
Constructing a Triangle Ensemble of Pt Clusters for Enhanced Direct-Pathway Electrocatalysis of Formic Acid Oxidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
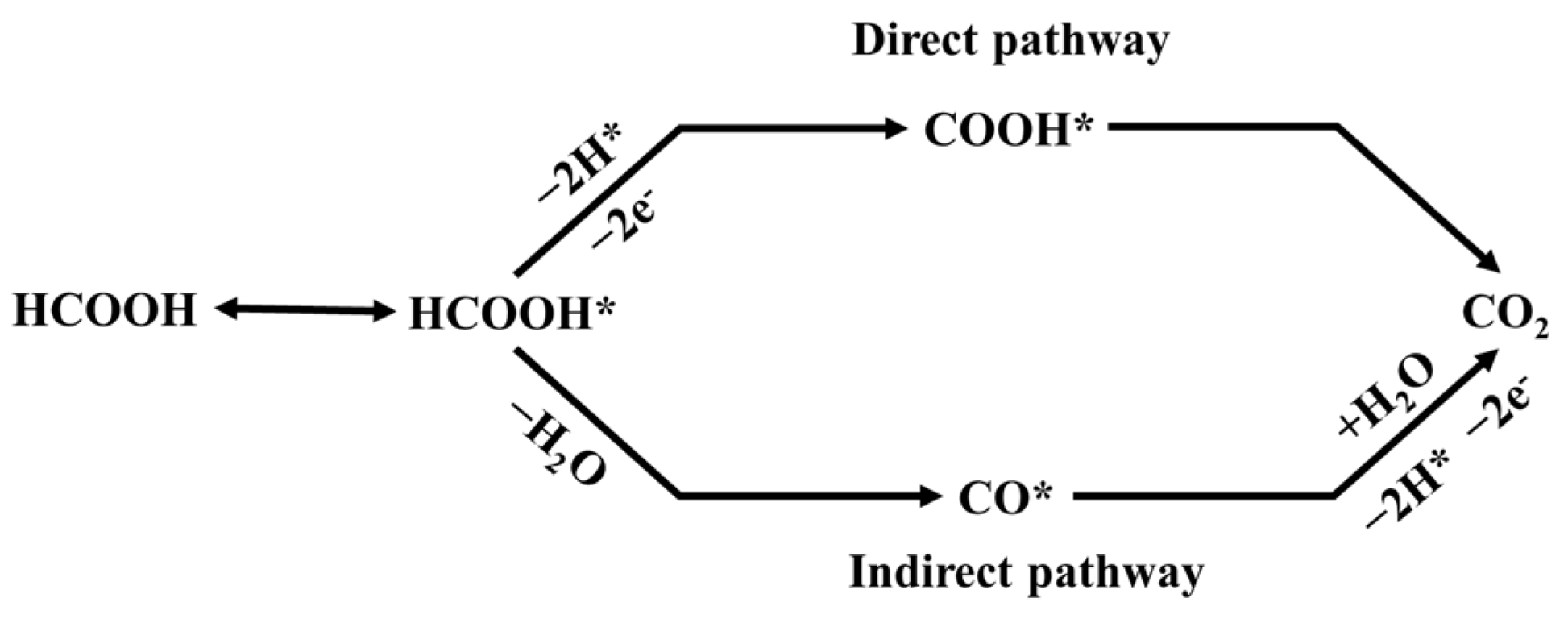
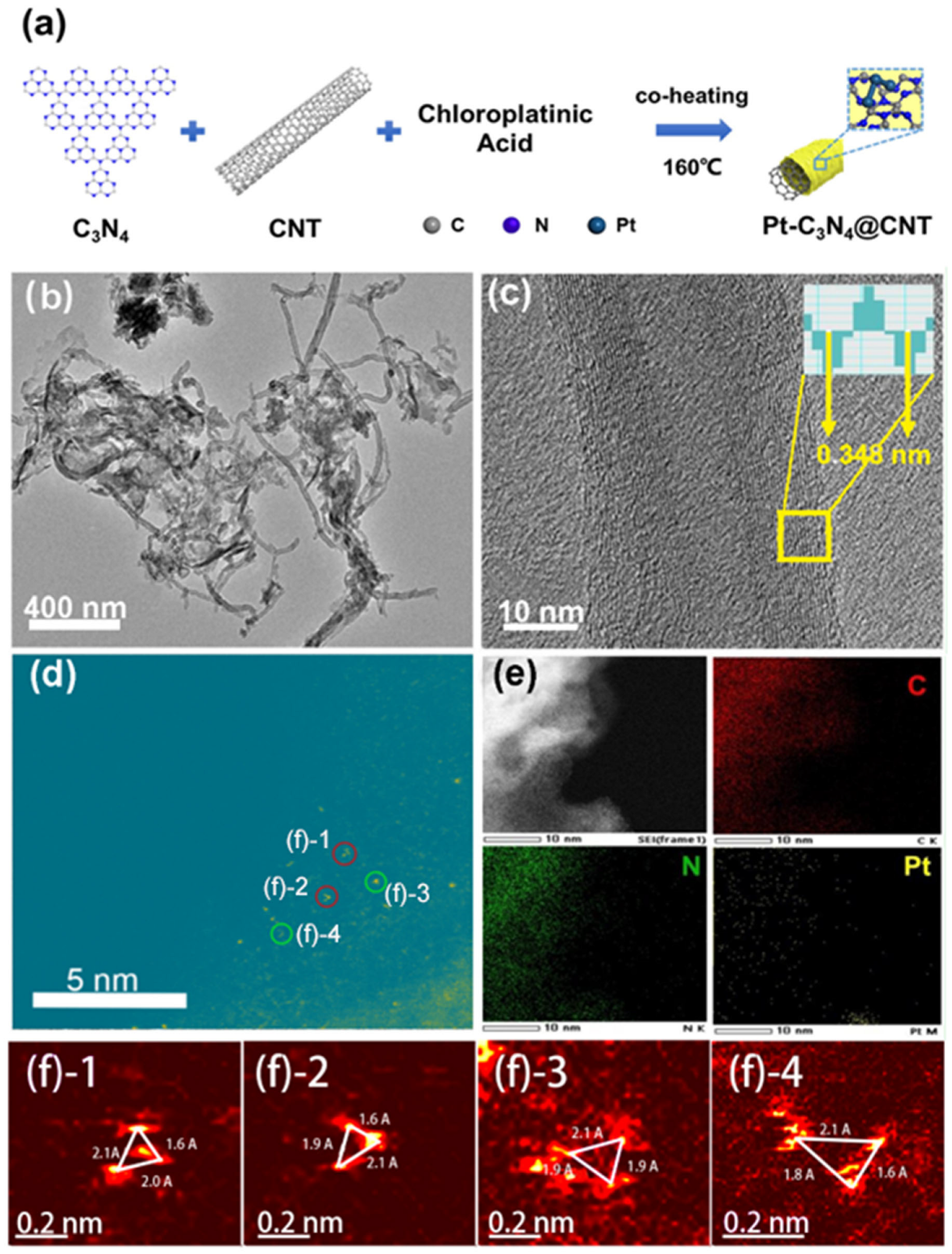
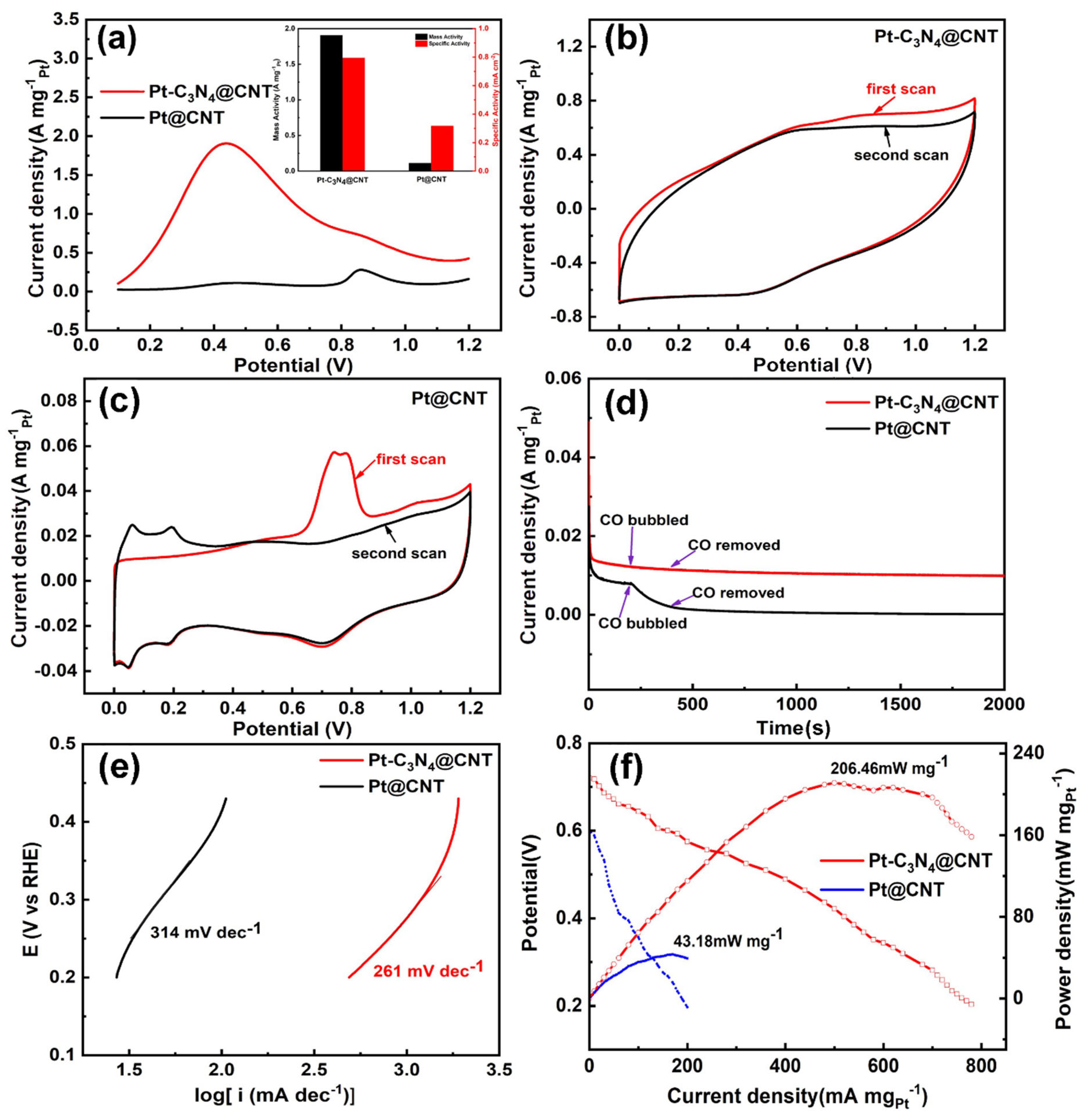
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lai, J.; Niu, W.; Li, S.; Wu, F.; Luque, R.; Xu, G. Concave and duck web-like platinum nanopentagons with enhanced electrocatalytic properties for formic acid oxidation. J. Mater. Chem. A 2016, 4, 807–812. [Google Scholar] [CrossRef]
- Vidal-Iglesias, F.J.; Lopez-Cudero, A.; Solla-Gullon, J.; Feliu, J.M. Towards more active and stable electrocatalysts for formic acid electrooxidation: Antimony-decorated octahedral platinum nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 964–967. [Google Scholar] [CrossRef]
- Su, N.; Chen, X.; Ren, Y.; Yue, B.; Wang, H.; Cai, W.; He, H. The facile synthesis of single crystalline palladium arrow-headed tripods and their application in formic acid electro-oxidation. Chem. Commun. 2015, 51, 7195–7198. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Dong, J.; Yao, Z.; Shen, C.; Shi, X.; Tian, Y.; Lin, S.; Zhang, X. One-pot synthesis of graphene-supported monodisperse Pd nanoparticles as catalyst for formic acid electro-oxidation. Sci. Rep. 2014, 4, 4501. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Dong, J.; Huang, Z.Q.; Xin, P.; Chen, W.; Wang, Y.; Li, Z.; Jin, Z.; Xing, W.; Zhuang, Z.; et al. Single-atom Rh/N-doped carbon electrocatalyst for formic acid oxidation. Nat. Nanotechnol. 2020, 15, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, Y.; Ji, S.; Tang, Y.; Chen, W.; Li, A.; Zhao, J.; Xiong, Y.; Wu, Y.; Gong, Y.; et al. Iridium single-atom catalyst on nitrogen-doped carbon for formic acid oxidation synthesized using a general host-guest strategy. Nat. Chem. 2020, 12, 764–772. [Google Scholar] [CrossRef]
- Park, S.; Xie, Y.; Weaver, M.J. Electrocatalytic Pathways on Carbon-Supported Platinum Nanoparticles: Comparison of Particle-Size-Dependent Rates of Methanol, Formic Acid, and Formaldehyde Electrooxidation. Langmuir 2002, 18, 5792–5798. [Google Scholar] [CrossRef]
