Org. Lett. 2012, 14, 1322–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgy, M.; Lesot, P.; Campagne, J.-M. Synthetic Studies on Macrolactin A: Construction of C4−C24 Fragment. J. Org. Chem. 2007, 72, 3543–3549. [Google Scholar] [CrossRef]
- Das, S.; Kuilya, T.K.; Goswami, R.K. Asymmetric Total Synthesis of Bioactive Natural Lipid Mycalol. J. Org. Chem. 2015, 80, 6467–6489. [Google Scholar] [CrossRef]
- Nageswara Rao, K.; Kumar, K.; Ghosh, S. Total Synthesis of the Anticancer Marine Natural Product Mycalol. Eur. J. Org. Chem. 2018, 2018, 398–412. [Google Scholar] [CrossRef]
- Trost, B.M.; Horne, D.B.; Woltering, M.J. Palladium-Catalyzed DYKAT of Vinyl Epoxides: Enantioselective Total Synthesis and Assignment of the Configuration of (+)-Broussonetine G. Angew. Chem. Int. Ed. 2003, 42, 5987. [Google Scholar] [CrossRef]
- Trost, B.M.; Horne, D.B.; Woltering, M.J. Palladium-Catalyzed DYKAT of Butadiene Monoepoxide: Enantioselective Total Synthesis of (+)-DMDP, (−)-Bulgecinine, and (+)-Broussonetine G. Chem. A Eur. J. 2006, 12, 6607–6620. [Google Scholar] [CrossRef]
- Seetharamsingh, B.; Rajamohanan, P.R.; Reddy, D.S. Total Synthesis and Structural Revision of Mycalol, an Anticancer Natural Product from the Marine Source. Org. Lett. 2015, 17, 1652–1655. [Google Scholar] [CrossRef]
- Oliveira, J.M.; Freitas, J.C.R.; Comasseto, J.V.; Menezes, P.H. Synthesis of substituted α,β-unsaturated δ-lactones from vinyl tellurides. Tetrahedron 2011, 67, 3003–3009. [Google Scholar] [CrossRef]
- Svenningsen, E.B.; Ottosen, R.N.; Jørgensen, K.H.; Nisavic, M.; Larsen, C.K.; Hansen, B.K.; Wang, Y.; Lindorff-Larsen, K.; Tørring, T.; Hacker, S.M.; et al. The covalent reactivity of functionalized 5-hydroxy-butyrolactams is the basis for targeting of fatty acid binding protein 5 (FABP5) by the neurotrophic agent MT-21. RSC Chem. Biol. 2022, 3, 1216–1229. [Google Scholar] [CrossRef]
