
Advances in Research on Semi-Synthesis, Biotransformation and Biological Activities of Novel Derivatives from Maslinic Acid



Abstract
1. Introduction
2. Biotransformations of MA (1) and Bioactivities of Derivatives
3. Chemical Modifications of MA (1) and Bioactivities
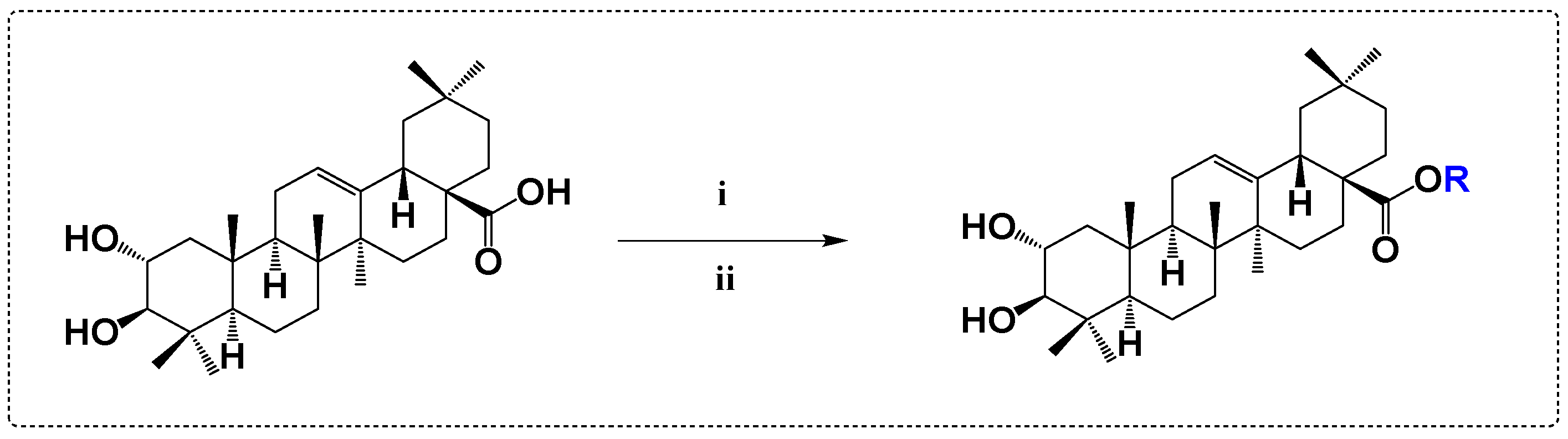
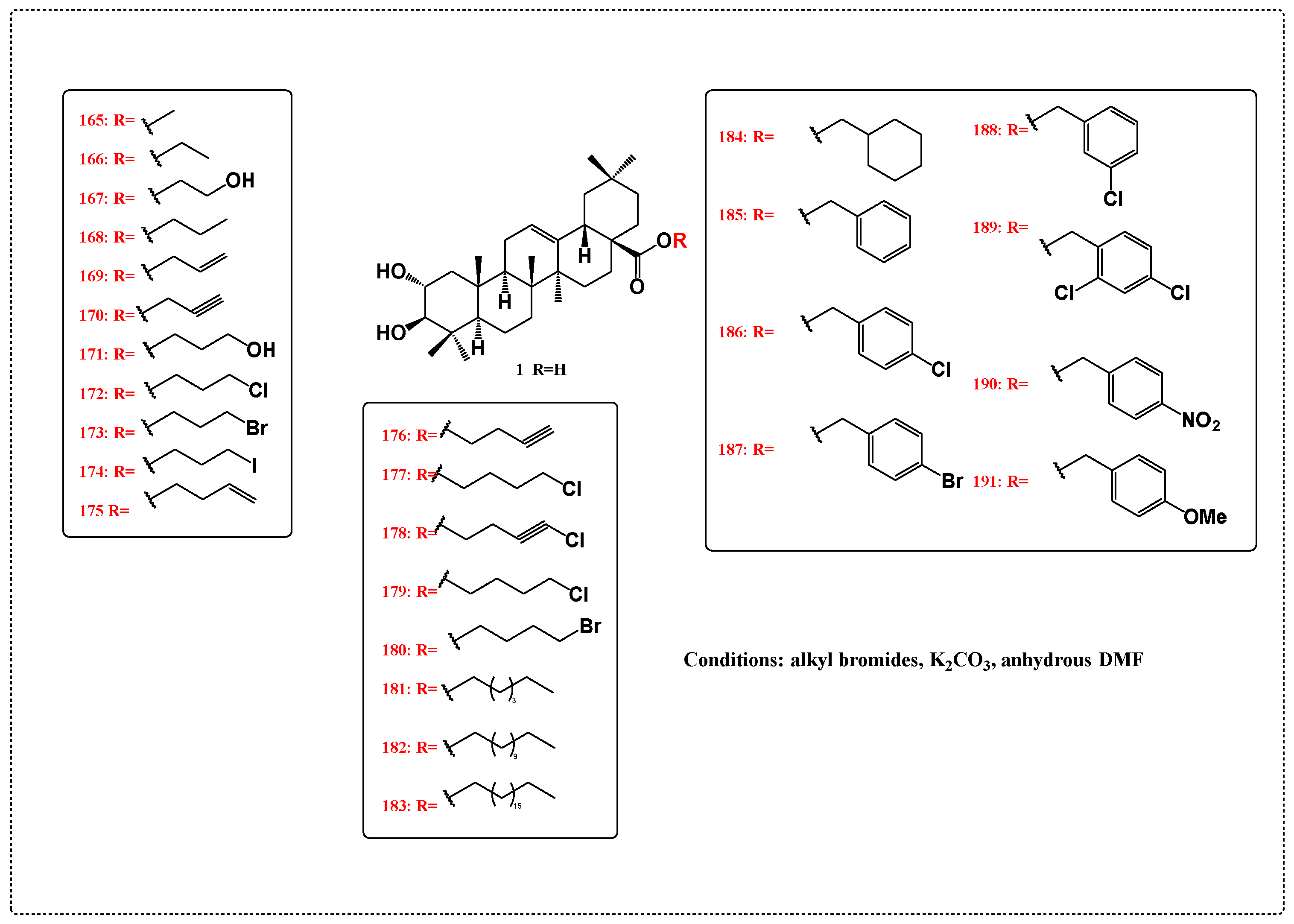
3.1. Anticancer Derivatives
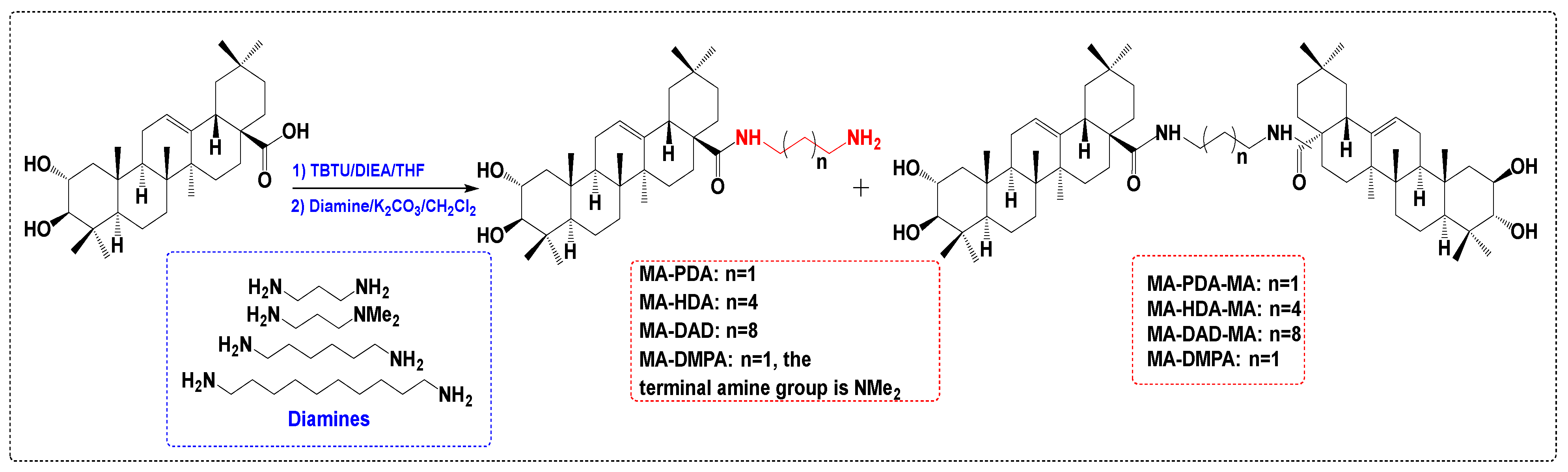
3.2. Anti-Inflammatory Derivatives
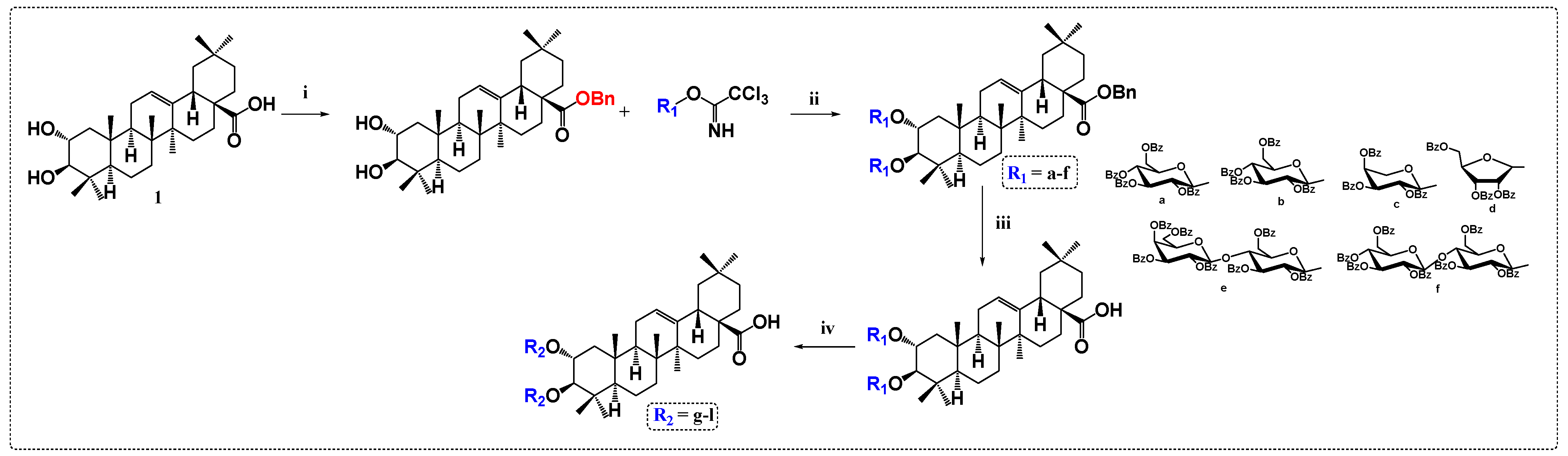
3.3. Glycogen Phosphorylase Inhibitors Derivatives
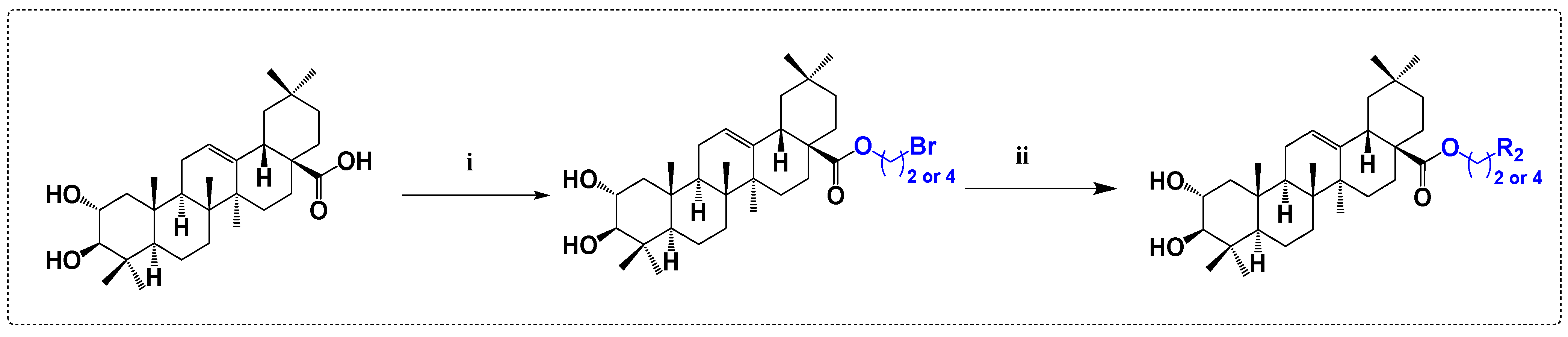
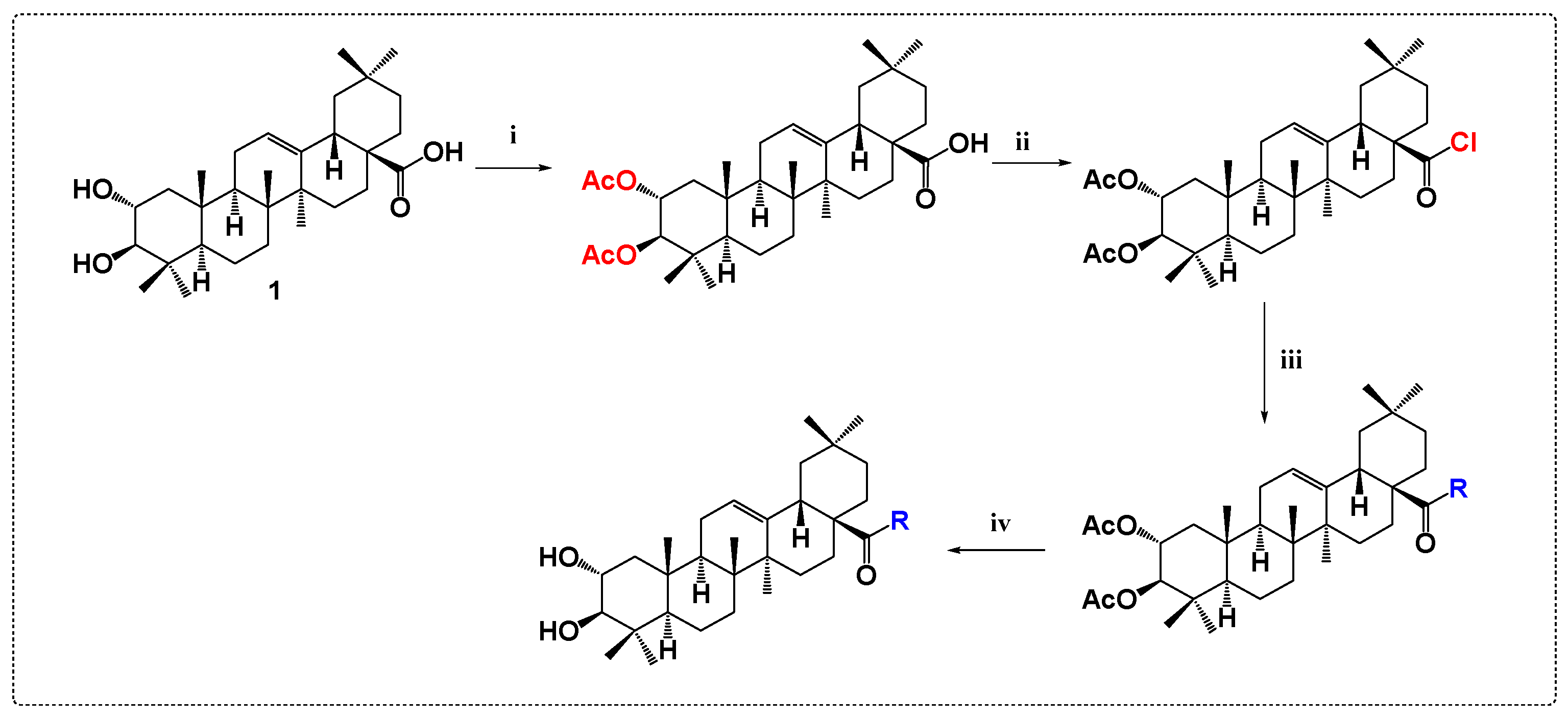
3.4. Antibacterial and Antifungal Derivatives
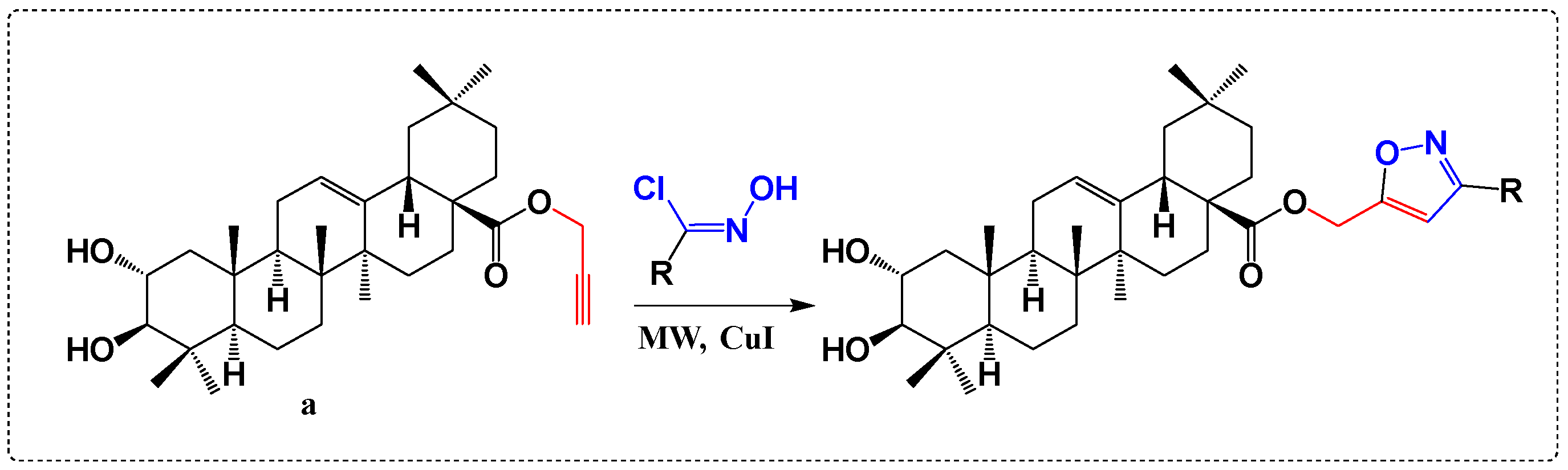
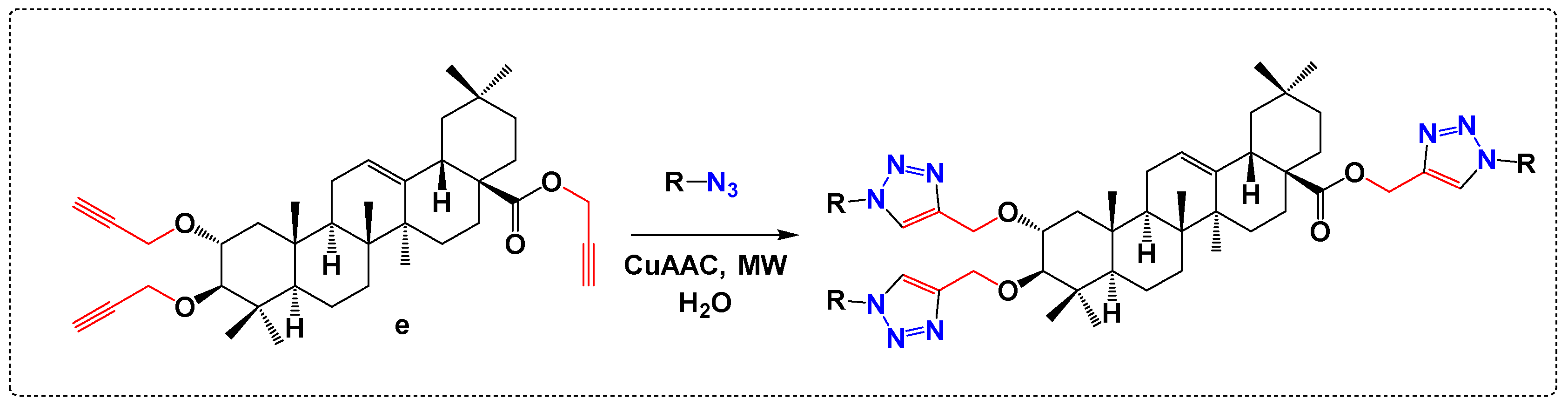
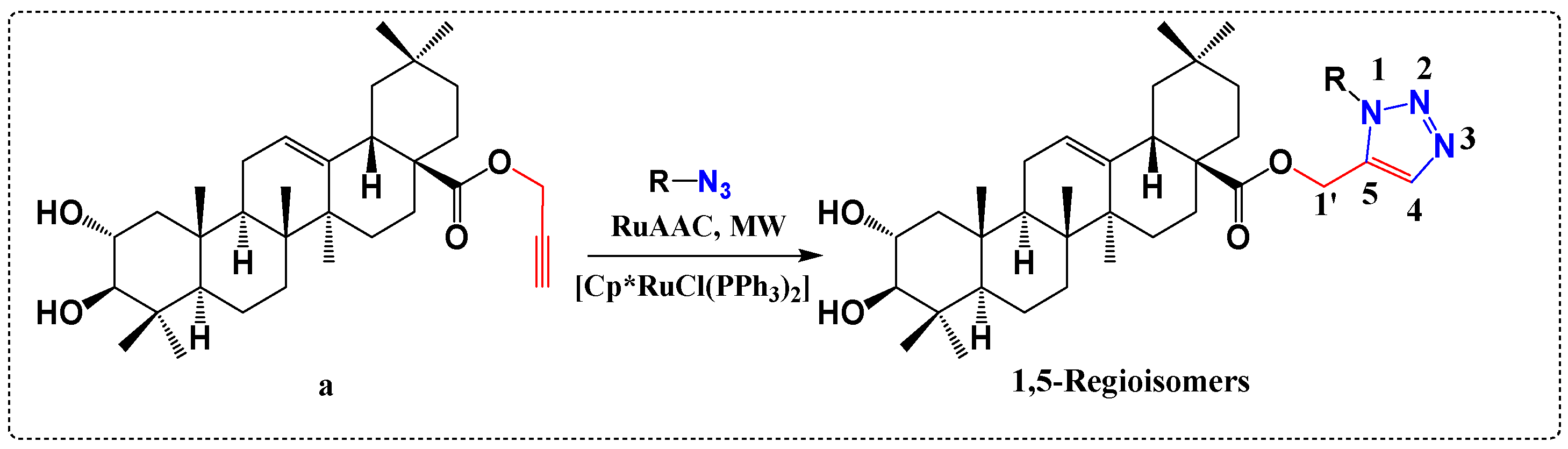
3.5. Derivatives with Herbicide Activity
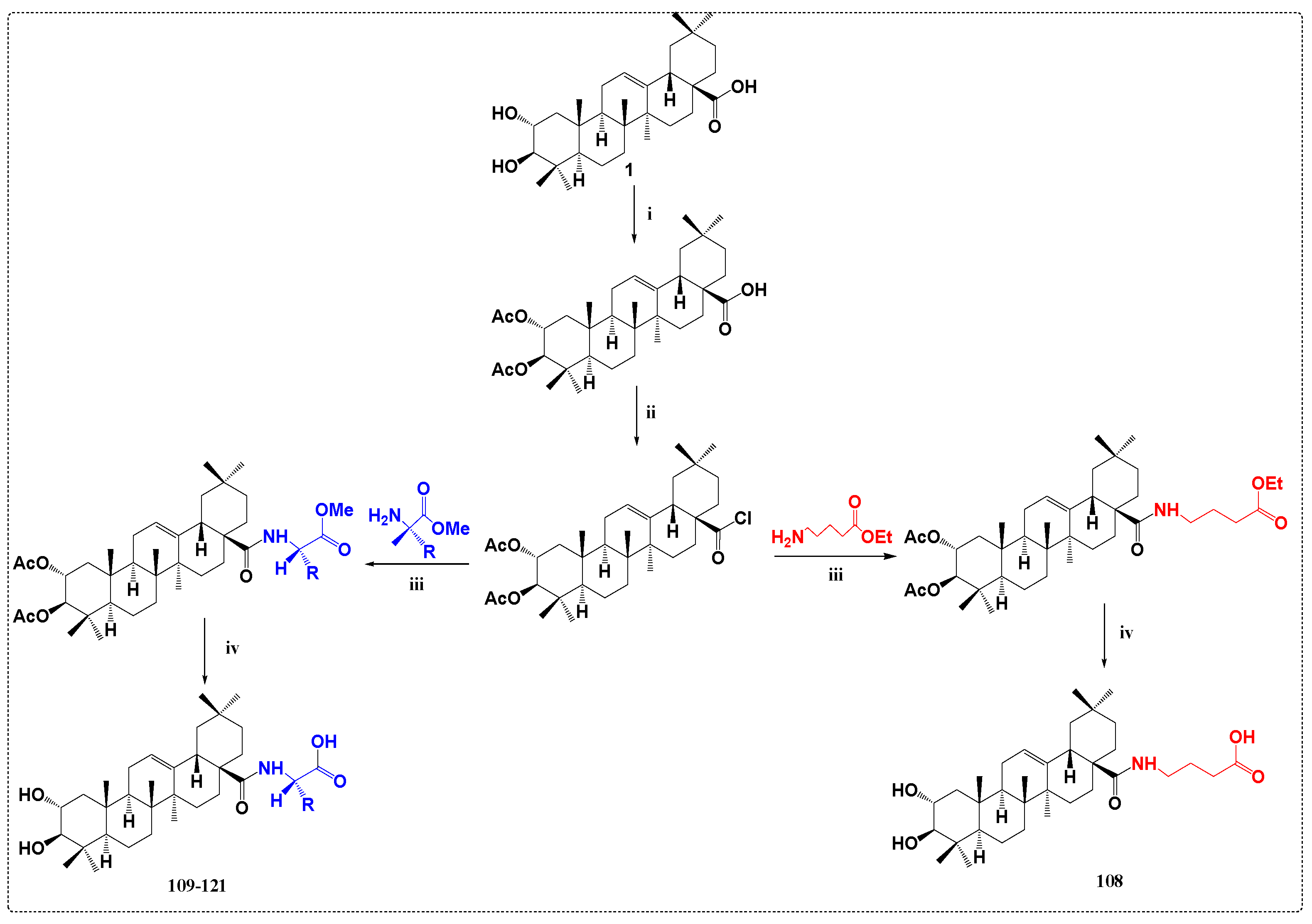
3.6. Anti-Diabetic Derivatives
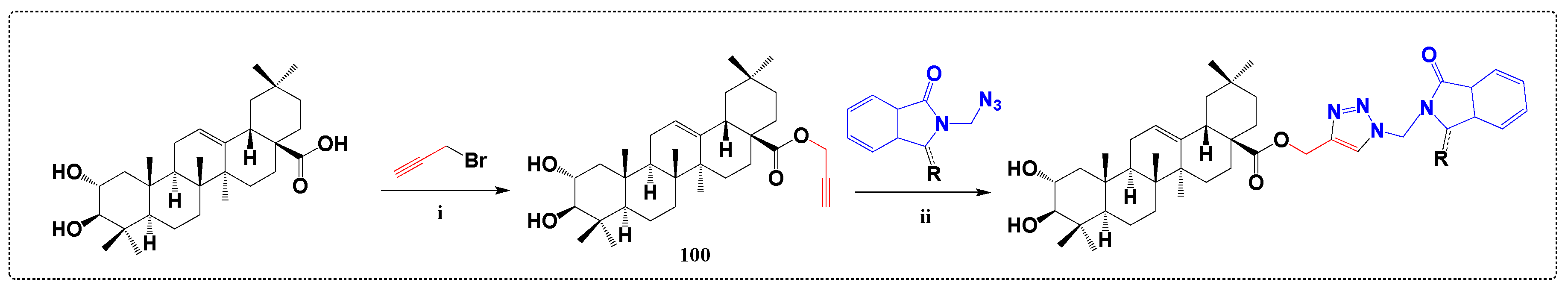
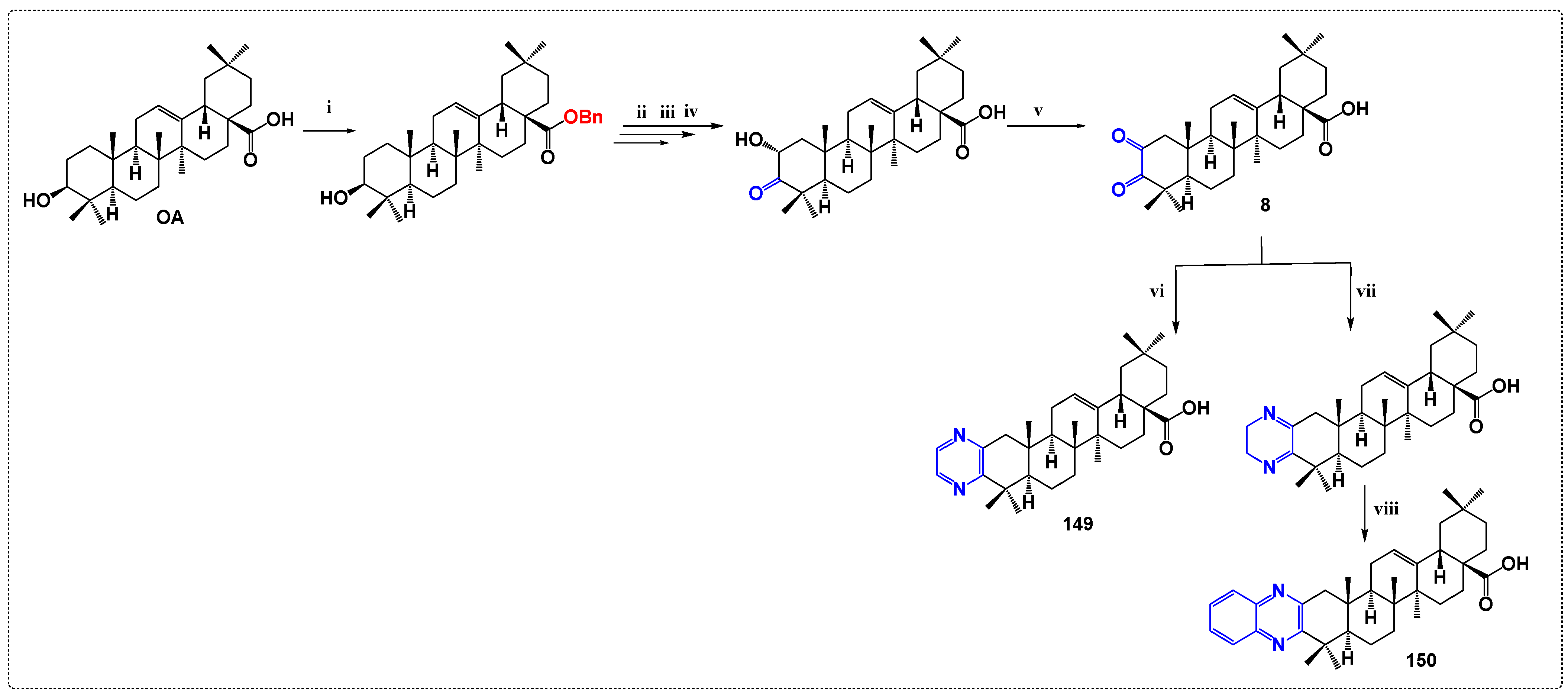
3.7. Inhibitors of Protein Tyrosine Phosphatase 1B Derivatives
3.8. Derivatives as Inhibitors of Acylcholinesterases
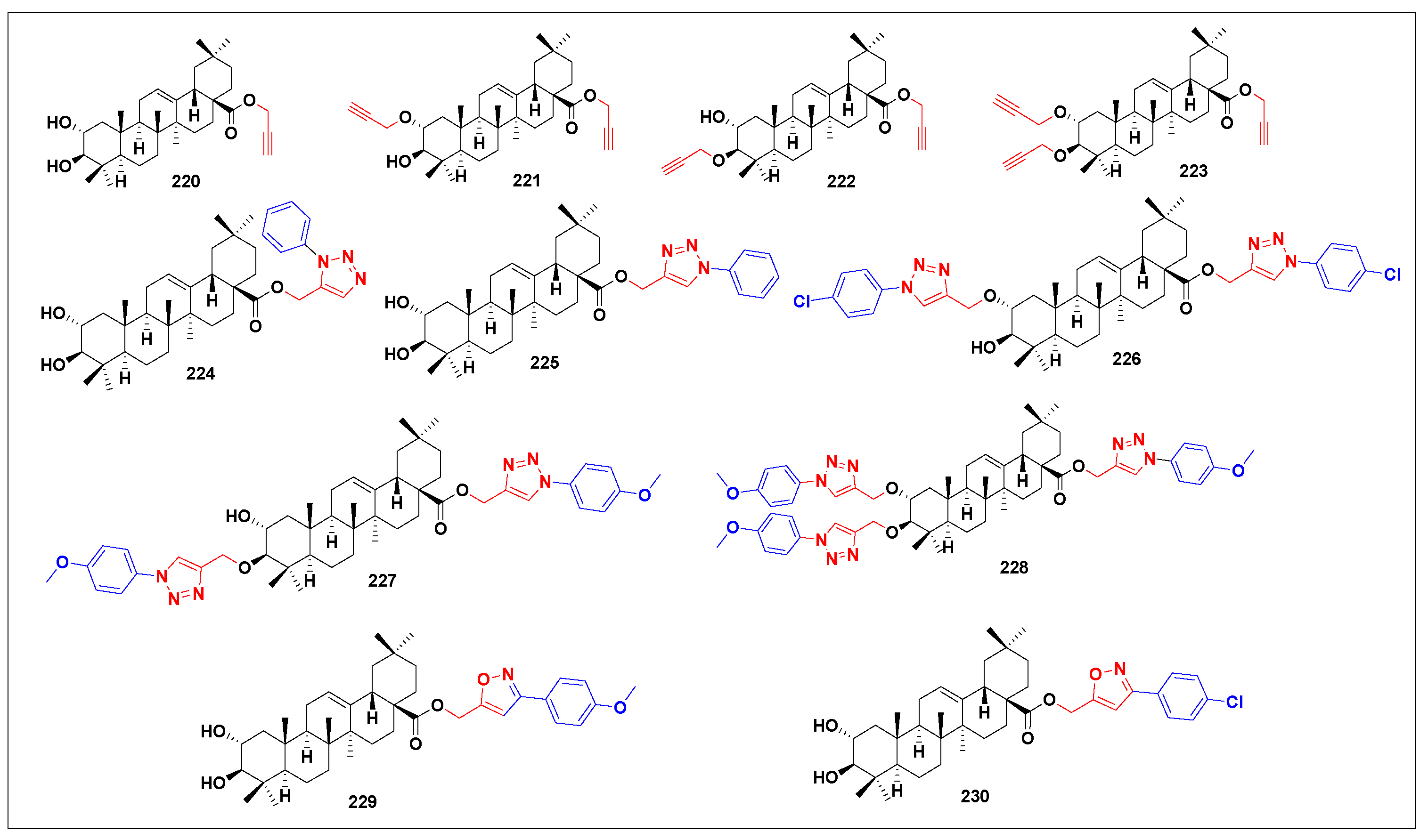
3.9. Antiviral Derivatives
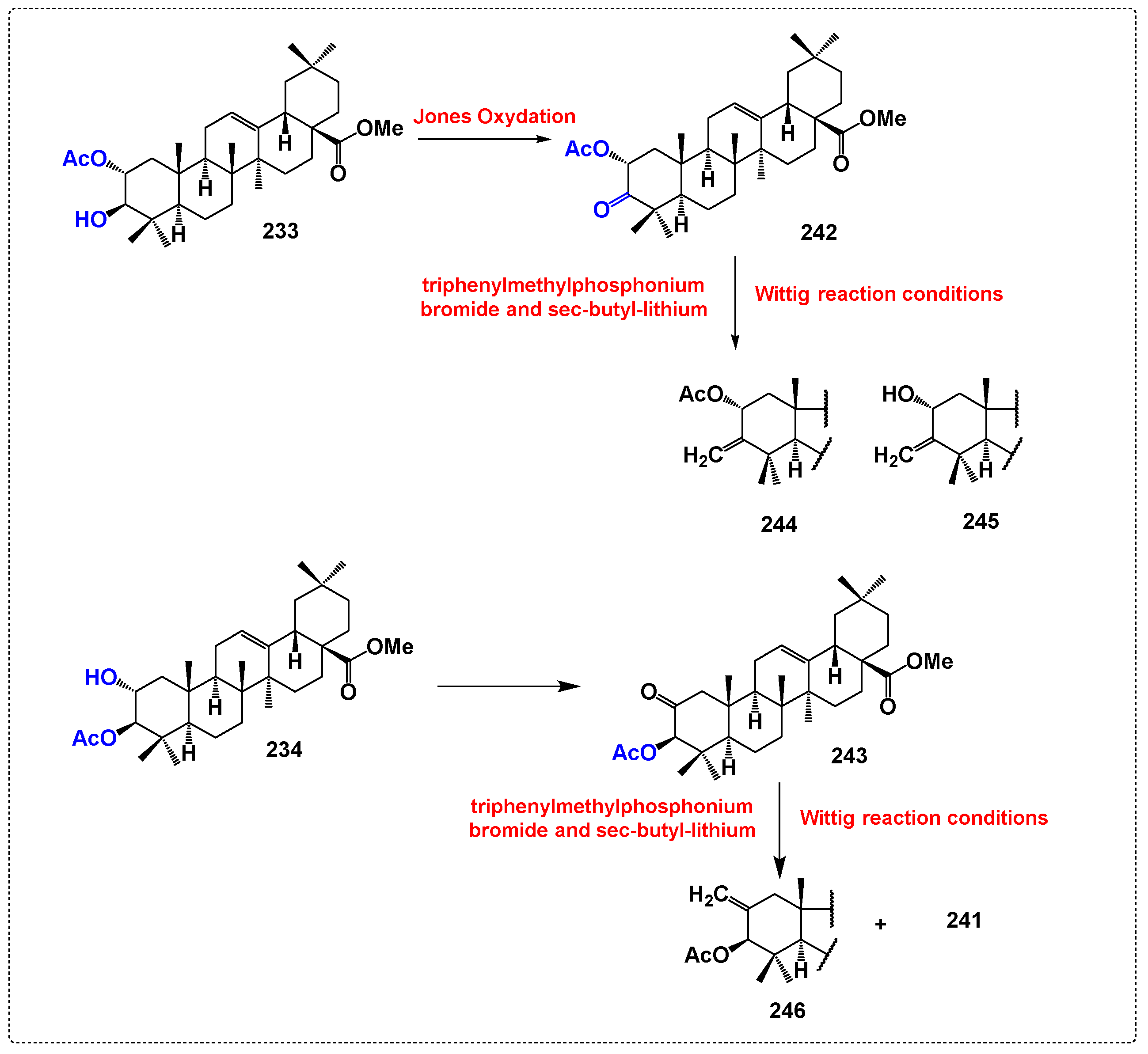
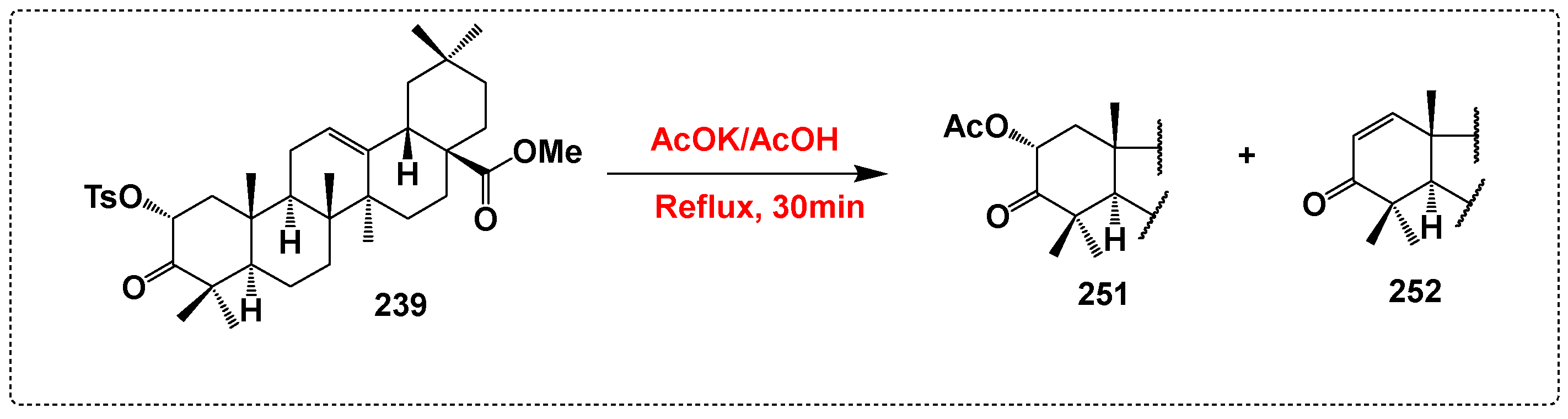
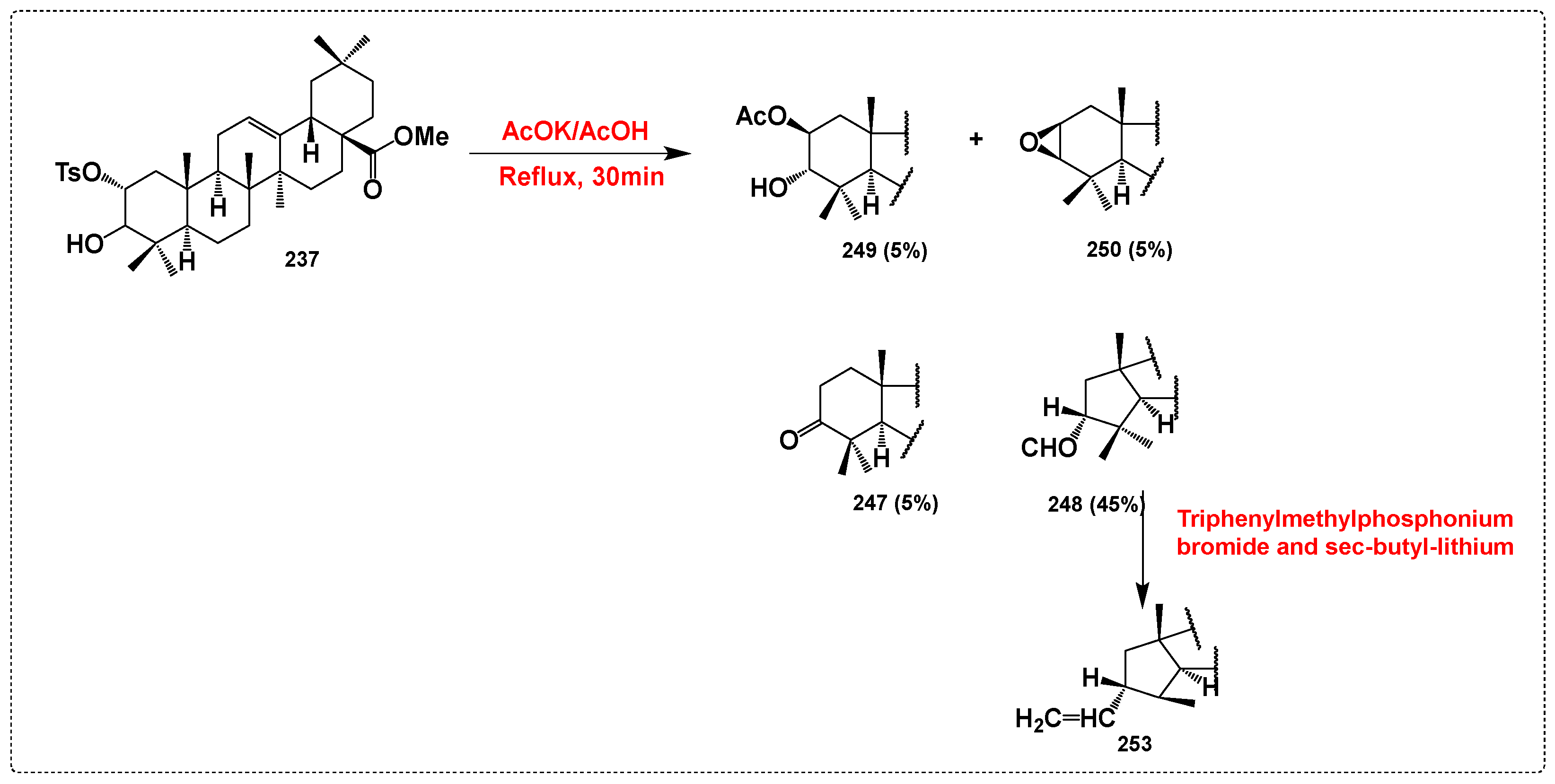
3.10. Derivatives Synthesized with No Activity Assessed
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heard, S.C.; Wu, G.; Winter, J.M. Antifungal Natural Products. Curr. Opin. Biotechnol. 2021, 69, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The Birth of Artemisinin. Pharmacol. Ther. 2020, 216, 107658. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, F.; Lotfalizadeh, M.; Badpeyma, M.; Shakeri, A.; Soheili, V. From Plants to Antimicrobials: Natural Products against Bacterial Membranes. Phytother. Res. 2022, 36, 33–52. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Ahmed, S.; Brankov, N.; Perloff, M. Triterpenoids as potential agents for the chemoprevention and therapy of breast cancer. Front. Biosci. 2011, 16, 980. [Google Scholar] [CrossRef]
- Stiti, N.; Triki, S.; Hartmann, M.A. Formation of triterpenoids throughout Olea europaea fruit ontogeny. Lipids 2007, 42, 55–67. [Google Scholar] [CrossRef]
- García, A.; Brenes, M.; Dobarganes, M.C.; Romero, C.; Ruiz-Méndez, M.V. Enrichment of pomace olive oil in triterpenic acids during storage of “Alpeorujo” olive paste. Eur. J. Lipid Sci. Technol. 2008, 110, 1136–1141. [Google Scholar] [CrossRef]
- Sun, Z.; Li, Z.; Zuo, L.; Wang, Z.; Zhou, L.; Shi, Y.; Kang, J.; Zhu, Z.; Zhang, X. Qualitative and quantitative determination of YiXinShu Tablet using ultra high performance liquid chromatography with Q Exactive hybrid quadrupole orbitrap high-resolution accurate mass spectrometry. J. Sep. Sci. 2017, 40, 4453–4466. [Google Scholar] [CrossRef]
- Jing, Z.; Rui, W.; Ruihua, L.; Hao, Y.; Hengtong, F. Review of the Biological Activity of Maslinic Acid. Curr. Drug. Targets. 2021, 22, 1496–1506. [Google Scholar] [CrossRef]
- Wang, S.-S.; Zhang, Q.-L.; Chu, P.; Kong, L.-Q.; Li, G.-Z.; Li, Y.-Q.; Yang, L.; Zhao, W.-J.; Guo, X.-H.; Tang, Z.-Y. Synthesis and Antitumor Activity of α,β-Unsaturated Carbonyl Moiety- Containing Oleanolic Acid Derivatives Targeting PI3K/AKT/mTOR Signaling Pathway. Bioorg. Chem. 2020, 101, 104036. [Google Scholar] [CrossRef]
- Wan, S.Z.; Liu, C.; Huang, C.K.; Luo, F.Y.; Zhu, X. Ursolic acid improves intestinal damage and bacterial dysbiosis in liver fibrosis mice. Front. Pharmacol. 2019, 10, 1321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xiao, Y.; Xu, L.; Liu, Y.; Jiang, G.; Wang, W.; Li, B.; Zhu, T.; Tan, Q.; Tang, L.; et al. Glycyrrhizic Acid Nanoparticles as Antiviral and Anti-Inflammatory Agents for COVID-19 Treatment. ACS Appl. Mater. Interfaces 2021, 13, 20995–21006. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Huang, X.; Lai, Y.; Liu, X.; Jiang, Y.; Zhan, S. Glycyrrhizic acid for COVID-19: Findings of targeting pivotal inflammatory pathways triggered by SARS-CoV-2. Front. Pharmacol. 2021, 12, 631206. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.R.; Cobos, E.J.; Entrena, J.M.; Parra, A.; García-Granados, A.; Baeyens, J.M. Antiallodynic and analgesic effects of maslinic acid, a pentacyclic triterpenoid from Olea europaea. J. Nat. Prod. 2013, 76, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.K.; Kim, D.H.; Lee, S.Y.; Kim, K.R.; Choi, S.U.; Hong, J.K.; Lee, J.H.; Park, Y.H.; Lee, K.R. Triterpenoic Acids of Prunella Vulgaris Var. Lilacina and Their Cytotoxic Activities In Vitro. Arch. Pharm. Res. 2008, 31, 1578–1583. [Google Scholar] [CrossRef]
