
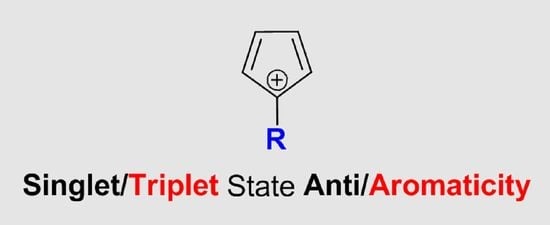
Singlet/Triplet State Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect

Abstract
:
1. Introduction
2. Computational Details and Methods
3. Results
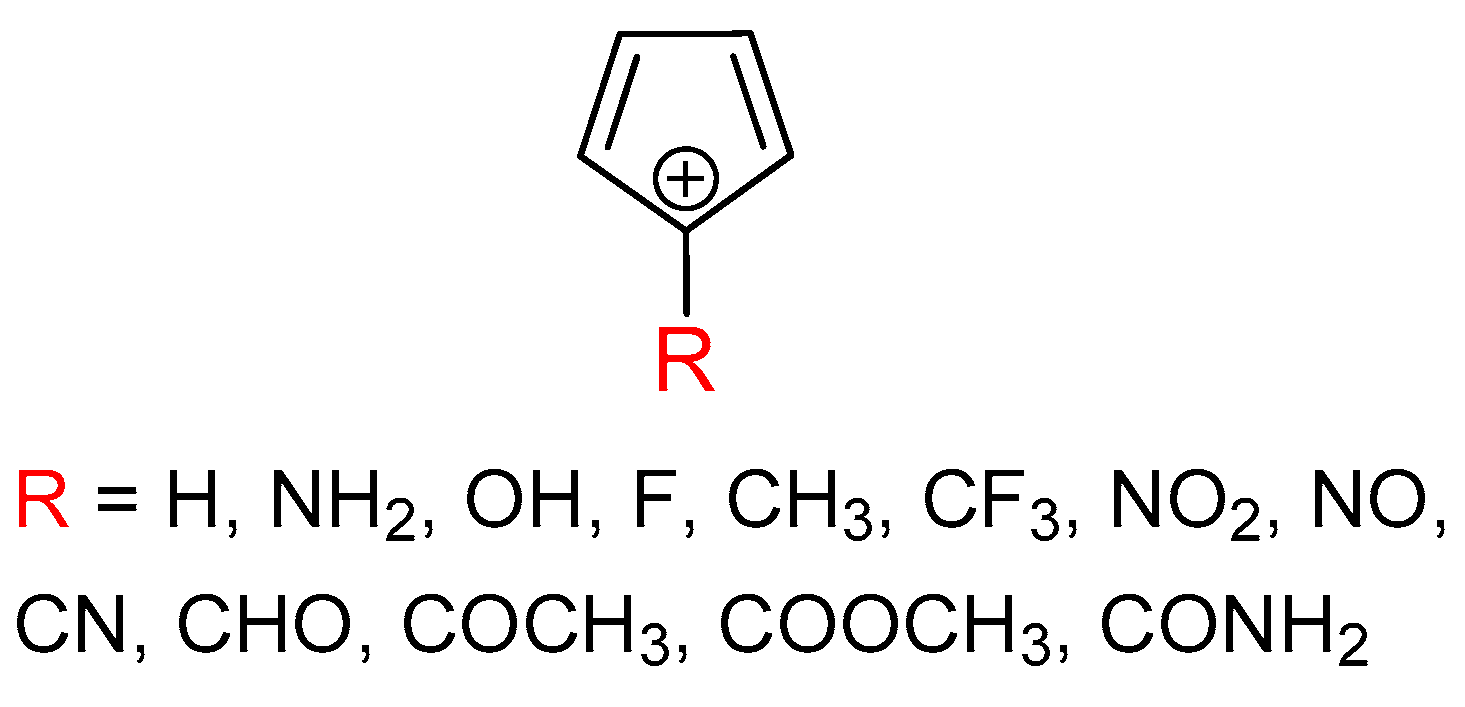
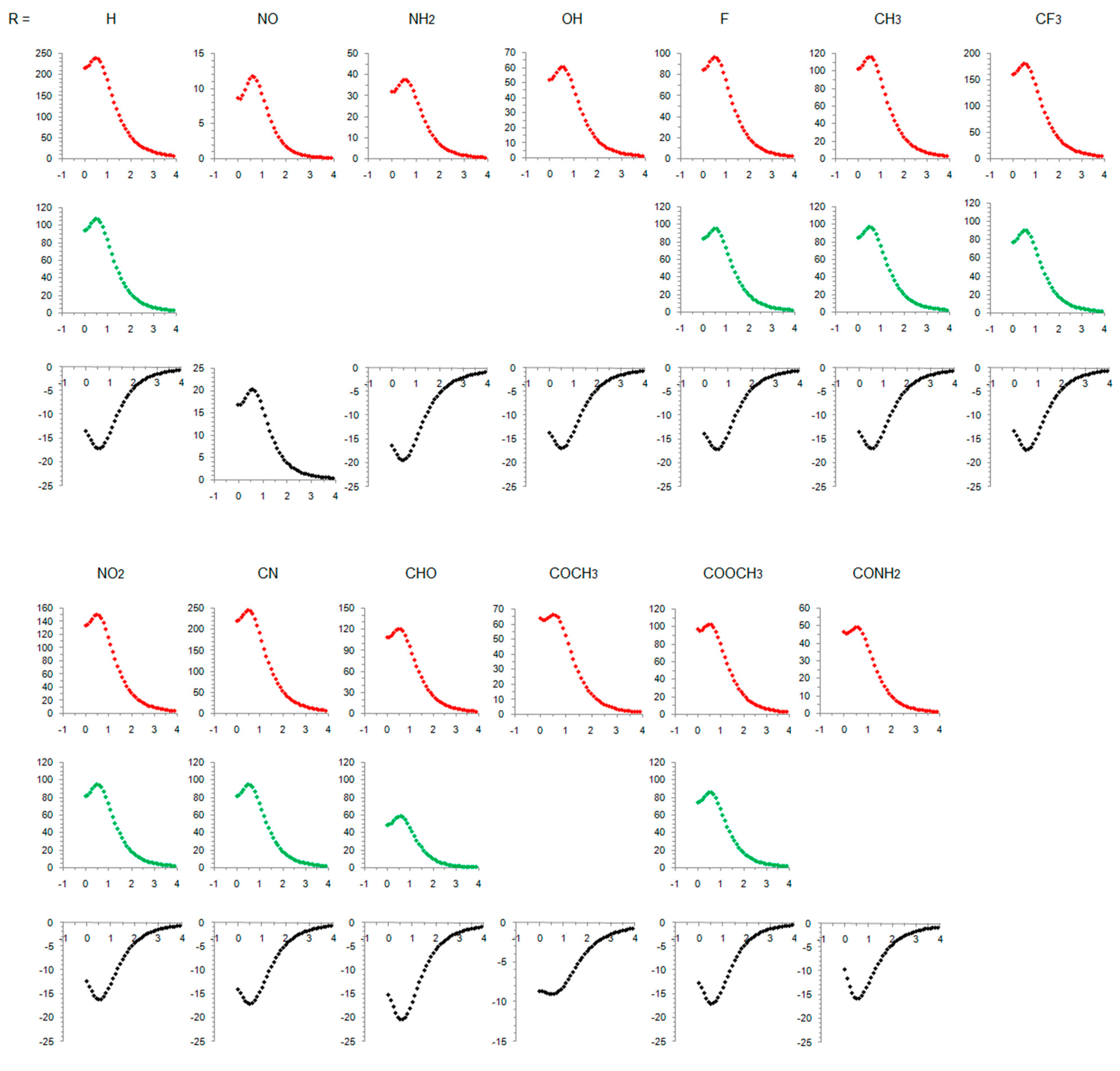
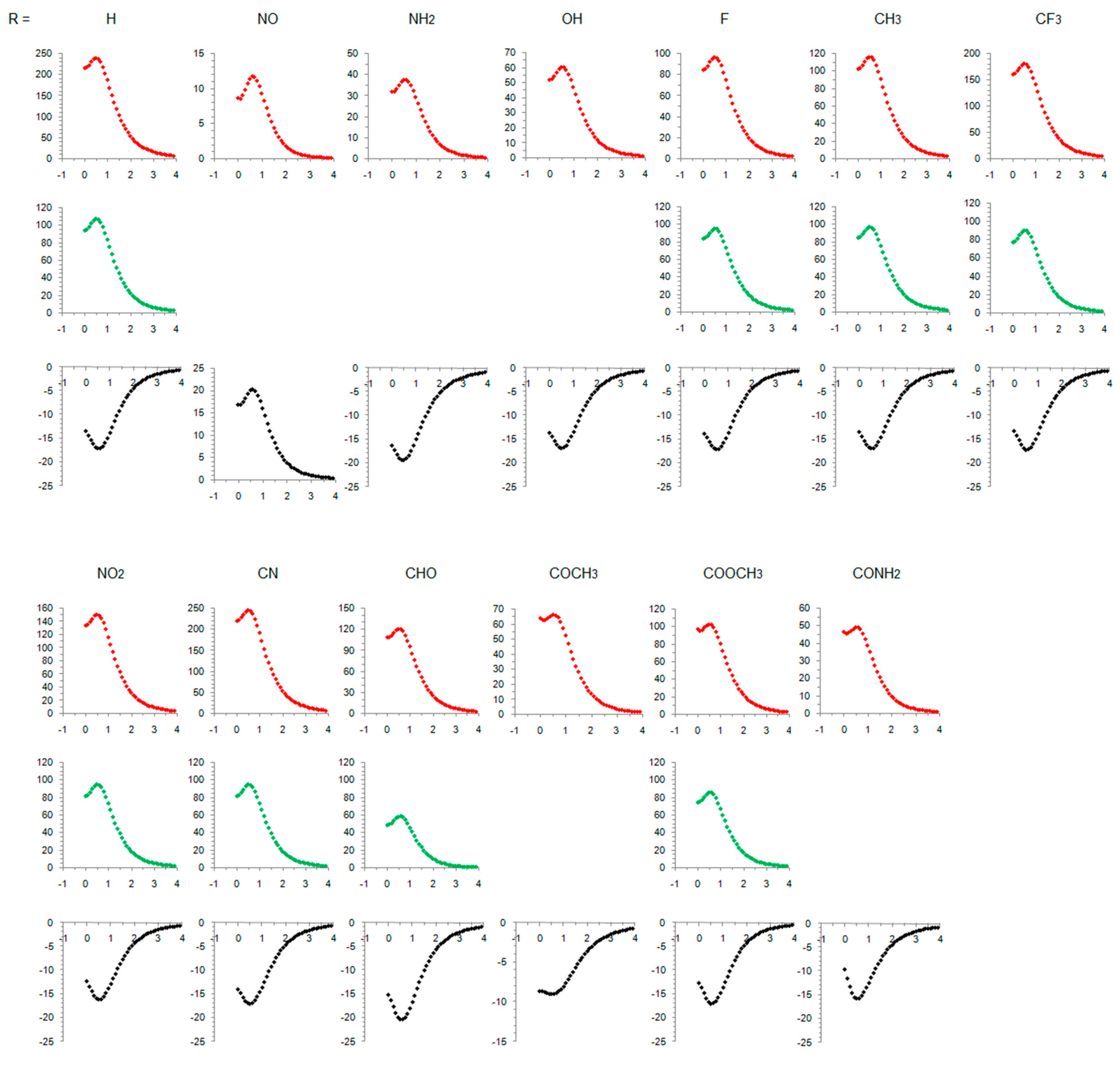
3.1. Cyclopentadienyl Cation
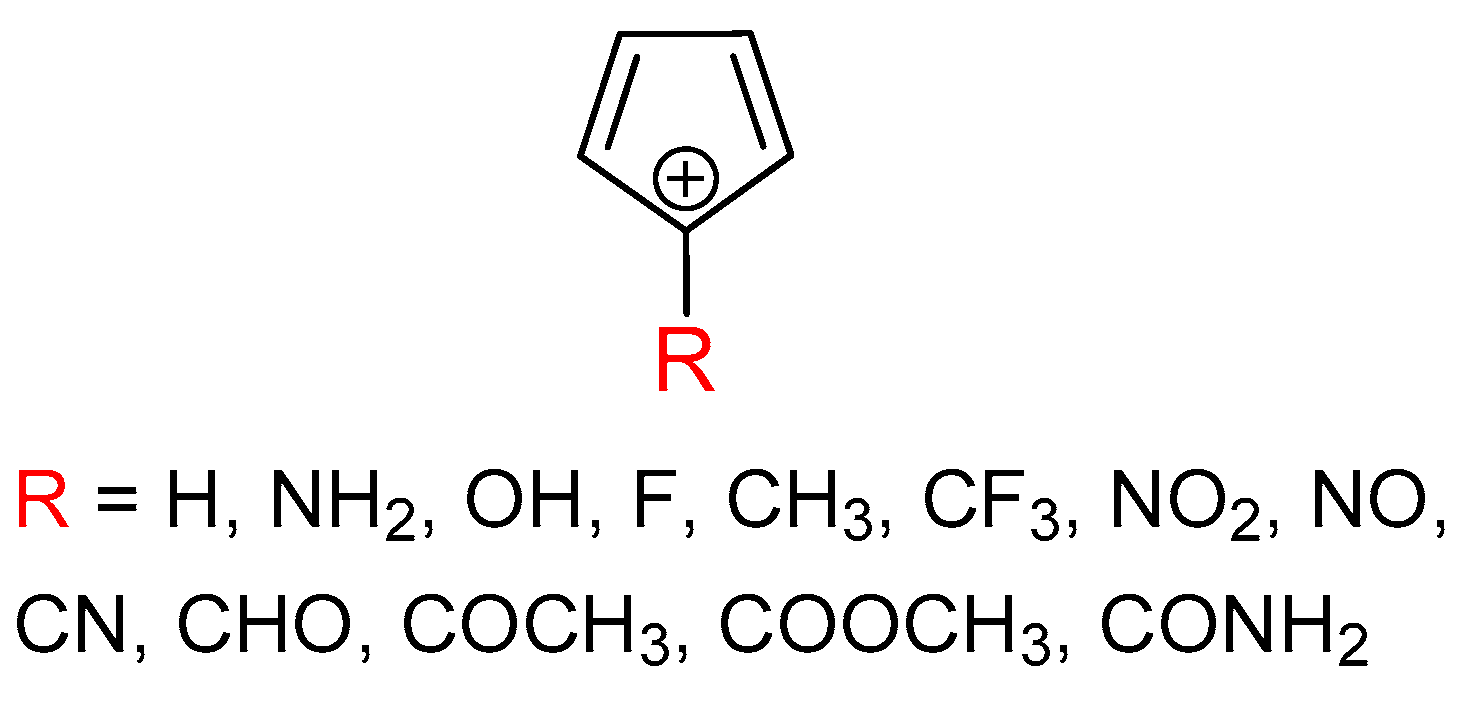
3.2. Singlet State of Substituted Cyclopentadienyl Cations