- Zhong, W.H.; Wang, R.Y.; Zhang, D.J.; Liu, C.B. Theoretical Study of the Oxidation of Formic Acid on the PtAu(111) Surface in the Continuum Water Solution Phase. J. Phys. Chem. C 2012, 116, 24143–24150. [Google Scholar] [CrossRef]
- Gao, W.; Song, E.H.; Jiang, Q.; Jacob, T. Revealing the active intermediates in the oxidation of formic acid on Au and Pt(111). Chemistry 2014, 20, 11005–11012. [Google Scholar] [CrossRef]
- Xu, J.; Yuan, D.; Yang, F.; Mei, D.; Zhang, Z.; Chen, Y.X. On the mechanism of the direct pathway for formic acid oxidation at a Pt(111) electrode. Phys. Chem. Chem. Phys. 2013, 15, 4367–4376. [Google Scholar] [CrossRef]
- Yue, C.M.Y.; Lim, K.H. Adsorption of Formic Acid and its Decomposed Intermediates on (100) Surfaces of Pt and Pd: A Density Functional Study. Catal. Lett. 2008, 128, 221–226. [Google Scholar] [CrossRef]
- Geng, J.R.; Zhu, Z.; Bai, X.X.; Li, F.J.; Chen, J. Hot-Injection Synthesis of PtCu3 Concave Nanocubes with High-Index Facets for Electrocatalytic Oxidation of Methanol and Formic Acid. ACS Appl. Energy Mater. 2020, 3, 1010–1016. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Fu, G.; Wu, X.; Liu, Y.; Zhang, M.; Sun, D.; Xu, L.; Tang, Y. Ultrathin AgPt alloy nanowires as a high-performance electrocatalyst for formic acid oxidation. Nano Res. 2017, 11, 499–510. [Google Scholar] [CrossRef]
- Scofield, M.E.; Koenigsmann, C.; Wang, L.; Liu, H.Q.; Wong, S.S. Tailoring the composition of ultrathin, ternary alloy PtRuFe nanowires for the methanol oxidation reaction and formic acid oxidation reaction. Energ. Environ. Sci. 2015, 8, 350–363. [Google Scholar] [CrossRef]
- Gao, X.; Li, Y.; Zhang, Q.; Li, S.; Chen, Y.; Lee, J.-M. Polyethyleneimine-assisted synthesis of high-quality platinum/graphene hybrids: The effect of molecular weight on electrochemical properties. J. Mater. Chem. A 2015, 3, 12000–12004. [Google Scholar] [CrossRef]