- Ho, H.Y.; Lin, W.C.; Kitanaka, S.; Chang, C.T.; Wu, J.B. Analysis of bioactive triterpenes in Eriobotrya japonica Lindl. by high-performance liquid chromatography. J. Food. Drug. Anal. 2008, 16, 5. [Google Scholar] [CrossRef]
- Xu, J.; Liu, T.; Li, Y.; Yuan, C.; Ma, H.; Seeram, N.P.; Liu, F.; Mu, Y.; Huang, X.; Li, L. Hypoglycemic and hypolipidemic effects of triterpenoid-enriched Jamun (Eugenia jambolana Lam.) fruit extract in streptozotocin-induced type 1 diabetic mice. Food Funct. 2018, 9, 3330–3337. [Google Scholar] [CrossRef]
- Masullo, M.; Montoro, P.; Autore, G.; Marzocco, S.; Pizza, C.; Piacente, S. Quali-quantitative determination of triterpenic acids of Ziziphus jujuba fruits and evaluation of their capability to interfere in macrophages activation inhibiting NO release and iNOS expression. Food Res. Int. 2015, 77, 109–117. [Google Scholar] [CrossRef]
- Hossain, M.A.; Ismail, Z. Isolation and characterization of triterpenes from the leaves of Orthosiphon stamineus. Arab. J. Chem. 2013, 6, 295–298. [Google Scholar] [CrossRef]
- Yang, Z.-G.; Li, H.-R.; Wang, L.-Y.; Li, Y.-H.; Lu, S.-G.; Wen, X.-F.; Wang, J.; Daikonya, A.; Kitanaka, S. Triterpenoids from Hippophae rhamnoides L. and their nitric oxide production-inhibitory and DPPH radical-scavenging activities. Chem. Pharm. Bull. 2007, 55, 15–18. [Google Scholar] [CrossRef]
- Mooi, L.Y.; Wahab, N.A.; Lajis, N.H.; Ali, A.M. Chemopreventive properties of phytosterols and maslinic acid extracted from Coleus tuberosus in inhibiting the expression of EBV early-antigen in Raji cells. Chem. Biodivers. 2010, 7, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka-Oliveira, Â.M.; Radai, J.A.S.; Leitão, M.M.; Lima Cardoso, C.A.; Silva-Filho, S.E.; Leite Kassuya, C.A. Anti-inflammatory and anti-arthritic activity in extract from the leaves of Eriobotrya japónica. J. Ethnopharmacol. 2020, 249, 112418. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Liu, Q.; Kim, S.B.; Ahn, J.H.; Ahn, M.-J.; Hwang, B.Y.; Lee, M.K. Maslinic acid, a triterpenoid from the root barks of Ulmus davidiana var. japonica, affects the viability of HSC-T6 hepatic stellate cells. Nat. Prod. Sci. 2011, 17, 216–220. [Google Scholar]
- Zong, W.; Xia, W.; Cui, B. Determination of Corosolic and Maslinic Acids in Lagerstroemia Speciosa Leaves by TLC/HPLC. Method. Pharm. Chem. J. 2007, 41, 222–224. [Google Scholar] [CrossRef]
- Pérez-Camino, M.C.; Cert, A. Quantitative determination of hydroxy pentacyclic triterpene acids in vegetable oils. J. Agric. Food Chem. 1999, 47, 1558–1562. [Google Scholar] [CrossRef]
- Sánchez-González, M.; Lozano-Mena, G.; Juan, M.E.; García-Granados, A.; Planas, J.M. Assessment of the safety of maslinic acid, a bioactive compound from Olea europaea L. Mol. Nutr. Food. Res. 2013, 57, 339–346. [Google Scholar] [CrossRef]
- Gupta, N.; Singh, A.T. Toxicological Evaluation Of Oleanolic Acid (Pentacyclic Triterpenoid) Extracted From Lantana Camara Roots Following Oral Exposure In Wistar Rats. Asian J. Pharm. Clin. Res. 2024, 17, 5. [Google Scholar] [CrossRef]
- Lozano-Mena, G.; Sánchez-González, M.; Juan, M.E.; Planas, J.M. Maslinic acid, a natural phytoalexin-type triterpene from olives—a promising nutraceutical? Molecules 2014, 19, 11538–11559. [Google Scholar] [CrossRef]
- Kim, K.; Kim, J.; Baek, M.; Bae, J. Novel Factor Xa Inhibitor, Maslinic Acid, with Antiplatelet Aggregation Activity. Cell. Physiol. 2020, 235, 9445–9456. [Google Scholar] [CrossRef]
- Pavel, I.Z.; Csuk, R.; Danciu, C.; Avram, S.; Baderca, F.; Cioca, A.; Moacă, E.-A.; Mihali, C.-V.; Pinzaru, I.; Muntean, D.M. Assessment of the Antiangiogenic and Anti-Inflammatory Properties of a Maslinic Acid Derivative and Its Potentiation Using Zinc Chloride. Int. J. Mol. Sci. 2019, 20, 2828. [Google Scholar] [CrossRef]
- Liou, C.J.; Dai, Y.W.; Wang, C.L.; Fang, L.W.; Huang, W.C. Maslinic Acid Protects against Obesity-Induced Nonalcoholic Fatty Liver Disease in Mice through Regulation of the Sirt1/AMPK Signaling Pathway. FASEB. J. 2019, 33, 11791. [Google Scholar] [CrossRef] [PubMed]