- Yoo, E.; Schulze, C.J.; Stokes, B.H.; Onguka, O.; Yeo, T.; Mok, S.; Gnädig, N.F.; Zhou, Y.; Kurita, K.; Foe, I.T.; et al. The Antimalarial Natural Product Salinipostin A Identifies Essential α/β Serine Hydrolases Involved in Lipid Metabolism in P. falciparum Parasites. Cell Chem. Biol. 2020, 27, 143–157.e145. [Google Scholar] [CrossRef] [PubMed]
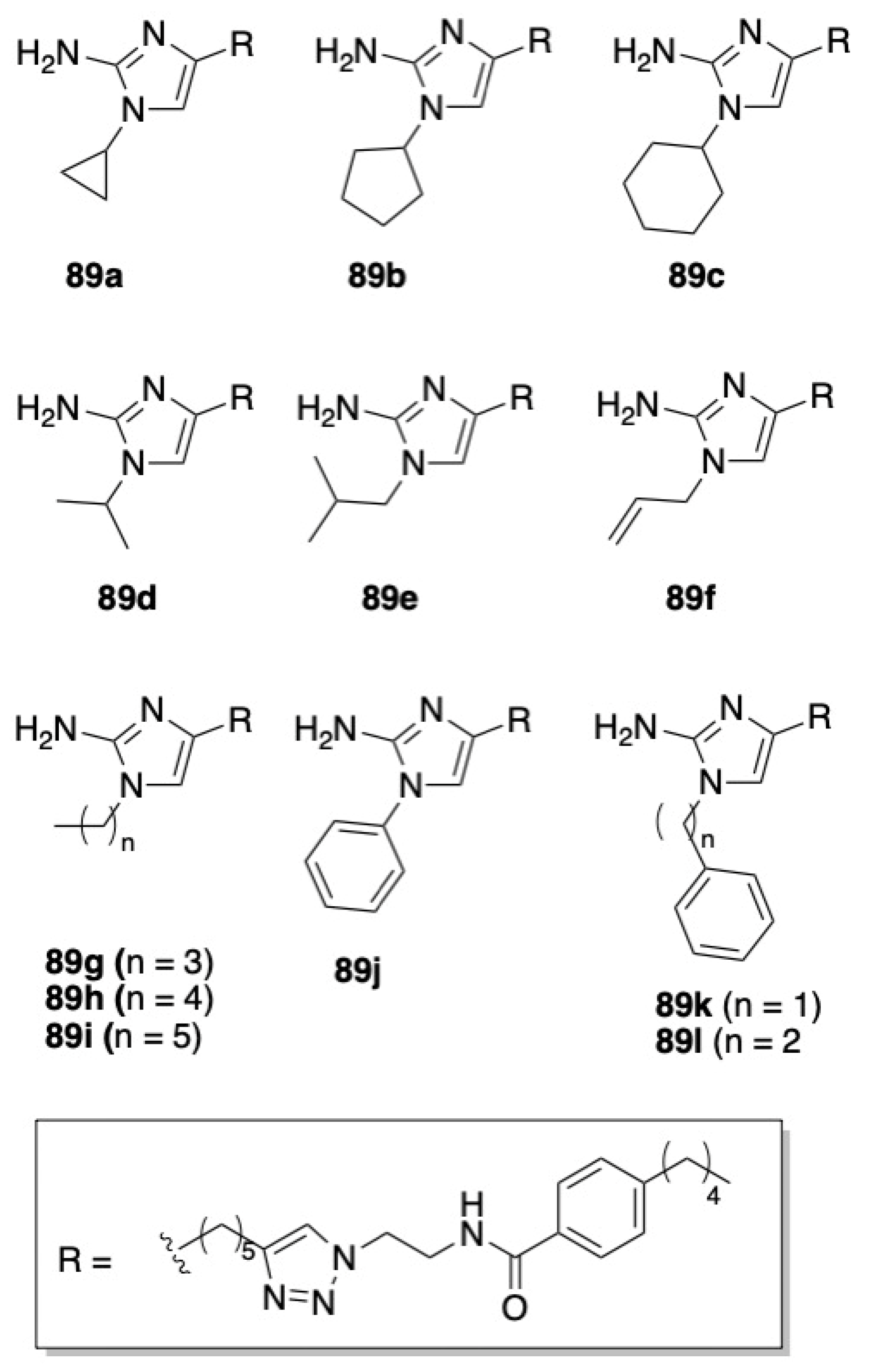
- Furlani, R.E.; Yeagley, A.A.; Melander, C. A flexible approach to 1,4-di-substituted 2-aminoimidazoles that inhibit and disperse biofilms and potentiate the effects of β-lactams against multi-drug resistant bacteria. Eur. J. Med. Chem. 2013, 62, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yeagley, A.A.; Su, R.; Peng, L.; Melander, C. Structural Studies on 4,5-Disubstituted 2-Aminoimidazole-Based Biofilm Modulators that Suppress Bacterial Resistance to β-Lactams. ChemMedChem 2012, 7, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Kulyashova, A.E.; Sorokoumov, V.N.; Popik, V.V.; Balova, I.A. An acetylene zipper—Sonogashira reaction sequence for the efficient synthesis of conjugated arylalkadiynols. Tetrahedron Lett. 2013, 54, 2235–2238. [Google Scholar] [CrossRef]
- Balova, I.A.; Morozkina, S.N.; Knight, D.W.; Vasilevsky, S.F. A one-pot synthesis of 1-arylalka-1,3-diynes by sequential acetylene zipper and Sonogashira reactions. Tetrahedron Lett. 2003, 44, 107–109. [Google Scholar] [CrossRef]
- Govdi, A.I.; Kulyashova, A.E.; Vasilevsky, S.F.; Balova, I.A. Functionalized buta-1,3-diynyl-N-methylpyrazoles by sequential “diacetylene zipper” and Sonogashira coupling reactions. Tetrahedron Lett. 2017, 58, 762–765. [Google Scholar] [CrossRef]