- Zhou, Z.Y.; Ren, J.; Kang, X.; Song, Y.; Sun, S.G.; Chen, S. Butylphenyl-functionalized Pt nanoparticles as CO-resistant electrocatalysts for formic acid oxidation. Phys. Chem. Chem. Phys. 2012, 14, 1412–1417. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, C.; Liao, J.; Lu, T.; Xing, W. Platinum-macrocycle co-catalysts for electro-oxidation of formic acid. J. Power Sources 2008, 179, 481–488. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, H.X.; Zou, S.; Cai, W.B. Electrocatalysis of formic acid on palladium and platinum surfaces: From fundamental mechanisms to fuel cell applications. Phys. Chem. Chem. Phys. 2014, 16, 20360–20376. [Google Scholar] [CrossRef]
- Yang, N.T.; Zhao, Y.H.; Wu, P.; Liu, G.J.; Sun, F.F.; Ma, J.Y.; Jiang, Z.; Sun, Y.H.; Zeng, G.F. Defective C3N4 frameworks coordinated diatomic copper catalyst: Towards mild oxidation of methane to C1 oxygenates. Appl. Catal. B 2021, 299, 120682. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, S.; Feng, J.; Tong, Y.; Zhou, F.; Li, Q.; Song, L.; Chen, S.; Wu, K.H.; Su, P.; et al. Confined Fe-Cu Clusters as Sub-Nanometer Reactors for Efficiently Regulating the Electrochemical Nitrogen Reduction Reaction. Adv. Mater. 2020, 32, e2004382. [Google Scholar] [CrossRef]
- Bagchi, R.; Elshazly, M.; N’Diaye, J.; Yu, D.; Howe, J.Y.; Lian, K. Effects of Carboxyl Functionalized CNT on Electrochemical Behaviour of Polyluminol-CNT Composites. Chemistry 2022, 4, 1561–1575. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, J.; Yue, L.; Xu, Z.; Dong, K.; Liu, Q.; Luo, Y.; Li, T.; Cheng, X.; Cui, G.; et al. N-Doped Carbon Nanotubes Supported CoSe2 Nanoparticles: A Highly Efficient and Stable Catalyst for H2O2 Electrosynthesis in Acidic Media. Nano Res. 2022, 15, 304–309. [Google Scholar] [CrossRef]
- Han, Q.; Wang, B.; Gao, J.; Cheng, Z.; Zhao, Y.; Zhang, Z.; Qu, L. Atomically Thin Mesoporous Nanomesh of Graphitic C3N4 for High-Efficiency Photocatalytic Hydrogen Evolution. ACS Nano 2016, 10, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Ai, Z.; Zhang, L. Efficient anoxic pollutant removal with oxygen functionalized graphitic carbon nitride under visible light. RSC Adv. 2014, 4, 5553. [Google Scholar] [CrossRef]