- Phang, S.W.; Ooi, B.K.; Ahemad, N.; Yap, W.H. Maslinic Acid Suppresses Macrophage Foam Cells Formation: Regulation of Monocyte Recruitment and Macrophage Lipids Homeostasis. Vasc. Pharmacol. 2020, 128, 106675. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Kong, C.Y.; Song, P.; Zhou, H.; Zhao, X.S.; Tang, Q.Z. Maslinic Acid Protects against Pressure Overload-Induced Cardiac Hypertrophy in Mice. J. Pharmacol. Sci. 2018, 138, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yan, D.; Wu, C.; Xuan, J.; Jin, C.; Hu, X.; Bao, G.; Bian, Y.; Hu, Z.; Shen, Z.; et al. Maslinic Acid Prevents IL-1β-induced Inflammatory Response in Osteoarthritis via PI3K/AKT/NF-κB Pathways. J. Cell. Physiol. 2021, 236, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Siewert, B.; Pianowski, E.; Csuk, R. Esters and Amides of Maslinic Acid Trigger Apoptosis in Human Tumor Cells and Alter Their Mode of Action with Respect to the Substitution Pattern at C-28. Eur. J. Med. Chem. 2013, 70, 259–272. [Google Scholar] [CrossRef]
- Gao, H.; Wu, L.; Kuroyanagi, M.; Harada, K.; Kawahara, N.; Nakane, T.; Umehara, K.; Hirasawa, A.; Nakamura, Y. Antitumor-Promoting Constituents from Chaenomeles sinensis K OEHNE and Their Activities in JB6 Mouse Epidermal Cells. Chem. Pharm. Bull. 2003, 51, 1318–1321. [Google Scholar] [CrossRef]
- Taniguchi, S.; Imayoshi, Y.; Kobayashi, E.; Takamatsu, Y.; Ito, H.; Hatano, T.; Sakagami, H.; Tokuda, H.; Nishino, H.; Sugita, D.; et al. Production of bioactive triterpenes by Eriobotrya japonica calli. Phytochemistry 2002, 2002 59, 315–323. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Xia, N.; Li, B.; Jiang, X. Maslinic acid potentiates the antitumor activities of gemcitabine in vitro and in vivo by inhibiting NF-κB-mediated survival signaling pathways in human gallbladder cancer cells. Oncol. Rep. 2015, 33, 1683–1690. [Google Scholar] [CrossRef]
- Lee, W.; Lee, H.; Lee, T.; Park, E.K.; Bae, J.-S. Inhibitory Functions of Maslinic Acid, a Natural Triterpene, on HMGB1-Mediated Septic Responses. Phytomedicine 2020, 69, 153200. [Google Scholar] [CrossRef]
- Blanco-Cabra, N.; Vega-Granados, K.; Moya-Andérico, L.; Vukomanovic, M.; Parra, A.; Álvarez De Cienfuegos, L.; Torrents, E. Novel Oleanolic and Maslinic Acid Derivatives as a Promising Treatment against Bacterial Biofilm in Nosocomial Infections: An in Vitro and in Vivo Study. ACS Infect. Dis. 2019, 5, 1581–1589. [Google Scholar] [CrossRef]
- Hung, Y.; Yang, H.; Yin, M. Asiatic Acid and Maslinic Acid Protected Heart via Anti-Glycative and Anti-Coagulatory Activities in Diabetic Mice. Food. Funct. 2015, 6, 2967–2974. [Google Scholar] [CrossRef] [PubMed]
- Mkhwanazi, B.N.; Van Heerden, F.R.; Mavondo, G.A.; Mabandla, M.V.; Musabayane, C.T. Triterpene Derivative Improves the Renal Function of Streptozotocin-Induced Diabetic Rats: A Follow-up Study on Maslinic Acid. Ren. Fail. 2019, 41, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.P.; Zhang, X.H.; Sun, L.N.; Xing, W.F.; Wang, Y.; Sun, S.Y.; Ma, M.Y.; Cheng, Z.P.; Wu, Z.D.; Xing, C.; et al. Corosolic Acid and Its Structural Analogs: A Systematic Review of Their Biological Activities and Underlying Mechanism of Action. Phytomedicine 2021, 91, 153696. [Google Scholar] [CrossRef] [PubMed]
- Hodon, J.; Borkova, L.; Pokorny, J.; Kazakova, A.; Urban, M. Design and Synthesis of Pentacyclic Triterpene Conjugates and Their Use in Medicinal Research. Eur. J. Med. Chem. 2019, 182, 111653. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Liu, L.; Huang, X.; Quan, Z.S.; Shen, Q.K.; Guo, H.Y. Bioactivities and structure-activity relationships of maslinic acid derivatives: A review. Chem. Biodivers. 2024, 21, e202301327. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, P.; Jiang, X.; Zhu, Y.; Ye, J.; Raj, R.; Xu, S.; Wang, W.; Yu, B.; Zhang, J. Microbial Transformation of Maslinic Acid for Potential Food Supplements against Sterile Inflammation. ACS. Food. Sci. Technol. 2023, 3, 808–815. [Google Scholar] [CrossRef]
- Martinez, A.; Rivas, F.; Perojil, A.; Parra, A.; Garcia-Granados, A.; Fernandez-Vivas, A. Biotransformation of Oleanolic and Maslinic Acids by Rhizomucor Miehei. Phytochemistry 2013, 94, 229–237. [Google Scholar] [CrossRef]
- Xu, F.; Luan, J.; Guo, F.F.; Li, D.P.; Chu, Z.Y. Microbial Transformation of Maslinic Acid by Cunninghamella Blakesleana. J. Mol. Catal. B Enzym. 2012, 82, 127–130. [Google Scholar] [CrossRef]
- Hu, F.; Chen, J.; Zhang, Y.; Sun, Y.; Liu, Y.; Yu, Y.; Xu, K.; Cai, H. Novel Biotransformation of Maslinic Acid to MA-2-O-β-D-Glucoside by UDP-Glycosyltransferases from Bacillus Subtilis. Catalysts 2022, 12, 884. [Google Scholar] [CrossRef]
- Falev, D.I.; Kosyakov, D.S.; Ul’yanovskii, N.V.; Ovchinnikov, D.V. Rapid Simultaneous Determination of Pentacyclic Triterpenoids by Mixed-Mode Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. A 2020, 1609, 460458. [Google Scholar] [CrossRef]