3.3. Triplet State of Substituted Cyclopentadienyl Cations
3.4. Comparison of the Results with the CAM-B3LYP Calculations
4. Discussion
5. Conclusions
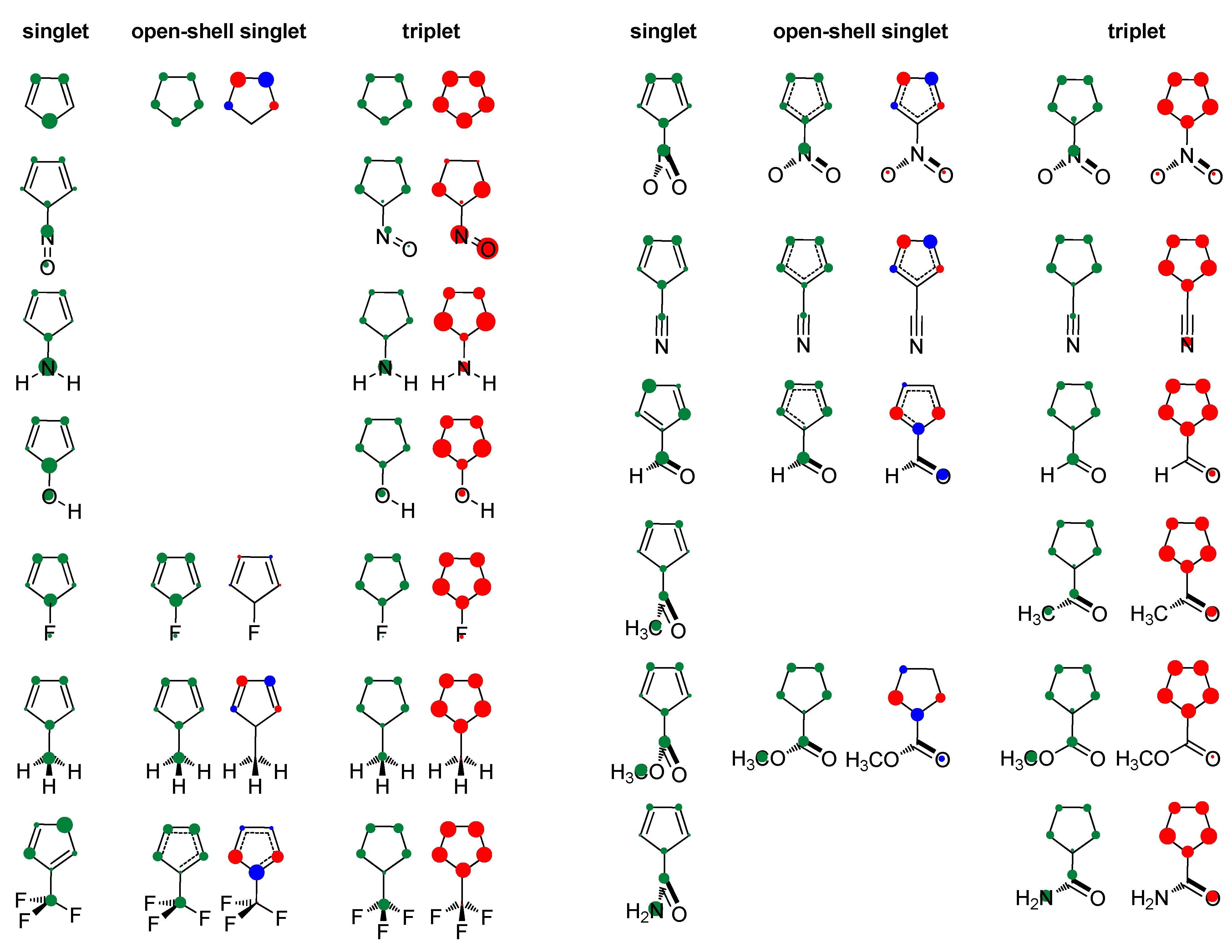
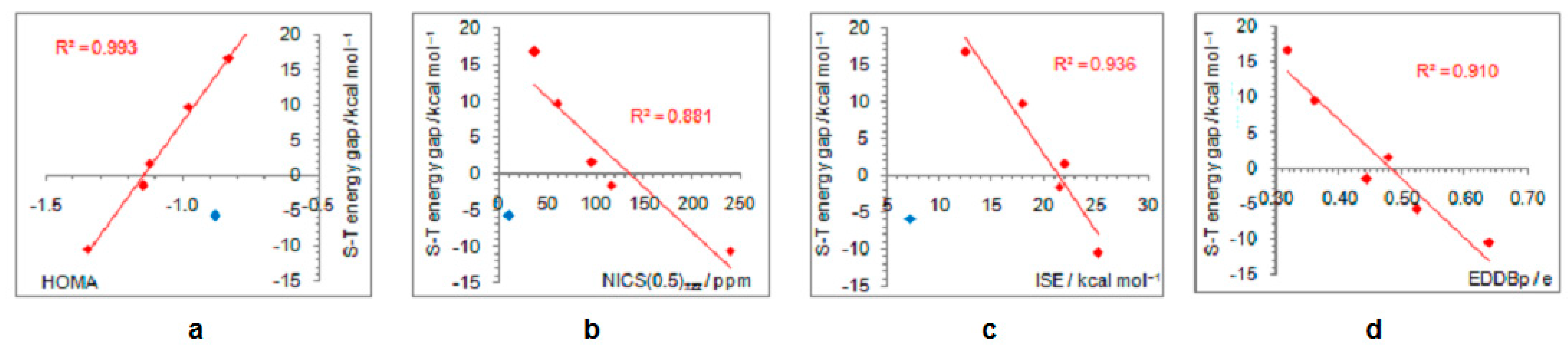
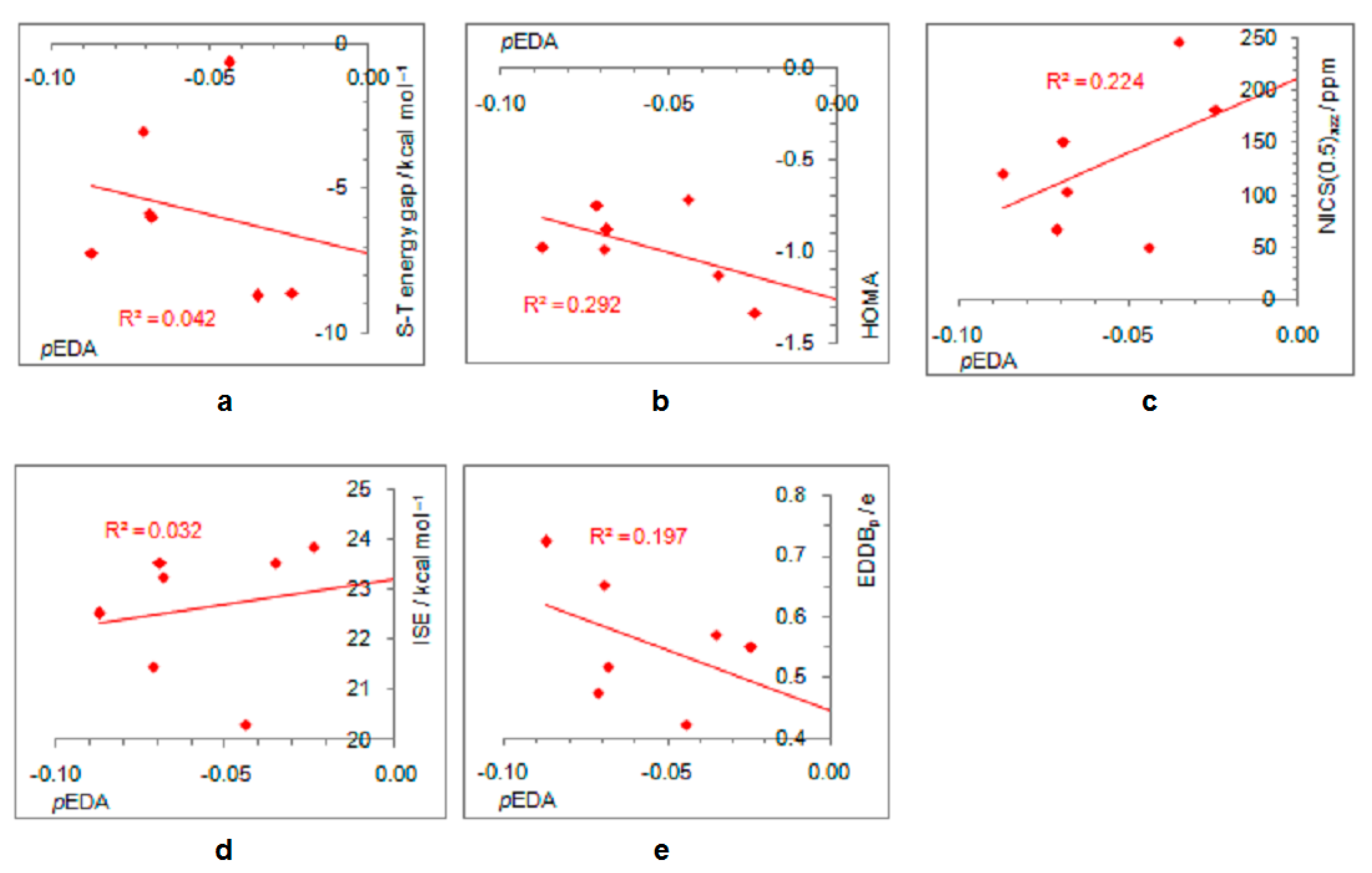
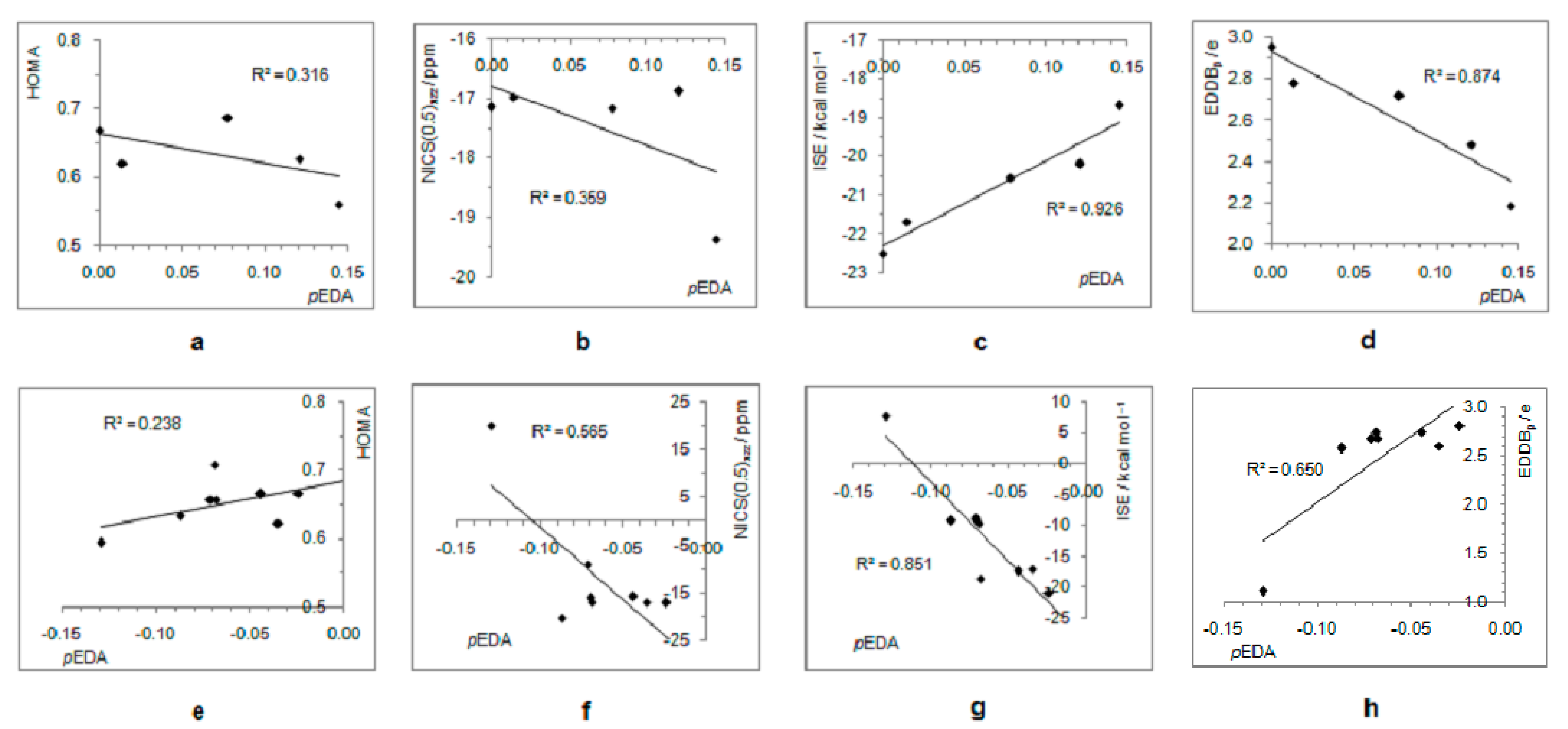
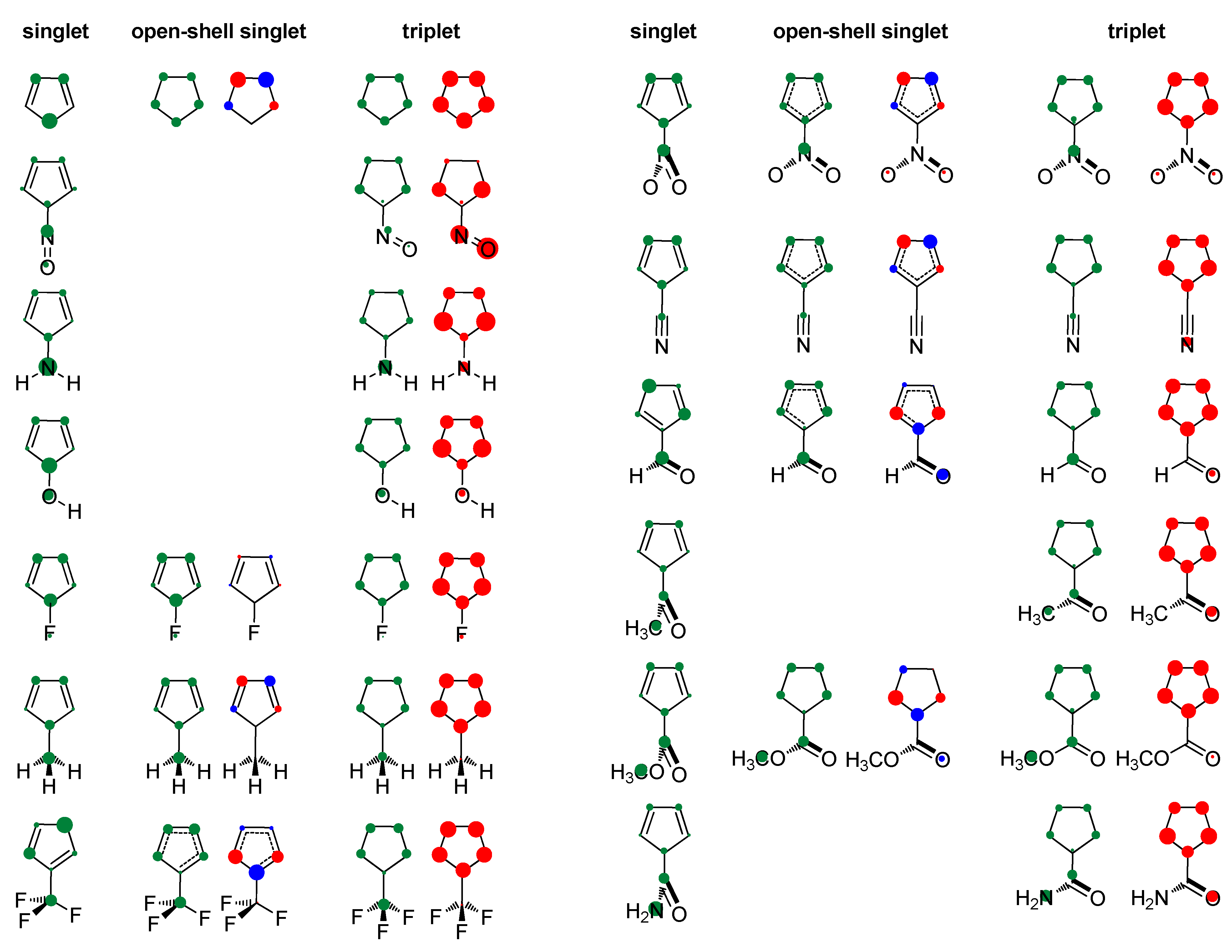
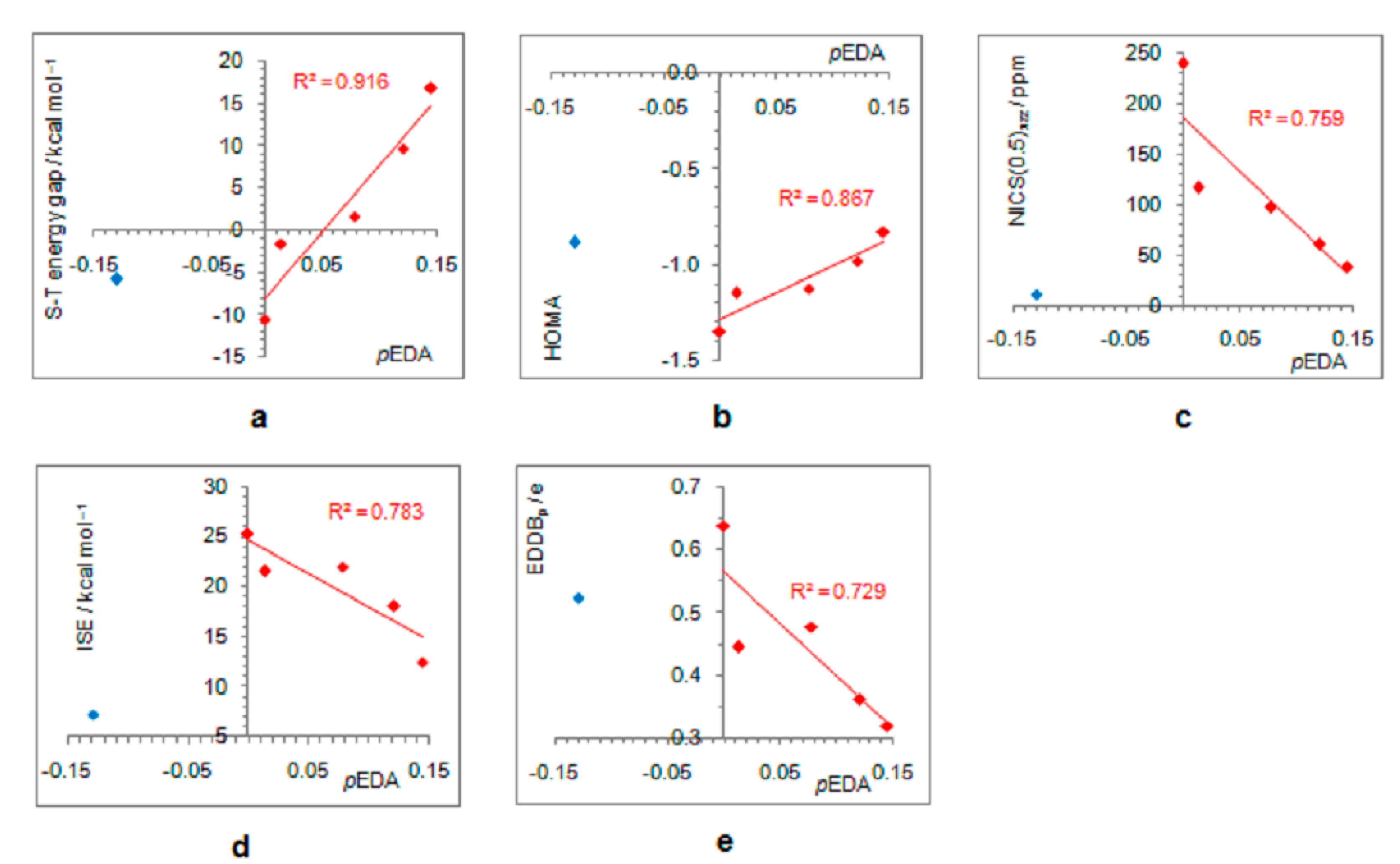
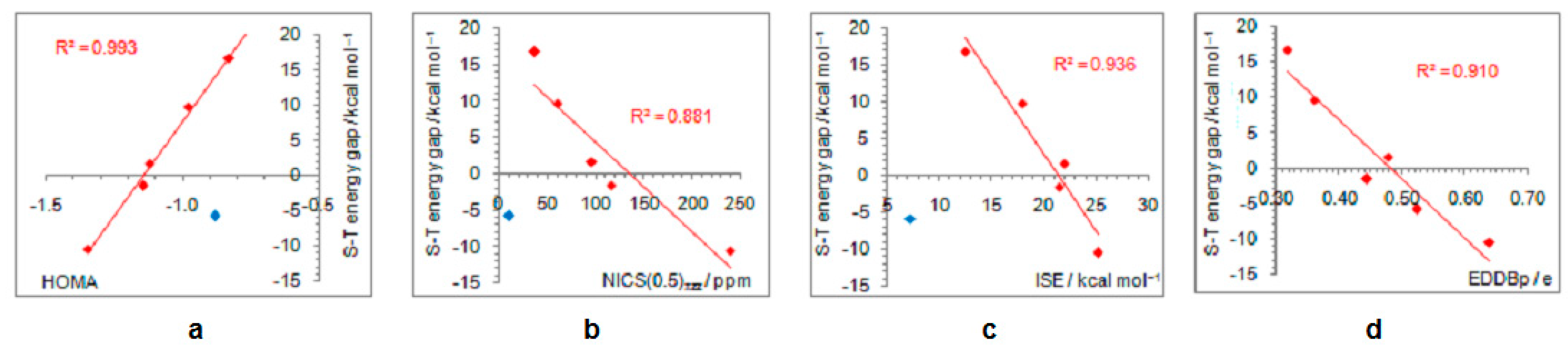
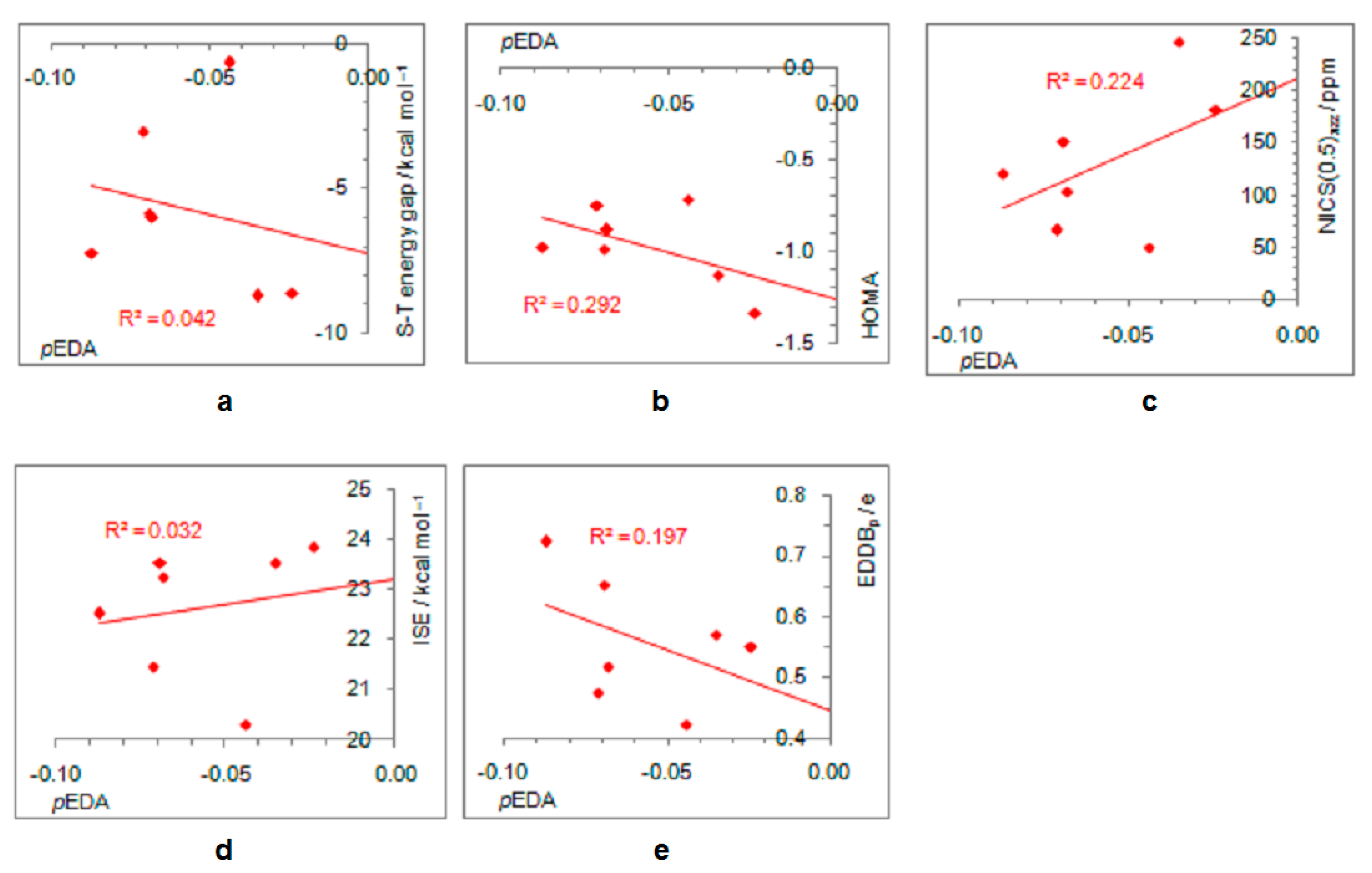