- Li, J.; Shen, B.; Hong, Z.; Lin, B.; Gao, B.; Chen, Y. A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity. Chem. Commun. 2012, 48, 12017–12019. [Google Scholar] [CrossRef]
- Zhang, L.; Long, R.; Zhang, Y.; Duan, D.; Xiong, Y.; Zhang, Y.; Bi, Y. Direct Observation of Dynamic Bond Evolution in Single-Atom Pt/C3N4 Catalysts. Angew. Chem. Int. Ed. 2020, 59, 6224–6229. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Lin, J.; Wang, X. Thermally-induced desulfurization and conversion of guanidine thiocyanate into graphitic carbon nitride catalysts for hydrogen photosynthesis. J. Mater. Chem. A 2014, 2, 2942–2951. [Google Scholar] [CrossRef]
- Liu, D.; Chen, D.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. Surface Engineering of g-C3N4 by Stacked BiOBr Sheets Rich in Oxygen Vacancies for Boosting Photocatalytic Performance. Angew. Chem. Int. Ed. 2020, 59, 4519–4524. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Y.; Guo, Y.; Guo, W.; Wang, M.; Guo, Y.; Huo, M. Preparation and enhanced visible-light photocatalytic activity of graphitic carbon nitride/bismuth niobate heterojunctions. J. Hazard. Mater. 2013, 261, 235–245. [Google Scholar] [CrossRef]
- Chen, K.; Chai, Z.; Li, C.; Shi, L.; Liu, M.; Xie, Q.; Zhang, Y.; Xu, D.; Manivannan, A.; Liu, Z. Catalyst-Free Growth of Three-Dimensional Graphene Flakes and Graphene/g-C3N4 Composite for Hydrocarbon Oxidation. ACS Nano 2016, 10, 3665–3673. [Google Scholar] [CrossRef]
- Kim, J.; Roh, C.-W.; Sahoo, S.K.; Yang, S.; Bae, J.; Han, J.W.; Lee, H. Highly Durable Platinum Single-Atom Alloy Catalyst for Electrochemical Reactions. Adv. Energy Mater. 2018, 8, 1701476. [Google Scholar] [CrossRef]
- Iyyamperumal, R.; Zhang, L.; Henkelman, G.; Crooks, R.M. Efficient electrocatalytic oxidation of formic acid using Au@Pt dendrimer-encapsulated nanoparticles. J. Am. Chem. Soc. 2013, 135, 5521–5524. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Escudero, M.; Lanova, B.; Baltruschat, H. Cyclic voltammetry, FTIRS, and DEMS study of the electrooxidation of carbon monoxide, formic acid, and methanol on cyanide-modified Pt(111) electrodes. Langmuir 2009, 25, 6500–6507. [Google Scholar] [CrossRef] [PubMed]