- Parra, A.; Rivas, F.; Martin-Fonseca, S.; Garcia-Granados, A.; Martinez, A. Maslinic Acid Derivatives Induce Significant Apoptosis in B16f10 Murine Melanoma Cells. Eur. J. Med. Chem. 2011, 46, 5991–6001. [Google Scholar] [CrossRef] [PubMed]
- Chouaïb, K.; Delemasure, S.; Dutartre, P.; Jannet, H.B. Microwave-Assisted Synthesis, Anti-Inflammatory and Anti-Proliferative Activities of New Maslinic Acid Derivatives Bearing 1,5- and 1,4-Disubstituted Triazoles. J. Enzyme Inhib. Med. Chem. 2016, 31, 130–147. [Google Scholar] [CrossRef] [PubMed]
- Siewert, B.; Csuk, R. Membrane Damaging Activity of a Maslinic Acid Analog. Eur. J. Med. Chem. 2014, 74, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Martinez, A.; Martin-Fonseca, S.; Garcia-Granados, A.; Ferrer-Martín, R.M.; Lupianez, J.A.; Parra, A. Semi-Synthesis and Antiproliferative Evaluation of PEGylated Pentacyclic Triterpenes. Eur. J. Med. Chem. 2016, 118, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Martinez, A.; Lupiañez, J.A.; Parra, A. Diamine and PEGylated-Diamine Conjugates of Triterpenic Acids as Potential Anticancer Agents. Eur. J. Med. Chem. 2018, 148, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Pavel, I.Z.; Danciu, C.; Oprean, C.; Dehelean, C.A.; Muntean, D.; Csuk, R.; Muntean, D.M. In Vitro Evaluation of the Antimicrobial Ability and Cytotoxicity on Two Melanoma Cell Lines of a Benzylamide Derivative of Maslinic Acid. Anal. Cell. Pathol. 2016, 2016, 2787623. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. Jnci-J. Natl. Cancer. I 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kuhfs, J.; Csuk, R. Urea Derivates of Ursolic, Oleanolic and Maslinic Acid Induce Apoptosis and Are Selective Cytotoxic for Several Human Tumor Cell Lines. Eur. J. Med. Chem. 2016, 119, 1–16. [Google Scholar] [CrossRef]
- Serbian, I.; Siewert, B.; AlHarrasi, A.; Csuk, R. 2-O-(2-Chlorobenzoyl) Maslinic Acid Triggers Apoptosis in A2780 Human Ovarian Carcinoma Cells. Eur. J. Med. Chem. 2019, 180, 457–464. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine B Conjugates of Triterpenoic Acids Are Cytotoxic Mitocans Even at Nanomolar Concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef]
- Vega-Granados, K.; Medina-O’Donnell, M.; Rivas, F.; Reyes-Zurita, F.J.; Martinez, A.; Alvarez De Cienfuegos, L.; Lupiañez, J.A.; Parra, A. Synthesis and Biological Activity of Triterpene–Coumarin Conjugates. J. Nat. Prod. 2021, 84, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Chouaïb, K.; Romdhane, A.; Delemasure, S.; Dutartre, P.; Elie, N.; Touboul, D. Regiospecific Synthesis, Anti-Inflammatory and Anticancer Evaluation of Novel 3, 5-Disubstituted Isoxazoles from the Natural Maslinic and Oleanolic Acids. Ind. Crops. Prod. 2016, 85, 287–299. [Google Scholar] [CrossRef]
- Wen, X.; Zhang, P.; Liu, J.; Zhang, L.; Wu, X.; Ni, P.; Sun, H. Pentacyclic Triterpenes. Part 2: Synthesis and Biological Evaluation of Maslinic Acid Derivatives as Glycogen Phosphorylase Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Ignasiak, K.; Maxwell, A. Galleria Mellonella (Greater Wax Moth) Larvae as a Model for Antibiotic Susceptibility Testing and Acute Toxicity Trials. BMC. Res. Notes 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Chouaïb, K.; Hichri, F.; Nguir, A.; Daami-Remadi, M.; Elie, N.; Touboul, D.; Ben Jannet, H.; Hamza, M.A. Semi-Synthesis of New Antimicrobial Esters from the Natural Oleanolic and Maslinic Acids. Food Chem. 2015, 183, 8–17. [Google Scholar] [CrossRef]
- Nejma, A.B.; Znati, M.; Daich, A.; Othman, M.; Lawson, A.M.; Jannet, H.B. Design and Semisynthesis of New Herbicide as 1, 2, 3-Triazole Derivatives of the Natural Maslinic Acid. Steroids 2018, 138, 102–107. [Google Scholar] [CrossRef]
- Burke, I.C.; Bell, J.L. Plant Health Management: Herbicides. In Encyclopedia of Agriculture and Food Systems; Van Alfen, N.K., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 425–440. [Google Scholar]
- Zeng, Z.; Yin, X.; Wang, X.; Yang, W.; Liu, X.; Hong, Y. Synthesis of Water Soluble Pentacyclic Dihydroxyterpene Carboxylic Acid Derivatives Coupled Amino Acids and Their Inhibition Activities on α-Glucosidase. Bioorg. Chem. 2019, 86, 277–287. [Google Scholar] [CrossRef]
- Huang, J.; Zang, X.; Yang, W.; Yin, X.; Huang, J.; Wu, S.; Hong, Y. Pentacyclic Triterpene Carboxylic Acids Derivatives Integrated Piperazine-Amino Acid Complexes for α-Glucosidase Inhibition in Vitro. Bioorg. Chem. 2021, 115, 105212. [Google Scholar] [CrossRef]
- Liu, X.; Zang, X.; Yin, X.; Yang, W.; Huang, J.; Huang, J.; Yu, C.; Ke, C.; Hong, Y. Semi-Synthesis of C28-Modified Triterpene Acid Derivatives from Maslinic Acid or Corosolic Acid as Potential α-Glucosidase Inhibitors. Bioorg. Chem. 2020, 97, 103694. [Google Scholar] [CrossRef]