- Balova, I.A.; Morozkina, S.N.; Sorokoumov, V.N.; Vinogradova, O.V.; Vasilevskii, S.F. “Acetylene Zipper” Reactions and Pd-Cu-Catalyzed Cross-coupling in the Synthesis of Vicinal 1,3-Alkadiynylarylamines and Aminopyridines. Russ. J. Org. Chem. 2003, 39, 1613–1617. [Google Scholar] [CrossRef]
- Balova, I.A.; Sorokoumov, V.N.; Morozkina, S.N.; Vinogradova, O.V.; Knight, D.W.; Vasilevsky, S.F. A Convenient Synthesis of Functionalised 1-Aryl-1,3-alkadiynes. Eur. J. Org. Chem. 2005, 2005, 882–888. [Google Scholar] [CrossRef]
- Jõgi, A.; Mäeorg, U. Synthesis and isomerization of enyne-group containing compounds in NaEDA/EDA media. ARKIVOC 2001, 3, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Mäeorg, U.; Talu, L.; Kallas, K. Isomerization and Following Alkylation of (Z, E)-2-octen-4-yn-1-ol as a One Pot Procedure in Super Basic Media; Estonian Academy of Sciences Chemistry: Tallinn, Estonia, 1996; pp. 140–145. [Google Scholar]
- King, S.B.; Kobilka, B.M.; Kuczynski, J.; Wertz, J.T. Polyurethane Materials Formed From Unsaturated Plant Oils via an Alkyne Zipper Reaction. US Patent 9,732,181, 15 August 2017. [Google Scholar]
- Yu, W.; Du, M.; Zhang, D.; Lin, Y.; Zheng, Q. Influence of Dangling Chains on Molecular Dynamics of Polyurethanes. Macromolecules 2013, 46, 7341–7351. [Google Scholar] [CrossRef]
- Chao-Jun Li, B.M.T. (Ed.) Introduction. In Modern Alkyne Chemistry; Wiley and Sons: Hoboken, NJ, USA, 2014; pp. 1–8. [Google Scholar]
- Brown, C.A.; Negishi, E.-I. Novel synthesis of an ω-alkynylorganometallic reagent via triple bond isomerization with potassium 3-aminopropylamide: ω-(9-borabicyclo[3.3.1]nonan-9-yl)alkl-1-ynes. J. Chem. Soc. Chem. Commun. 1977, 9, 318–319. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef]