- Maurer, F.; Jelic, J.; Wang, J.; Gänzler, A.; Dolcet, P.; Wöll, C.; Wang, Y.; Studt, F.; Casapu, M.; Grunwaldt, J.-D. Tracking the formation, fate and consequence for catalytic activity of Pt single sites on CeO2. Nat. Catal. 2020, 3, 824–833. [Google Scholar] [CrossRef]
- Intikhab, S.; Rebollar, L.; Li, Y.; Pai, R.; Kalra, V.; Tang, M.H.; Snyder, J.D. Caffeinated Interfaces Enhance Alkaline Hydrogen Electrocatalysis. ACS Catal. 2020, 10, 6798–6802. [Google Scholar] [CrossRef]
- Zhang, S.; Shao, Y.; Yin, G.; Lin, Y. Electrostatic self-assembly of a Pt-around-Au nanocomposite with high activity towards formic acid oxidation. Angew. Chem. Int. Ed. 2010, 49, 2211–2214. [Google Scholar] [CrossRef]
- Ma, J.; Habrioux, A.; Morais, C.; Lewera, A.; Vogel, W.; Verde-Gómez, Y.; Ramos-Sanchez, G.; Balbuena, P.B.; Alonso-Vante, N. Spectroelectrochemical Probing of the Strong Interaction between Platinum Nanoparticles and Graphitic Domains of Carbon. ACS Catal. 2013, 3, 1940–1950. [Google Scholar] [CrossRef]
- Hammer, B.; Nielsen, O.H.; Nrskov, J.K. Structure sensitivity in adsorption: CO interaction with stepped and reconstructed Pt surfaces. Catal. Lett. 1997, 46, 31–35. [Google Scholar] [CrossRef]
- Kitchin, J.R.; Norskov, J.K.; Barteau, M.A.; Chen, J.G. Role of strain and ligand effects in the modification of the electronic and chemical properties of bimetallic surfaces. Phys. Rev. Lett. 2004, 93, 156801. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Wang, H.; Zhou, J.; Wang, J.; Duchesne, P.N.; Muir, D.; Zhang, P.; Han, N.; Zhao, F.; Zeng, M.; et al. Highly active and durable methanol oxidation electrocatalyst based on the synergy of platinum-nickel hydroxide-graphene. Nat. Commun. 2015, 6, 10035. [Google Scholar] [CrossRef] [Green Version]