- Xu, J.; Nie, X.; Hong, Y.; Jiang, Y.; Wu, G.; Yin, X.; Wang, C.; Wang, X. Synthesis of Water Soluble Glycosides of Pentacyclic Dihydroxytriterpene Carboxylic Acids as Inhibitors of α-Glucosidase. Carbohydr. Res. 2016, 424, 42–53. [Google Scholar] [CrossRef]
- Qiu, W.W.; Shen, Q.; Yang, F.; Wang, B.; Zou, H.; Li, J.Y.; Li, J.; Tang, J. Synthesis and biological evaluation of heterocyclic ring-substituted maslinic acid derivatives as novel inhibitors of protein tyrosine phosphatase 1B. Bioorg. Med. Chem. Lett. 2009, 19, 6618–6622. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Loesche, A.; Lucas, S.D.; Sommerwerk, S.; Serbian, I.; Siewert, B.; Pianowski, E.; Csuk, R. Converting maslinic acid into an effective inhibitor of acylcholinesterases. Eur. J. Med. Chem. 2015, 103, 103–445. [Google Scholar] [CrossRef] [PubMed]
- Parra, A.; Rivas, F.; Lopez, P.E.; Garcia-Granados, A.; Martinez, A.; Albericio, F.; Marquez, N.; Muñoz, E. Solution-and solid-phase synthesis and anti-HIV activity of maslinic acid derivatives containing amino acids and peptides. Bioorg. Med. Chem. 2009, 17, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Soltane, R.; Chrouda, A.; Mostafa, A.; Al-Karmalawy, A.A.; Chouaïb, K.; Dhahri, A.; Pashameah, R.A.; Alasiri, A.; Kutkat, O.; Shehata, M.; et al. Strong inhibitory activity and action modes of synthetic maslinic acid derivative on highly pathogenic coronaviruses: COVID-19 drug candidate. Pathogens 2021, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Serra, C.; Lampis, G.; Pompei, R.; Pinza, M. Antiviral activity of new triterpenic derivatives. Pharmacol. Res. 1994, 29, 359–366. [Google Scholar] [CrossRef]
- Garcia-Granados, A.; Dueñasa, J.; Melguizo, E.; Moliza, J.N.; Parraa, A.; Péreza, F.L.; Dobadob, J.A.; Molina, M. Semi-synthesis of triterpene A-ring derivatives from oleanolic and maslinic acids. Part II. Theoretical and experimental 13C chemical shifts. J. Chem. Res. 2000, 2, 211–212. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Content (g/Kg) | Ref. |
|---|---|---|
| Olive pomace | 6.6 | [14] |
| Plinia edulis (Vell.) Sobral fruits | 6.5 | [17] |
| Fruits of Zizyphus jujuba | 8.65 | [18] |
| Leaves of Orthosiphon stamineus | 0.03 | [19] |
| Hippophae rhamnoides L | 0.09 | [20] |
| Coleus tuberosus | 0.043 | [21] |
| Leaves of Eriobotrya japonica | 1.7 | [22] |
| Ulmus davidiana var. japonica | 0.042 | [23] |
| Lagerstroemia speciosa leaves | 0.53 | [24] |
| Eriobotrya japonica | 0.012 | [16] |
| Prunella vulgaris | 0.007 | [15] |
| Microorganism | Resulting Products | Yield (%) | Anti-Inflammatory Activity: Inhibitory Effects on NO Production in LPS-Induced or HMGB1-Induced RAW 264.7 Cells 1 | Ref. | ||
|---|---|---|---|---|---|---|
| IC50 (μM) | Cell Viability % (50 μM) | |||||
| LPS | HMGB1 | |||||
| Bacillus megaterium CGMCC 1.1741 |  | 28 | >100 2 | 79.2 | 100.38 | [46] |
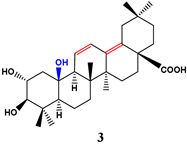 | 12 | 24.40 | 46 | 99.07 | ||
| Streptomyces olivaceus CICC 23628 |  | 26 | >100 2 | 16.41 | 104.19 | |
| Penicillium griseofulvum CICC 40293 |  | 5 | >100 2 | 49.11 | 98.47 | |
 | 12 | >100 2 | 10.77 | 94.69 | ||
| Streptomyces griseus ATCC 1 | 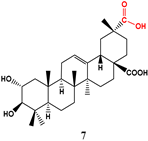 | 10 | >100 2 | 81.00 | 86.09 | |
 | 15 | >100 2 | 11.52 | 96.55 | ||
| Bacillus subtilis ATCC 6633 | 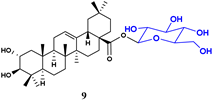 | 56 | >100 2 | >100 2 | 97.84 | |
| Rhizomucor miehei | 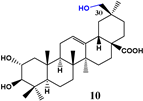 | 11 | - | - | - | [47] |
 | 0.5 | - | - | - | ||
 | 1 | - | - | - | ||
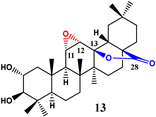 | 0.2 | - | - | - | ||
| Cunninghamella blakesleeana |  | 37 | - | - | - | [48] |
 | 3 | - | - | - | ||
 | 3 | - | - | - | ||
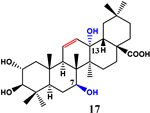 | 2 | - | - | - | - | |
| Bacillus subtilis (UDP-glycosyltransferases UGTs : YojK, YjiC, and UGT109A3) |  | 52 (YojK) 40 (YjiC) 39 (UGT109A3) | - | - | - | [49] |
| Derivative | Synthetic Route |
|---|---|
| 19 |  |
| 20 | 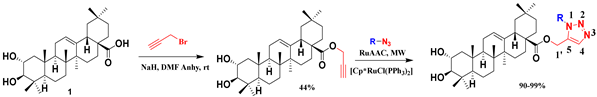 |
| 21 | 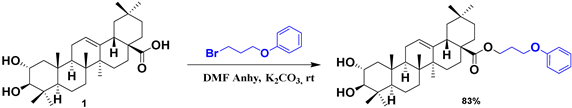 |
| 22 | 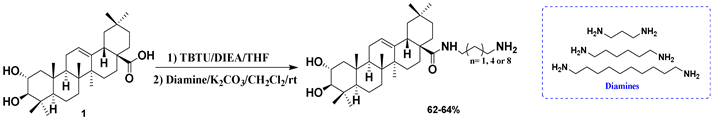 |
| 23 (a–d) | 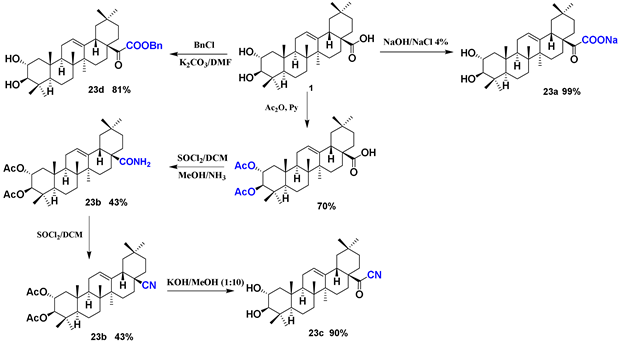 |
| 24 | 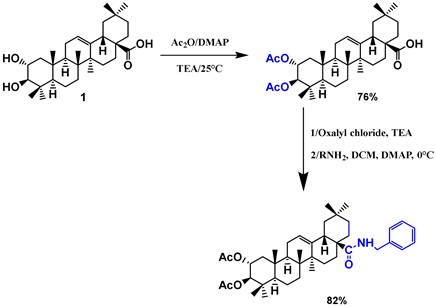 |
| 25 | 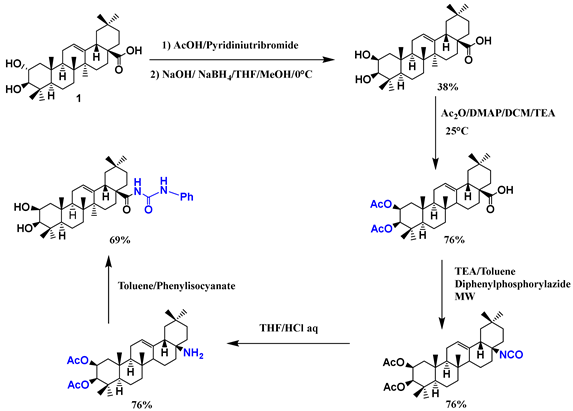 |
| 26 |  |
| 27 (a–c) |  |
| 27d, 27e |  |
 | ||||
|---|---|---|---|---|
| Derivatives | Method A (82%) 1 | (%) 2 | Method B (99%) 1 | (%) 2 |
| a | NaH (2 eq) 3 Propargyl bromide (3 eq) 3 2 h | 44 | NaH (4 eq) 3 Propargyl bromide (4 eq) 3 8 h | 25 |