- In its closed-shell singlet state, the positively charged and antiaromatic cyclopentadienyl cation strongly interacts with electron-donating substituents, such as NH2, OH, F, CH3, and NO, but does not interact with electron-withdrawing groups, which orient themselves in such a position to diminish or fully avoid interaction with the π system of the ring. As a consequence, the extent of antiaromaticity nicely correlates with the π–electron substituent effect, represented by the pEDA values, for electron-donors but not for electron acceptors. Although the NO group is basically an electron-withdrawing substituent, it strongly donates its lone pair to the cyclopentadienyl ring, diminishing its antiaromaticity to the largest extent.
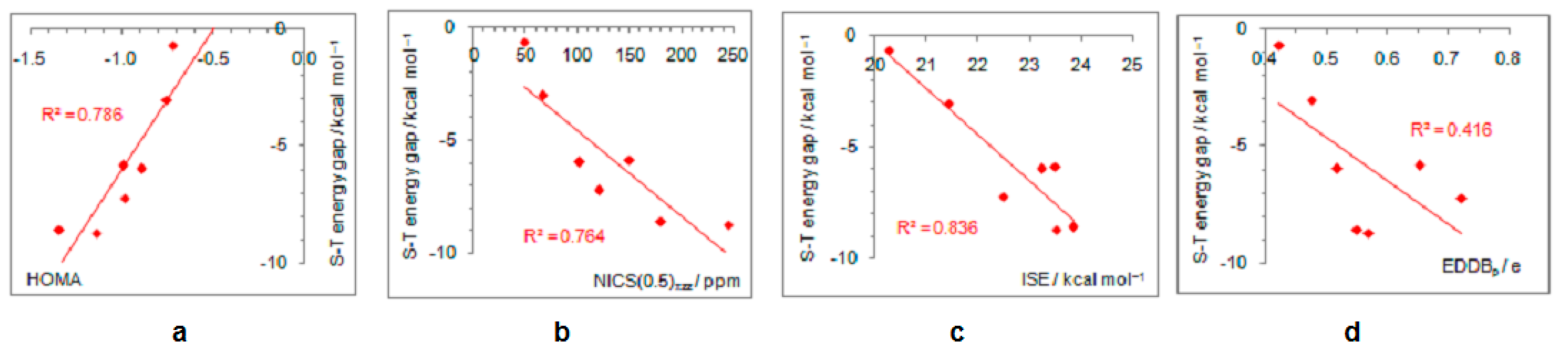
- The singlet–triplet energy gaps, favoring triplet in the parent molecule, show a good correlation with the extent of antiaromaticity. They increase with a decrease in antiaromaticity and favor singlet state for strong donors, such as NH2 and OH.
- The formation of an open-shell singlet state has little effect on energy. The slight change in MO extensions and drop in EDDBp values indicates that the decrease in the extent of antiaromaticity, as evidenced from the decrease in NICS values, should have its source in the weaker density of delocalized electrons.