- Uwitonze, N.; Zhou, D.; Lei, J.; Chen, W.; Zuo, X.Q.; Cai, J.; Chen, Y.-X. The high Tafel slope and small potential dependence of activation energy for formic acid oxidation on a Pd electrode. Electrochim. Acta 2018, 283, 1213–1222. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, B.; Gao, Y.; Tang, Y.; Lu, T.; Xing, W.; Liu, C. Kinetic study of formic acid oxidation on carbon supported Pd electrocatalyst. J. Power Sources 2009, 192, 372–375. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, X.; Zheng, L.; Shui, J. The Solid-Phase Synthesis of an Fe-N-C Electrocatalyst for High-Power Proton-Exchange Membrane Fuel Cells. Angew. Chem. Int. Ed. 2018, 57, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Li, H.; Xu, A.; Xia, F.; Zhang, L.; Zhang, J.; Ma, D.; Wu, J.; Yue, Q.; Yang, X.; et al. Phase Engineering of Intermetallic PtBi2 Nanoplates for Formic Acid Electrochemical Oxidation. Nano Lett. 2023, 23, 5467–5474. [Google Scholar] [CrossRef]
- Shi, H.; Liao, F.; Zhu, W.; Shao, C.; Shao, M. Effective PtAu Nanowire Network Catalysts with Ultralow Pt Content for Formic Acid Oxidation and Methanol Oxidation. Int. J. Hydrogen Energy 2020, 45, 16071–16079. [Google Scholar] [CrossRef]
- Luo, S.; Chen, W.; Cheng, Y.; Song, X.; Wu, Q.; Li, L.; Wu, X.; Wu, T.; Li, M.; Yang, Q.; et al. Trimetallic Synergy in Intermetallic PtSnBi Nanoplates Boosts Formic Acid Oxidation. Adv. Mater. 2019, 31, 1903683. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Chen, W.; Luo, S.; Wu, X.; Fan, X.; Liao, Y.; Song, X.; Cheng, Y.; Li, L.; Tan, L.; et al. Trace Pd Modified Intermetallic PtBi Nanoplates towards Efficient Formic Acid Electrocatalysis. J. Mater. Chem. A 2021, 9, 9602–9608. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Shen, C.; Zheng, Z.; Chen, H.; Jiang, Y.; Xie, Z. Heterogeneous fcc-Pt/hcp-PtBi Thick-Edge Nanoplates with Enhanced Activity for Formic Acid Oxidation. ACS Appl. Energy Mater. 2021, 4, 9190–9197. [Google Scholar] [CrossRef]
- Kang, Y.; Qi, L.; Li, M.; Diaz, R.E.; Su, D.; Adzic, R.R.; Stach, E.; Li, J.; Murray, C.B. Highly Active Pt3Pb and Core–Shell Pt3Pb–Pt Electrocatalysts for Formic Acid Oxidation. ACS Nano 2012, 6, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shao, Y.; Yin, G.; Lin, Y. Facile Synthesis of PtAu Alloy Nanoparticles with High Activity for Formic Acid Oxidation. J. Power Sources 2010, 195, 1103–1106. [Google Scholar] [CrossRef]