| b | 5 | 0 | ||
| c | 15 | 23 | ||
| d | 11 | 20 | ||
| e | 7 | 31 | ||
| Concentration (µM) | % PBMCs 1 | % IL-1β Production 2 | Ref. | ||||
|---|---|---|---|---|---|---|---|
| DMSO 0.33% + LPS | 100 | 100 | [62] | ||||
| DEXA 3 + LPS | 1 | 81 | 32 | ||||
| PRED 4 + LPS | 70 | 67 | 43 | ||||
| ZVAD 5+ LPS | 5 | 110 | 42 | ||||
| Code | |||||||
| MA | 100 | 14 | Nd 6 | ||||
| 30 | 53 | 109 | |||||
| R | Time (min) | Yield (%) | |||||
| 28 | 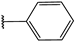 | 5 | 96 | 100 | 89 | 82 | |
| 30 | 32 | Nd | |||||
| 29 |  | 6 | 90 | 100 | 87 | 55 | |
| 30 | 74 | Nd | |||||
| 30 | 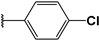 | 6 | 92 | 100 | 23 | Nd | |
| 30 | 57 | 33 | |||||
| 31 | 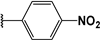 | 8 | 88 | 100 | 44 | Nd | |
| 30 | 72 | 52 | |||||
| 32 | 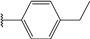 | 5 | 95 | 100 | 58 | Nd | |
| 30 | 42 | 44 | |||||
| 33 | 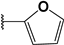 | 10 | 84 | 100 | 78 | 4 | |
| 30 | 85 | Nd | |||||
| Concentration (µM) | % PBMCs 1 | % IL-1β Production 2 | Ref. | ||||
|---|---|---|---|---|---|---|---|
| DMSO 0.33% + LPS | 100 | 100 | [52] | ||||
| DEXA 3 + LPS | 1 | 81 | 32 | ||||
| PRED 4 + LPS | 70 | 67 | 43 | ||||
| ZVAD 5+ LPS | 5 | 110 | 42 | ||||
| Code | |||||||
| MA | 100 | 14 | |||||
| 30 | 53 | ||||||
| R | Time (min) | Yield (%) | |||||
| 34 | 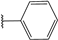 | 4 | 96 | 100 | 92 | 71 | |
| 30 | 92 | Nd 6 | |||||
| 35 |  | 6 | 94 | 100 | 98 | 56 | |
| 30 | 94 | Nd | |||||
| 36 |  | 6 | 94 | 100 | 66 | 100 | |
| 30 | 93 | Nd | |||||
| 37 | 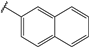 | 5 | 98 | 100 | 97 | 95 | |
| 30 | 80 | Nd |
| Concentration (µM) | % PBMCs 1 | % IL-1β Production 2 | Ref. | ||||
|---|---|---|---|---|---|---|---|
| DMSO 0.33% + LPS | 100 | 100 | [52] | ||||
| DEXA 3 + LPS | 1 | 81 | 32 | ||||
| PRED 4 + LPS | 70 | 67 | 43 | ||||
| ZVAD 5+ LPS | 5 | 110 | 42 | ||||
| Code | |||||||
| MA | 100 | 14 | |||||
| 30 | 53 | ||||||
| R | Time (min) | Yield (%) | |||||
| 38 | 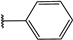 | 4 | 96 | 100 | 88 | 82 | |
| 30 | 82 | Nd 6 | |||||
| 39 | 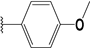 | 4 | 98 | 100 | 74 | 34 | |
| 30 | 88 | Nd | |||||
| 40 |  | 7 | 90 | 100 | 71 | 74 | |
| 30 | 91 | Nd | |||||
| 41 | 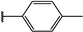 | 6 | 96 | 100 | 81 | 52 | |
| 30 | 83 | Nd | |||||
| 42 | 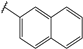 | 6 | 94 | 100 | 95 | 91 | |
| 30 | 93 | Nd |
| Concentration (µM) | % PBMCs 1 | % IL-1β Production 2 | Ref. | ||||
|---|---|---|---|---|---|---|---|
| DMSO 0.33% + LPS | 100 | 100 | [52] | ||||
| DEXA 3 + LPS | 1 | 81 | 32 | ||||
| PRED 4 + LPS | 70 | 67 | 43 | ||||
| ZVAD 5+ LPS | 5 | 110 | 42 | ||||
| Code | |||||||
| MA | 100 | 14 | |||||
| 30 | 53 | ||||||
| R | Time (min) | Yield (%) | |||||
| 43 | 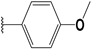 | 5 | 98 | 100 | 73 | 43 | |
| 30 | 96 | Nd | |||||
| 44 | 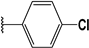 | 8 | 95 | 100 | 83 | 47 | |
| 30 | 85 | Nd 6 | |||||
| 45 |  | 10 | 96 | 100 | 40 | Nd | |
| 30 | 63 | 23 | |||||
| 46 | 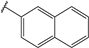 | 8 | 98 | 100 | 31 | Nd | |
| 30 | 72 | 34 |
| Concentration (µM) | % PBMCs 1 | % IL-1β Production 2 | Ref. | ||||
|---|---|---|---|---|---|---|---|
| DMSO 0.33% + LPS | 100 | 100 | [52] | ||||
| DEXA 3 + LPS | 1 | 81 | 32 | ||||
| PRED 4 + LPS | 70 | 67 | 43 | ||||
| ZVAD 5+ LPS | 5 | 110 | 42 | ||||
| Code | |||||||
| MA | 100 | 14 | |||||
| 30 | 53 | ||||||
| R | Time (min) | Yield (%) | |||||
| 47 | 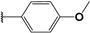 | 4 | 98 | 100 | 87 | 91 | |
| 30 | 93 | Nd 6 | |||||
| 48 | 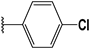 | 6 | 95 | 100 | 76 | 60 | |
| 30 | 74 | Nd | |||||
| 49 |  | 4 | 96 | 100 | 94 | 61 | |
| 30 | 87 | Nd | |||||
| 50 | 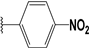 | 8 | 83 | 100 | 75 | 42 | |
| 30 | 93 | Nd | |||||
| 51 | 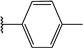 | 6 | 95 | 100 | 51 | 46 | |
| 30 | 99 | Nd | |||||
| 52 | 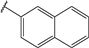 | 6 | 96 | 100 | 93 | 76 | |
| 30 | 95 | Nd |
| Concentration (µM) | % PBMCs 1 | % IL-1β Production 2 | Ref. | ||||
|---|---|---|---|---|---|---|---|
| DMSO 0.33% + LPS | 100 | 100 | [52] | ||||
| DEXA 3 + LPS | 1 | 81 | 32 | ||||
| PRED 4 + LPS | 70 | 67 | 43 | ||||
| ZVAD 5+ LPS | 5 | 110 | 42 | ||||
| Code | |||||||
| MA | 100 | 14 | |||||
| 30 | 53 | ||||||
| R | Time (min) | Yield (%) | |||||
| 53 |  | 2 | 98 | 100 | 92 | 62 | |
| 30 | 91 | Nd 6 | |||||
| 54 |  | 2 | 98 | 100 | 71 | 65 | |