- The triplet state is characterized as aromatic in all but one case (R = NO), which conforms to Baird’s rule. Increased electron delocalization, that is, aromaticity, decreases substituent–ring electronic interactions. Thus, in the absence of steric effects, all substituents adopt a conformation that allows their interaction with the π system of the ring, but it is weak so that the triplet state aromaticity of this chosen model compound is just weakly sensitive to substituent effects. This can be explained by the fact that this triplet state aromaticity is not as large as that of singlet benzene, the aromaticity of which almost resists substituent effects.
- The spin density distribution determines whether the cyclopentadienyl ring is triplet state aromatic or antiaromatic: if it is accumulated in the ring, the system is aromatic, but if it is accumulated at the substituent, the system is antiaromatic. The latter was found for the NO group only so that 5-nitrosocyclopentadienyl cation, according to our calculations, is antiaromatic in both singlet and triplet states. Similarly, our previous calculations indicated that nitrosobenzene would be aromatic in both singlet and triplet states [44]. This is reminiscent of adaptive aromaticity [31,32,33], which here, was enabled by the introduction of a substituent.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hückel, E. Quantum Theoretical Contributions to the Benzene Problem. Z. Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Hückel, E. Broad Theory of Unsaturated and Aromatic Compounds; Verlag Chemie: Berlin, Germany, 1938; pp. 77–85. [Google Scholar]
- Von Eggers Doering, W.; Detert, F.L. Cycloheptatrienylium Oxide. J. Am. Chem. Soc. 1951, 73, 876–877. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, the Gold Book; IUPAC: Zurich, Switzerland, 2014; Available online: https://goldbook.iupac.org/files/pdf/goldbook.pdf (accessed on 30 January 2020).
- Breslow, R.; Brown, J.; Gajewski, J.J. Antiaromaticity of CyclopropenylAnions. J. Am. Chem. Soc. 1967, 89, 4383–4390. [Google Scholar] [CrossRef]
- Breslow, R. Antiaromaticity. Acc. Chem. Res. 1973, 6, 393–398. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. Quantifying Aromaticity with Electron Delocalization Measures. Chem. Soc. Rev. 2015, 44, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the Viewpoint of Molecular Geometry: Application to Planar Systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef] [PubMed]
- Cyrański, M.K. Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef]
- Mo, Y.; von Ragué Schleyer, P. An Energetic Measure of Aromaticity and Antiaromaticity Based on the Pauling–Wheland Resonance Energies. Chem. Eur. J. 2006, 12, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Gershoni-Poranne, R.; Stanger, A. Magnetic Criteria of Aromaticity. Chem. Soc. Rev. 2015, 44, 6597–6615. [Google Scholar] [CrossRef]
- Baird, C. Quantum Organic Photochemistry. II. Resonance and Aromaticity in the Lowest 3ππ* State of Cyclic Hydrocarbons. J. Am. Chem. Soc. 1972, 94, 4941–4948. [Google Scholar] [CrossRef]
- Karadakov, P. Ground- and Excited-State Aromaticity and Antiaromaticity in Benzene and Cyclobutadiene. J. Phys. Chem. A 2008, 112, 7303–7309. [Google Scholar] [CrossRef]
- Villaume, S.; Fogarty, H.A.; Ottosson, H. Triplet-State Aromaticity of 4nπ-Electron Monocycles: Analysis of Bifurcation in the π Contribution to the Electron Localization Function. Chem. Phys. Chem. 2008, 9, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Matito, E.; Solà, M.; Poater, J. Patterns of π-Electron Delocalization in Aromatic and Antiaromatic Organic Compounds in the Light of Hückel’s 4n + 2 rule. Phys. Chem. Chem. Phys. 2010, 12, 7126–7137. [Google Scholar] [CrossRef] [Green Version]
- Feixas, F.; Vandenbussche, J.; Bultinck, P.; Matito, E.; Solà, M. Electron Delocalization and Aromaticity in Low-Lying Excited States of Archetypal Organic Compounds. Phys. Chem. Chem. Phys. 2011, 13, 20690–20703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogonea, V.; von Ragué Schleyer, P.; Schreiner, P.R. Consequences of Triplet Aromaticity in 4nπ-Electron Annulenes: Calculation of Magnetic Shieldings for Open-Shell Species. Angew. Chem. Int. Ed. 1998, 37, 1945–1948. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyrański, M.K. Two Sources of the Decrease of Aromaticity: Bond Length Alternation and Bond Elongation. Part II. An Analysis Based on Geometry of the Singlet and Triplet States of 4nπ Annulenes: C4H4, C5H5 +, C6H62+, C7H7−, C8H8, C9H9+. Tetrahedron 1999, 55, 11143–11148. [Google Scholar] [CrossRef]
- Zhu, J.; An, K.; von Ragué Schleyer, P. Evaluation of Triplet Aromaticity by the Isomerization Stabilization Energy. Org. Lett. 2013, 15, 2442–2445. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Zhu, J. Evaluation of Triplet Aromaticity by the Indene–Isoindene Isomerization Stabilization Energy Method. Eur. J. Org. Chem. 2014, 2764–2769. [Google Scholar] [CrossRef]
- Baranac-Stojanović, M. Triplet-State Structures, Energies, and Antiaromaticity of BN Analogues of Benzene and Their Benzo-Fused Derivatives. J. Org. Chem. 2019, 84, 13582–13594. [Google Scholar] [CrossRef]
- Fowler, P.W.; Steiner, E.; Jenneskens, L.W. Ring-Current Aromaticity in Triplet States of 4nπ Electron Monocycles. Chem. Phys. Lett. 2003, 371, 719–723. [Google Scholar] [CrossRef] [Green Version]
- Karadakov, P.B. Aromaticity and Antiaromaticity in the Low-Lying Electronic States of Cyclooctatetraene. J. Phys. Chem. A 2008, 112, 12707–12713. [Google Scholar] [CrossRef] [PubMed]
- Karadakov, P.B.; Hearnshaw, P.; Horner, K.E. Magnetic Shielding, Aromaticity, Antiaromaticity, and Bonding in the Low-Lying Electronic States of Benzene and Cyclobutadiene. J. Org. Chem. 2016, 81, 11346–11352. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; An, K.; Zhu, J. Triplet State Aromaticity: NICS Criterion, Hyperconjugation, and Charge Effects. Chem. Asian J. 2016, 11, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, R.; Li, H.; Bergman, J.; Lundstedt, A.; Jorner, K.; Ayub, R.; Haldar, S.; Jahn, B.O.; Denisova, A.; Zietz, B.; et al. Metal-Free Photochemical Silylations and Transfer Hydrogenations of Benzenoid Hydrocarbons and Graphene. Nat. Commun. 2016, 7, 12962. [Google Scholar] [CrossRef]
- Ueda, M.; Jorner, K.; Sung, Y.M.; Mori, T.; Xiao, Q.; Kim, D.; Ottosson, H.; Aida, T.; Itoh, Y. Energetics of Baird Aromaticity Supported by Inversion of Photoexcited Chiral [4n] Annulene Derivatives. Nat. Commun. 2017, 8, 346. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Sung, Y.M.; Hong, Y.; Kim, D. Spectroscopic Diagnosis of Excited-State Aromaticity: Capturing Electronic Structures and Conformations upon Aromaticity Reversal. Acc. Chem. Res. 2018, 51, 1349–1358. [Google Scholar] [CrossRef]
- Rosenberg, M.; Dahlstrand, C.; Kilså, K.; Ottosson, H. Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations. Chem. Rev. 2014, 114, 5379–5425. [Google Scholar] [CrossRef]
- Papadakis, R.; Ottosson, H. The Excited State Antiaromatic Benzene Ring: A Molecular Mr Hyde? Chem. Soc. Rev. 2015, 44, 6472–6493. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Chen, D.; Lin, L.; Zhu, J. Dual Aromaticity in Both the T0 and S1 States: Osmapyridinium with Phosphonium Substituents. J. Am. Chem. Soc. 2019, 141, 5720–5727. [Google Scholar] [CrossRef]