- Liang, M.; Xia, T.; Gao, H.; Zhao, K.; Cao, T.; Deng, M.; Ren, X.; Li, S.; Guo, H.; Wang, R. Modulating Reaction Pathways of Formic Acid Oxidation for Optimized Electrocatalytic Performance of PtAu/CoNC. Nano Res. 2022, 15, 1221–1229. [Google Scholar] [CrossRef]
- Xia, B.Y.; Wu, H.B.; Yan, Y.; Lou, X.W.; Wang, X. Ultrathin and Ultralong Single-Crystal Platinum Nanowire Assemblies with Highly Stable Electrocatalytic Activity. J. Am. Chem. Soc. 2013, 135, 9480–9485. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, H.; Chung, H.-S.; Sohn, Y.; Rhee, C.K. Pt-Bi Co-Deposit Shell on Au Nanoparticle Core: High Performance and Long Durability for Formic Acid Oxidation. Catalysts 2021, 11, 1049. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Tang, Z.; Shi, L.; Li, Y.; Ji, Y.; Zhang, K.; Yang, Z.; Yan, Y.-M. Constructing a Triangle Ensemble of Pt Clusters for Enhanced Direct-Pathway Electrocatalysis of Formic Acid Oxidation. Chemistry 2023, 5, 1621-1633. https://doi.org/10.3390/chemistry5030111
Li C, Tang Z, Shi L, Li Y, Ji Y, Zhang K, Yang Z, Yan Y-M. Constructing a Triangle Ensemble of Pt Clusters for Enhanced Direct-Pathway Electrocatalysis of Formic Acid Oxidation. Chemistry. 2023; 5(3):1621-1633. https://doi.org/10.3390/chemistry5030111
Chicago/Turabian StyleLi, Cheng, Zheng Tang, Lanlan Shi, Yongjia Li, Yingjie Ji, Kaixin Zhang, Zhiyu Yang, and Yi-Ming Yan. 2023. "Constructing a Triangle Ensemble of Pt Clusters for Enhanced Direct-Pathway Electrocatalysis of Formic Acid Oxidation" Chemistry 5, no. 3: 1621-1633. https://doi.org/10.3390/chemistry5030111
APA StyleLi, C., Tang, Z., Shi, L., Li, Y., Ji, Y., Zhang, K., Yang, Z., & Yan, Y.-M. (2023). Constructing a Triangle Ensemble of Pt Clusters for Enhanced Direct-Pathway Electrocatalysis of Formic Acid Oxidation. Chemistry, 5(3), 1621-1633. https://doi.org/10.3390/chemistry5030111