| 30 | 75 | Nd | |||||
| 55 | 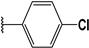 | 4 | 94 | 100 | 83 | 82 | |
| 30 | 55 | Nd | |||||
| 56 | 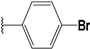 | 4 | 94 | 100 | 92 | 21 | |
| 30 | 74 | Nd | |||||
| 57 |  | 2 | 96 | 100 | 88 | 59 | |
| 30 | 97 | Nd | |||||
| 58 | 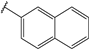 | 2 | 95 | 100 | 97 | 53 | |
| 30 | 88 | Nd |
| Code | R | Yield (%) | GPa Inhibition Assay Results |
|---|---|---|---|
| RMGPa 1 IC50 (µM) | |||
| 59 | -CH3 | 95 | 393 |
| 60 | 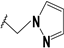 | - | 66 |
| 61 | -CH2COOC2H5 | - | 19 |
| 62 | -CH2COOH | - | 1651 |
| Code | R | Yield (%) | GPa Inhibition Assay Results |
|---|---|---|---|
| RMGPa 1 IC50 (µM) | |||
| 63 | -Br n = 2 | - | 50 |
| 64 | -Br n = 4 | - | 7 |
| 65 | -OH n = 2 | 78 | 153 |
| 66 | -N(C2H5)2 n = 2 | 82 | 51 |
| 67 | 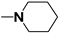 n = 2 | - | 31 |
| 68 | 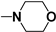 n = 2 | - | 43 |
| 69 | -N(C2H5)2 n = 4 | - | 62 |
| 70 | 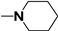 n = 4 | - | 580 |
| Code | R | Yield (%) | GPa Inhibition Assay Results |
|---|---|---|---|
| RMGPa 1 IC50 (µM) | |||
| 72 | -Ac | - | 29 |
| 73 | -TBDMS | - | Nd 2 |
| 74 | -Ac | 75 | 80 |
| 75 | -TBDMS | 74 | 1707 |
| 76 | -Ac | 14 | 63 |
| 77 | -TBDMS | 19 | 580 |
| Code | Compound | Yield (%) | MIC50 (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gram-Positive | Gram-Negative | ||||||||||
| S. aureus | S. aureus MRSA | S. epidermidis | S. mutans | S. faecalis | E. coli | P. aeruginosa | CC50 1 (µM/mL) | LD50 2 (µM/mL) | |||
| 1 | MA | - | 0.03 20.9/504 4 | 0.15 | 0.05 12.5/302 | 0.03 20.9/504 | 0.03 20.9/504 | NA 3 | NA | 0.66 | 0.64 |
| 78 | MA-PDA | 63 | 0.37 | 0.37 | NA | NA | NA | NA | 0.37 | 0.58 | - |
| 79 | MA-HDA | 64 | 0.02 17.9/661 | 0.04 10.7/397 | 0.03 13.4/496 | 0.09 5.4/198 | 0.14 | NA | NA | 0.50 | 0.75 |
| 80 | MA-DAD | 62 | 0.03 10.5 | 0.03 8.4 | 0.01 21 | 0.03 8.4 | 0.03 8.4 | NA | NA | 0.39 | - |
| 81 | MA-DMPA | 90 | 0.13 | 0.15 | 0.13 | 0.15 | 0.13 | NA | NA | NC 5 | - |
| 82 | MA-PDA MA | 27 | NA | NA | NA | NA | NA | NA | NA | 0.11 | - |
| 83 | MA-HDA-MA | 28 | NA | NA | 100 | 100 | NA | NA | NA | 0.29 | - |
| 84 | MA-DAD-MA | 30 | NA | NA | 0.04 | NA | NA | NA | NA | 0.30 | - |
| Code | Compound | Yield (%) | MIC50 (µM) 1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gram-Positive | Gram-Negative | Zone of Inhibition (mm) | |||||||||
| S. aureus | S. faecalis | E. coli | P. aeruginosa | A. flavus | A. niger | P. digitatum | P. italicum | T. harzianum | |||
| 1 | MA | - | 15 | 30 | 15 | 15 | NI 2 | 9 | NI | 13 | 9.5 |
| 85 |  | 59 | 5 | 10 | 10 | 25 | 9 | 13.5 | 11.5 | 24 | 26 |
| 86 | 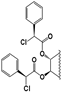 | 48 | 15 | 5 | 15 | 5 | NI | NI | NI | NI | 9.5 |
| 87 |  | 55 | 5 | 5 | 5 | 5 | NI | NI | 15 | NI | 9.5 |
| 88 |  | 45 | 5 | 5 | 5 | 5 | NI | NI | NI | NI | NI |
| 89 |  | 52 | 150 | 100 | 300 | 150 | NI | NI | NI | NI | NI |
| 90 | 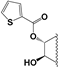 | 33 | 15 | 10 | 10 | 15 | NI | 19 | 15.5 | 14 | 16 |
| 91 | 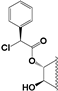 | 32 | 100 | 25 | 15 | 10 | NI | 19.0 | NI | NI | 13 |
| 92 | 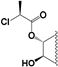 | 31 | 10 | 5 | 5 | 15 | 12 | NI | NI | NI | NI |
| 93 |  | 39 | 5 | 5 | 5 | 5 | NI | NI | NI | NI | 10 |
| 94 |  | 28 | NI | NI | NI | NI | NI | NI | NI | 13 | NI |
| 95 | 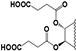 | 81 | 30 | 30 | 50 | 50 | NI | NI | NI | NI | 16 |
| 96 | 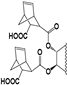 | 88 | 300 | 50 | 50 | 90 | NI | NI | 9 | NI | 9.5 |
| 97 | 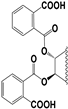 | 82 | 300 | 30 | 50 | 25 | 15 | NI | 13 | NI | NI |
| 98 | 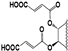 | 76 | 50 | 50 | 25 | 50 | 19 | NI | NI | NI | 17 |
| 99 |  | 78 | 50 | 100 | 100 | 50 | NI | NI | NI | NI | NI |
| IMIPENEM | - | 15 | 15 | 25 | 5 | - | - | - | - | - | |
| FONG 3 | - | - | - | - | - | 40 | 28 | 45 | 43 | 34 | |
| GT (1) | RL (2) | SL (3) | ||||
|---|---|---|---|---|---|---|
| Control | 01.89 | 98.57 | 98.94 | |||
| Code | ||||||
| MA | 5.56 | 97.81 | 95.70 | |||
| 100 | 0.00 | 100 | 100 | |||
| R | Yield (%) | |||||
| Condition (a) | Condition (b) | |||||
| 101 | -O | 36 | 56 | 91.79 | 09.0 | 10.00 |
| 102 | -OH | 34 | 55 | 96.00 | 03.85 | 04.50 |
| 103 | -OAc | 32 | 52 | 92.44 | 06.85 | 09.08 |
| 104 | 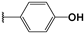 | 33 | 56 | 94.85 | 05.09 | 05.66 |
| 105 | 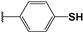 | 34 | 56 | 100.00 | 00.0 | 00.0 |