- Dai, C.; Chen, D.; Zhu, J. Achieving Adaptive Aromaticity in Cyclo [10] carbon by Screening Cyclo [n] carbon (n = 8–24). Chem. Asian J. 2020, 15, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, D.; Zhu, J. Adaptive Aromaticity in Metallasilapentalynes. Organometallics 2021, 40, 899–906. [Google Scholar] [CrossRef]
- Siodla, T.; Szatylowicz, H.; Varaksin, K.S.; Krygowski, T.M. Difference in pi-electron delocalization for monosubstituted olefinic and aromatic systems. RSC Adv. 2016, 6, 96527–96530. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Ejsmont, K.; Stepień, B.T.; Cyrański, M.K.; Poater, J.; Solà, M. Relation between the Substituent Effect and Aromaticity. J. Org. Chem. 2004, 69, 6634–6640. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Stępień, B.T. Sigma- and Pi-Electron Delocalization: Focus on Substituent Effects. Chem. Rev. 2005, 105, 3482–3512. [Google Scholar] [CrossRef] [PubMed]
- Omelchenko, I.V.; Shishkin, O.V.; Gorb, L.; Hill, F.C.; Leszczynksi, J. Properties, aromaticity, and substituents effects in poly nitro- and amino-substituted benzenes. Struct. Chem. 2012, 23, 1585–1597. [Google Scholar] [CrossRef]
- Palusiak, M.; Krygowski, T.M. Substituent Effects in Mono- and Disubstituted 1,3,5,7-Cyclooctatetraene Derivatives in Natural and Planar Conformations. New J. Chem. 2009, 33, 1753–1759. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Rozas, I.; Alkorta, I. Influence of Fluoro and Cyano Substituents in the Aromatic and Antiaromatic Characteristics of Cyclooctatetraene. Phys. Chem. Chem. Phys. 2015, 17, 14961–14971. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Halla, J.O.C.; Matito, E.; Solà, M.; Braunschweig, H.; Hörl, C.; Krummenacher, I.; Wahler, J. A Theoretical Study of the Aromaticity in Neutral and Anionic Borole Compounds. Dalton Trans. 2015, 44, 6740–6747. [Google Scholar] [CrossRef] [Green Version]
- Pincock, J.A.; Speed, A.W.H. The Aromatic Character and Resonance Stabilization Energies of Substituted Cyclopentadienyl and Indenyl Cations. Can. J. Chem. 2005, 83, 1287–1298. [Google Scholar] [CrossRef]
- Alonso, M.; Herradón, B. Substituent Effects on the Aromaticity of Carbocyclic Five-Membered Rings. Phys. Chem. Chem. Phys. 2010, 12, 1305–1317. [Google Scholar] [CrossRef]
- Palusiak, M.; Domagała, M.; Dominikowska, J.; Bickelhaupt, F.M. The Substituent Effect on Benzene Dications. Phys. Chem. Chem. Phys. 2014, 16, 4752–4763. [Google Scholar] [CrossRef] [Green Version]
- Baranac-Stojanović, M. Substituent Effect on Triplet State Aromaticity of Benzene. J. Org. Chem. 2020, 85, 4289–4297. [Google Scholar] [CrossRef]
- Breslow, R.; Hoffman, J.M., Jr. Antiaromaticity in the Parent Cyclopentadienyl Cation. Reaction of 5-Iodocyclopentadiene with Silver Ion. J. Am. Chem. Soc. 1972, 94, 2110–2111. [Google Scholar] [CrossRef]
- Saunders, M.; Berger, R.; Jaffe, A.; McBride, J.M.; O’Neill, J.; Breslow, R.; Hoffman, J.N., Jr.; Perchonock, C.; Wasserman, E.; Hutton, R.S.; et al. Unsubstituted Cyclopentadienyl Cation, a Ground State Triplet. J. Am. Chem. Soc. 1973, 95, 3017–3018. [Google Scholar] [CrossRef]
- Breslow, R.; Chang, H.W.; Yager, W.A. A Stable Triplet State of Pentaphenylcyclopentadienyl Cation. J. Am. Chem. Soc. 1963, 85, 2033–2034. [Google Scholar] [CrossRef]
- Breslow, R.; Hill, R.; Wasserman, E. Pentachlorocyclopentadienyl Cation, a Ground State Triplet. J. Am. Chem. Soc. 1964, 86, 5349–5350. [Google Scholar] [CrossRef]
- Sitzmann, H.; Bock, H.; Boese, R.; Dezember, T.; Havlas, Z.; Kaim, W.; Moscherosch, M.; Zanathy, L. Pentaisopropylcyclopentadienyl: Singlet Anion, Doublet Radical, and Triplet Cation of a Carbocyclic π System. J. Am. Chem. Soc. 1993, 115, 12003–12009. [Google Scholar] [CrossRef]
- Costa, P.; Trosien, I.; Mieres-Perez, J.; Sander, W. Isolation of an Antiaromatic Singlet Cyclopentadienyl Zwitterion. J. Am. Chem. Soc. 2017, 139, 13024–13030. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian, Inc.: Pittsburgh, CA, USA, 1996. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 (Revision D.01); Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Takano, Y.; Taniguchi, T.; Isobe, H.; Kubo, T.; Morita, Y.; Yamamoto, K.; Nakasuji, K.; Takui, T.; Yamaguchi, K. Hybrid Density Functional Theory Studies on the Magnetic Interactions and the Weak Covalent Bonding for the Phenalenyl Radical Dimeric Pair. J. Am. Chem. Soc. 2002, 124, 11122–11130. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Saito, T.; Yamaguchi, K. Approximate Spin Projection for Broken-Symmetry Method and Its Application. In Symmetry (Group Theory) and Mathematical Treatment in Chemistry; Akitsu, T., Ed.; IntechOpen: London, UK, 2018; Chapter 7. [Google Scholar] [CrossRef] [Green Version]
- Kruszewski, J.; Krygowski, T.M. Definition of Aromaticity Basing on the Harmonic Oscillator Model. Tetrahedron Lett. 1972, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyrański, M.K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1419. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Von Ragué Schleyer, P.; Puhlhofer, F. Recommendations for the Evaluation of Aromatic Stabilization Energies. Org. Lett. 2002, 4, 2873–2876. [Google Scholar] [CrossRef]
- Von Ragué Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shift: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; von Ragué Schleyer, P. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Von Ragué Schleyer, P.; Jiao, H.; van Eikema Hommes, N.J.R.; Malkin, V.G.; Malkina, O.L. An Evaluation of the Aromaticity of Inorganic Rings: Refined Evidence from Magnetic Properties. J. Am. Chem. Soc. 1997, 119, 12669–12670. [Google Scholar] [CrossRef]
- Von Ragué Schleyer, P.; Manoharan, M.; Wang, Z.-X.; Kiran, B.; Jiao, H.; Puchta, R.; van Eikema Hommes, N.J.R. Dissected Nucleus Independent Chemical Shift Analysis of π-Aromaticity and Antiaromaticity. Org. Lett. 2001, 3, 2465–2468. [Google Scholar] [CrossRef]
- Viglione, R.G.; Zanasi, R.; Lazzeretti, P. Are Ring Currents Still Useful to Rationalize the Benzene Proton Magnetic Shielding? Org. Lett. 2004, 6, 2265–2267. [Google Scholar] [CrossRef]
- Lazzeretti, P. Assessment of Aromaticity via Molecular Response Properties. Phys. Chem. Chem. Phys. 2004, 6, 217–223. [Google Scholar] [CrossRef]
- Faglioni, F.; Ligabue, A.; Pelloni, S.; Soncini, A.; Viglione, R.G.; Ferraro, M.B.; Zanasi, R.; Lazzeretti, P. Why Downfield Proton Chemical Shifts Are Not Reliable Aromaticity Indicators. Org. Lett. 2005, 7, 3457–3460. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Bagher-Shaidaei, H.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; von Ragué Schleyer, P. Which NICS Aromaticity Index for Planar π Rings is Best? Org. Lett. 2006, 8, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Stanger, A. Nucleus-Independent Chemical Shifts (NICS): Distance Dependence and Revised Criteria for Aromaticity and Antiaromaticity. J. Org. Chem. 2006, 71, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Halla, J.O.C.; Matito, E.; Robles, J.; Solà, M. Nucleus-Independent Chemical Shift (NICS) Profiles in a Series of Monocyclic Planar Inorganic Compounds. J. Organomet. Chem. 2006, 691, 4359–4366. [Google Scholar] [CrossRef]
- Stanger, A. Reexamination of NICSπzz: Height Dependence, Off-Center Values, and Integration. J. Phys. Chem. A 2019, 123, 3922–3927. [Google Scholar] [CrossRef]
- Ditchfeld, R. Self-Consistent Perturbation Theory of Diamagnetism. 1. Gauge-Invariant LCAO Method for N.M.R. Chemical Shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Rahalkar, A.; Stanger, A. Aroma. Available online: http://chemistry.technion.ac.il/members/amnon-stanger/ (accessed on 30 January 2020).