- Parker, C.G.; Pratt, M.R. Click chemistry in proteomic investigations. Cell 2020, 180, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Bratlie, K.M. Click chemistry and material selection for in situ fabrication of hydrogels in tissue engineering applications. ACS Biomater. Sci. Eng. 2018, 4, 2276–2291. [Google Scholar] [CrossRef] [PubMed]
- Arslan, M.; Acik, G.; Tasdelen, M.A. The emerging applications of click chemistry reactions in the modification of industrial polymers. Polym. Chem. 2019, 10, 3806–3821. [Google Scholar] [CrossRef]
- Badria, A. Click Chemistry: A Promising Tool for Building Hierarchical Structures. Polymers 2022, 14, 4077. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
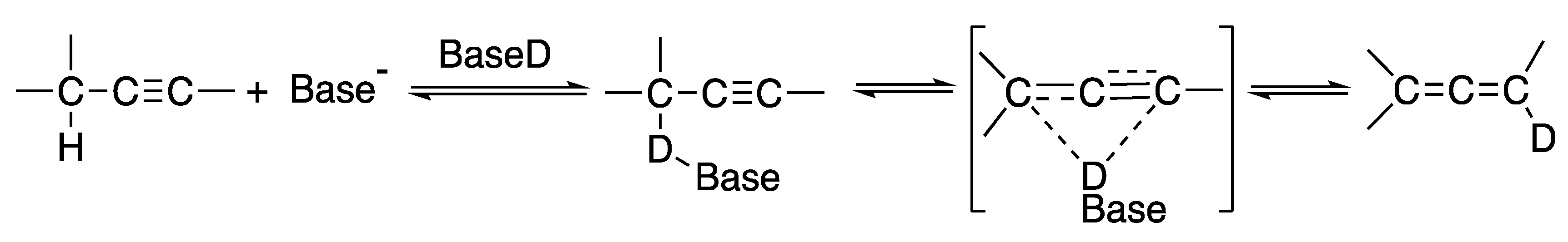{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
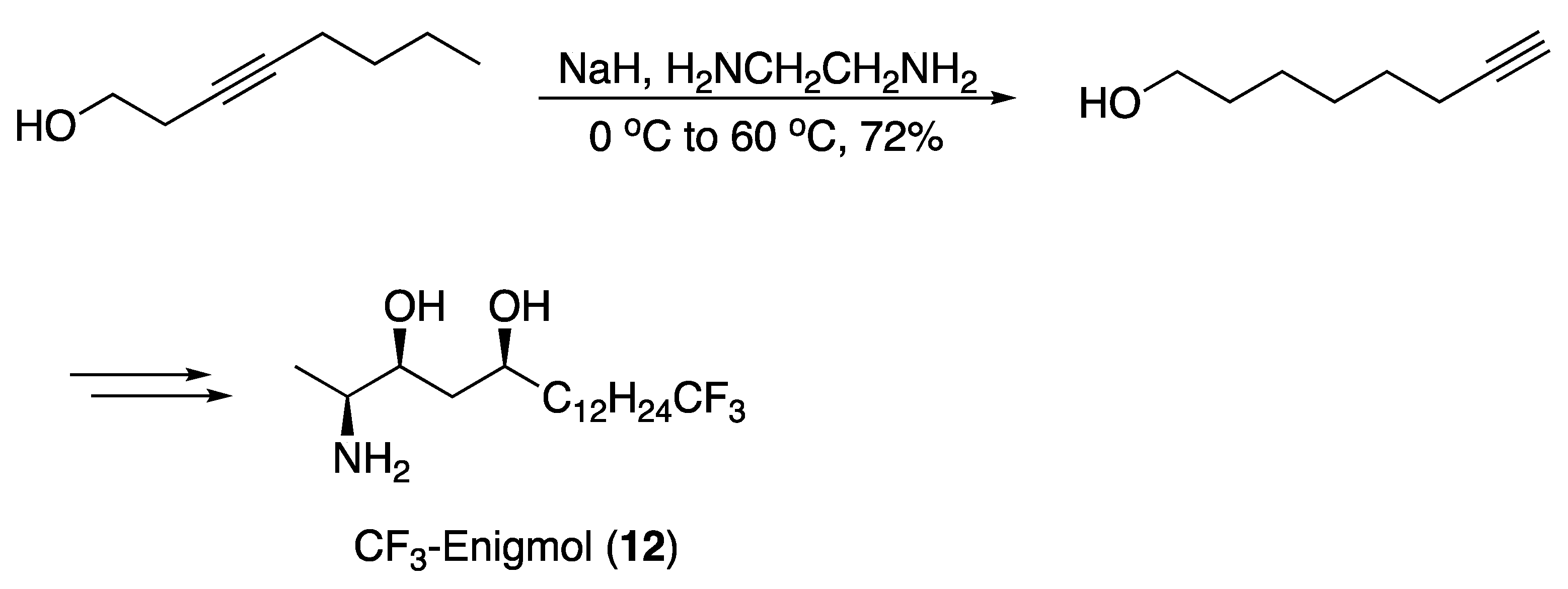{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


















































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sørskår, Å.M.; Stenstrøm, H.Ø.K.; Stenstrøm, Y.; Antonsen, S.G. The Alkyne Zipper Reaction: A Useful Tool in Synthetic Chemistry. Reactions 2023, 4, 26-65. https://doi.org/10.3390/reactions4010002
Sørskår ÅM, Stenstrøm HØK, Stenstrøm Y, Antonsen SG. The Alkyne Zipper Reaction: A Useful Tool in Synthetic Chemistry. Reactions. 2023; 4(1):26-65. https://doi.org/10.3390/reactions4010002
Chicago/Turabian StyleSørskår, Åshild Moi, Helge Ø. K. Stenstrøm, Yngve Stenstrøm, and Simen Gjelseth Antonsen. 2023. "The Alkyne Zipper Reaction: A Useful Tool in Synthetic Chemistry" Reactions 4, no. 1: 26-65. https://doi.org/10.3390/reactions4010002
APA StyleSørskår, Å. M., Stenstrøm, H. Ø. K., Stenstrøm, Y., & Antonsen, S. G. (2023). The Alkyne Zipper Reaction: A Useful Tool in Synthetic Chemistry. Reactions, 4(1), 26-65. https://doi.org/10.3390/reactions4010002