- Stanger, A. Obtaining Relative Induced Ring Currents Quantitatively from NICS. J. Org. Chem. 2010, 75, 2281–2288. [Google Scholar] [CrossRef]
- Herges, R.; Geuenich, D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Anisotropy of the Induced Current Density (ACID), a General Method to Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Cason, C.; Froehlich, T.; Lipka, C. POV-Ray 3.7.0.msvc10.win64. Persistence of Vision; Raytracer Pty. Ltd.: Fortitude Valley, Australia, 2013. [Google Scholar]
- Szczepanik, D.W.; Andrzejak, M.; Dyduch, K.; Żak, E.; Makowski, M.; Mazur, G.; Mrozek, J. A Uniform Approach to the Description of Multicenter Bonding. Phys. Chem. Chem. Phys. 2014, 16, 20514–20523. [Google Scholar] [CrossRef] [PubMed]
- Szczepanik, D.W. A New Perspective on Quantifying Electron Localization and Delocalization in Molecular Systems. Comput. Theor. Chem. 2016, 1080, 33–37. [Google Scholar] [CrossRef]
- Szczepanik, D.W.; Andrzejak, M.; Dominikowska, J.; Pawełek, B.; Krygowski, T.M.; Szatylowicz, H.; Solà, M. The Electron Density of Delocalized Bonds (EDDB) Applied for Quantifying Aromaticity. Phys. Chem. Chem. Phys. 2017, 19, 28970–28981. [Google Scholar] [CrossRef] [Green Version]
- Szczepanik, D.W.; Solà, M. The Electron Density of Delocalized Bonds(EDDBs) as a Measure of Local and Global Aromaticity. In Aromaticity. Modern Computational Methods and Applicattions; Fernandez, I., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 259–284. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1.; Gaussian Inc.: Pittsburgh, CA, USA, 2003. [Google Scholar]
- Available online: http://www.eddb.pl/runeddb/ (accessed on 8 July 2021).
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Iversen, K.J.; Wilson, D.J.D.; Dutton, J.L. A Computational Study on a Strategy for Isolating a Stable Cyclopentadienyl Cation. Chem. Eur. J. 2014, 20, 14132–14138. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.P.F.; Wright, T.G. A Study of the Lowest-Lying Triplet and Singlet States of the Cyclopentadienyl Cation (c-C5H5+). Phys. Chem. Chem. Phys. 1999, 1, 219–225. [Google Scholar] [CrossRef]
- Saito, T.; Nishihara, S.; Yamanaka, S.; Kitagawa, Y.; Kawakumi, T.; Yamada, S.; Isobe, H.; Okumura, M.; Yamaguchi, K. Symmetry and Broken Symmetry in Molecular Orbital Description of Unstable Molecules IV: Comparison Between Single- and Multi-Reference Computational Results for Antiaromatic Molecules. Theor. Chem. Acc. 2011, 130, 749–763. [Google Scholar] [CrossRef]
- Ozimiński, W.P.; Dobrowolski, J.C. σ- and π-Electron Contributions to the Substituent Effect: Natural Population Analysis. J. Phys. Org. Chem. 2009, 22, 769–778. [Google Scholar] [CrossRef]
- Szczepanik, D.W.; Solà, M.; Andrzejak, M.; Pawełek, B.; Dominikowska, J.; Kukułka, M.; Dyduch, K.; Krygowski, T.M.; Szatylowicz, H. The Role of the Long-Range Exchange Corrections in the Description of Electron Delocalization in Aromatic Species. J. Comput. Chem. 2017, 38, 1640–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, J.C.; Karpińska, G. Substituent Effect in the First Excited Triplet State of Monosubstituted Benzenes. ACS Omega 2020, 16, 9477–9490. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | Symmetry Point Group | R–Ring Dihedral Angle (°) | Relative Energy (kcal/mol) | HOMA | NICS(0.5)πzz (ppm) | ISE (kcal/mol) | EDDBp (e) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | T | OS | S | T | OS | S | T | OS | S | T | OS | S | T | OS | S | T | S | T | OS | |
| H | C2v | D5h | C2v | / | / | / | 0.0 | −10.5 | −1.4 | −1.346 | 0.668 | 0.630 | 239.2 | −17.2 | 107.2 | 25.2 | −22.5 | 0.639 | 2.955 | 0.472 |
| NO | C2v | Cs | / | 0 | 0 | / | 0.0 | −5.8 | / | −0.882 | 0.593 | / | 11.4 | 20.0 | / | 7.3 | 7.7 | 0.523 | 1.102 | / |
| NH2 | C2v | C2v | / | 0 | 0 | / | 0.0 | 16.7 | / | −0.828 | 0.560 | / | 37.5 | −19.4 | / | 12.5 | −18.7 | 0.318 | 2.183 | / |
| OH | Cs | Cs | / | 0 | 0 | / | 0.0 | 9.7 | / | −0.978 | 0.627 | / | 60.3 | −16.9 | / | 18.0 | −20.2 | 0.361 | 2.485 | / |
| F | C2v | C2v | C2v | 0 | 0 | 0 | 0.0 | 1.7 | 0.0 | −1.119 | 0.687 | −1.029 | 96.6 | −17.2 | 95.2 | 22.0 | −20.6 | 0.478 | 2.715 | 0.440 |
| CH3 | Cs | Cs | Cs | 89 1 | 89 1 | 89 1 | 0.0 | −1.5 | −0.1 | −1.146 | 0.620 | −0.229 | 116.3 | −17.0 | 96.9 | 21.5 | −21.7 | 0.445 | 2.777 | 0.768 |
| CF3 | Cs | Cs | Cs | 0 2 | 89 2 | 0 2 | 0.0 | −8.6 | −0.6 | −1.340 | 0.666 | 0.596 | 181.1 | −17.2 | 90.0 | 23.8 | −20.9 | 0.551 | 2.807 | 0.518 |
| NO2 | C2v | C2 | C2 | 90 | 45 | 75 | 0.0 | −5.9 | 1.0 | −0.991 | 0.707 | 0.398 | 150.7 | −16.2 | 94.7 | 23.5 | −9.6 | 0.653 | 2.750 | 0.462 |
| CN | C2v | C2v | C2v | 0 | 0 | 0 | 0.0 | −8.7 | 0.5 | −1.135 | 0.622 | 0.466 | 245.3 | −17.1 | 94.8 | 23.5 | −17.0 | 0.570 | 2.599 | 0.521 |
| CHO | C1 | Cs | C1 | 15 3 | 0 | 12 3 | 0.0 | −7.2 | −1.2 | −0.981 | 0.632 | 0.555 | 120.8 | −20.4 | 58.5 | 22.5 | −9.2 | 0.723 | 2.576 | 0.566 |
| COCH3 | Cs | C1 | / | 86 | 31 3 | / | 0.0 | −3.0 | / | −0.750 | 0.656 | / | 66.2 | −9.0 | / | 21.4 | −9.0 | 0.476 | 2.675 | / |
| COOCH3 | Cs | Cs | C1 | 86 | 0 | 19 3 | 0.0 | −6.0 | 2.0 | −0.888 | 0.656 | 0.628 | 102.6 | −17.0 | 85.7 | 23.2 | −18.6 | 0.519 | 2.687 | 0.503 |
| CONH2 | Cs | C1 | / | 88 | 55 3 | / | 0.0 | −0.7 | / | −0.721 | 0.664 | / | 49.2 | −15.8 | / | 20.3 | −17.4 | 0.423 | 2.752 | / |
| R | Relative Energy (kcal/mol) | HOMA | NICS(0.5)πzz (ppm) | ISE (kcal/mol) | EDDBp (e) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | T | OS | S | T | OS | S | T | OS | S | T | S | T | OS | |
| H | 0.0 0.0 | −10.5 −10.6 | −1.4 −4.1 | −1.346 −1.168 | 0.668 0.777 | 0.630 0.732 | 239.2 244.5 | −17.2 −17.4 | 107.2 98.3 | 25.2 23.8 | −22.5 −22.6 | 0.639 0.616 | 2.955 2.953 | 0.472 0.464 |
| NO | 0.0 0.0 | −5.8 −6.5 | / / | −0.882 −0.831 | 0.593 0.682 | / / | 11.4 10.6 | 20.0 / 1 | / / | 7.3 7.0 | 7.7 8.1 | 0.523 0.443 | 1.102 0.959 | / / |
| NH2 | 0.0 0.0 | 16.7 17.4 | / / | −0.828 −0.792 | 0.560 0.640 | / / | 37.5 35.2 | −19.4 −16.9 | / / | 12.5 11.5 | −18.7 −19.0 | 0.318 0.291 | 2.183 2.074 | / / |
| CHO | 0.0 0.0 | −7.2 −8.2 | / / | −0.981 −1.067 | 0.632 0.750 | / / | 120.8 176.0 | −20.4 / 1 | / / | 22.5 21.4 | −9.2 −10.4 | 0.723 0.654 | 2.576 2.610 | / / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stojanović, M.; Aleksić, J.; Baranac-Stojanović, M. Singlet/Triplet State Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect. Chemistry 2021, 3, 765-782. https://doi.org/10.3390/chemistry3030055
Stojanović M, Aleksić J, Baranac-Stojanović M. Singlet/Triplet State Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect. Chemistry. 2021; 3(3):765-782. https://doi.org/10.3390/chemistry3030055
Chicago/Turabian StyleStojanović, Milovan, Jovana Aleksić, and Marija Baranac-Stojanović. 2021. "Singlet/Triplet State Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect" Chemistry 3, no. 3: 765-782. https://doi.org/10.3390/chemistry3030055
APA StyleStojanović, M., Aleksić, J., & Baranac-Stojanović, M. (2021). Singlet/Triplet State Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect. Chemistry, 3(3), 765-782. https://doi.org/10.3390/chemistry3030055