| 106 | 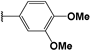 | 31 | 47 | 95.79 | 03.0 | 02.78 |
| 107 | 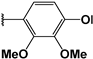 | 29 | 39 | 98.21 | 02.23 | 03.02 |
| Code | R | Yield (%) | IC50 (μM) | Ref. | |
|---|---|---|---|---|---|
| EtOH (5%)/H2O | DMSO (5%) | ||||
| 108 | - | 70 | 987 | - | [68] |
| 109 | -CH(CH3)C2H5 | 65 | 847 | - | |
| 110 | -CH(CH3)2 | 68 | NI 1 | NI | |
| 111 | -CHOHCH3 | 65 | 998 | - | |
| 112 | -CH2CH2COOH | 62 | 495 | 486 | |
| 113 | -CH2COOH | 64 | 382 | 438 | |
| 114 | -H | 75 | 1000 | - | |
| 115 | -CH2OH | 68 | 1321 | - | |
| 116 | -CH2CH(CH3)2 | 66 | 608 | - | |
| 117 | -CH2C6H5 | 63 | 798 | - | |
| 118 | -CH3 | 69 | 987 | - | |
| 119 | 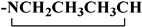 | 71 | NI | - | |
| 120 | -CH2C6H4-p-OH | 73 | 684 | - | |
| 121 | -CH2CH2SCH3 | 62 | 598 | - | |
| Acarbose | - | - | 484 | 447 | |
| MA | - | - | - | 283 | |
| Code | R | Yield (%) | IC50 (μM) | Ref. | |
|---|---|---|---|---|---|
| EtOH (5%)/H2O | DMSO (5%) | ||||
| 122 | -H | 83 | 3638 | - | [69] |
| 123 | -CH2C6H5 | 86 | 4072 | - | |
| 124 | -CH(CH3)2 | 85 | NI 1 | NI | |
| 125 | -CH3 | 88 | 2754 | - | |
| 126 | -CH2COOH | 87 | 591 | - | |
| 127 | -CH2CH2CH2NH2 | 89 | NI | NI | |
| 128 | -CH2OH | 87 | 2754 | - | |
| 129 | -CHOHCH3 | 89 | 2579 | - | |
| 130 | -H | 78 | 1505 | 1505 | |
| 131 | -CH3 | 72 | 1506 | 1193 | |
| Acarbose | - | - | 374 | 493 | |
| MA | - | - | - | 83 | |
| Code | R | Yield (%) | IC50 (μM) | Ref. | |
|---|---|---|---|---|---|
| EtOH (5%)/H2O | DMSO (5%) | ||||
| 132 | n = 2 | 52 | - | 2041.4 | [70] |
| 133 | n = 3 | 50 | - | 1468.4 | |
| 134 | n = 4 | 50 | - | 1718.4 | |
| 135 | n = 5 | 50 | - | 3660.4 | |
| 136 | n = 6 | 45 | - | NI 1 | |
| 137 | n = 8 | 47 | - | 3068.4 | |
| 138 | n = 10 | 50 | - | / 2 | |
| 139 | n = 12 | 54 | - | / 2 | |
| Acarbose | - | - | - | 606 | |
| MA | - | - | - | 2540.6 | |
| Code | R | Yield (%) | IC50 (μM) | Ref. | |
|---|---|---|---|---|---|
| EtOH (5%)/H2O | DMSO (5%) | ||||
| 140 | 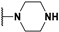 | 57 | - | NI 1 | [70] |
| 141 | 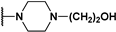 | 62 | - | 499.6 | |
| 142 |  | 66 | - | 768.5 | |
| Acarbose | - | - | - | 606 | |
| MA | - | - | - | 2540.6 | |
| Code | R | Yield (%) | IC50 (μM) | Ref. | |
|---|---|---|---|---|---|
| EtOH (5%)/H2O | DMSO (5%) | ||||
| 143 | 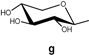 | 88.3 | 7865 | - | [71] |
| 144 | 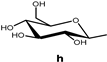 | 86.7 | 5362 | - | |
| 145 |  | 80.6 | 7837 | - | |
| 146 |  | 88.2 | NI 1 | NI | |
| 147 | 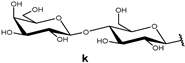 | 70.9 | 684 | 705 | |
| 148 | 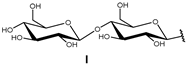 | 80.2 | 2940 | - | |
| MA | - | - | 103 | ||
| Acarbose | - | 478 | 449 | ||
| Code | Yield (%) | IC50 (μM) | TCPTP/PTP1B 1 | ||||
|---|---|---|---|---|---|---|---|
| PTP1B | TCPTP | LAR 2 | SHP-1 3 | SHP-2 4 | |||
| 1 | - | 5.93 | 19.47 | >40 | >40 | >40 | 3.3 |
| 149 | 56 | 1.43 | 5.88 | >40 | >40 | >40 | 4.1 |
| 150 | 28 | 1.79 | 8.31 | >40 | >40 | >40 | 4.6 |
| Code | Yield (%) | IC50 (μM) | TCPTP/PTP1B 1 | ||||
|---|---|---|---|---|---|---|---|
| PTP1B | TCPTP | LAR 2 | SHP-1 3 | SHP-2 4 | |||
| 151 | 12 | 5.44 | Nd 5 | - | - | - | - |
| 152 | 78 | 1.78 | 5.51 | - | - | - | 3.1 |
| 153 | 74 | 2.73 | 8.19 | >40 | >40 | >40 | 3.0 |
| 154 | 80 | 2.61 | 6.50 | - | - | - | 2.5 |
| Code | Yield (%) | IC50 (μM) | TCPTP/PTP1B 1 | ||||
|---|---|---|---|---|---|---|---|
| PTP1B | TCPTP | LAR 2 | SHP-1 3 | SHP-2 4 | |||
| 155 | 55 | 0.61 | 1.60 | - | - | - | 2.6 |
| 156 | 63 | 1.92 | 6.44 | >40 | >40 | >40 | 3.4 |
| 157 | 85 | 2.60 | 8.44 | >40 | >40 | >40 | 3.3 |
| 158 | 65 | 1.39 | 3.80 | >40 | >40 | >40 | 2.7 |
| 159 | 80 | 1.65 | 5.99 | >40 | >40 | >40 | 3.6 |
| 160 | 82 | 1.48 | 4.93 | - | - | - | 3.3 |
| 161 | 85 | 1.75 | 5.56 | - | - | - | 3.2 |
| 162 | 77 | 0.64 | 4.39 | - | - | - | 6.9 |
| 163 | 80 | 0.81 | 3.62 | - | - | - | 4.5 |
| 164 5 | - | 3.89 | 6.24 | - | - | - | 1.6 |
| Compound | MA | 168 | 173 | 174 | 177 | 178 | 181 | 189 |
|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | 16.6 | 13.4 | 21.6 | 12.3 | 12.9 | 33.1 | 14.1 | 17.3 |
| Ki (AChE) | >100 | 16.8 | 34.8 | 6.01 | 1.6 | 22.4 | 4.78 | 2.03 |
| Code | Dose (10 μM) | Dose (25 μM) | Ref. |
|---|---|---|---|
| Infection Percentage: Untreated Cells = 100% Infection | [74] | ||
| MA | 56.3 | 6.7 | |
| 208 | 75.5 | 56.3 | |
| 209 | 63.6 | 8.9 | |
| 210 | 72.0 | 11.1 | |
| 211 | 32.5 | 12.0 | |
| 212 | 69.6 | 36.8 | |
| 213 | 66.9 | 35.6 | |
| 214 | 162.1 | 130.7 | |
| 215 | 124.2 | 60.9 | |
| 216 | 102.8 | 121.2 | |
| 217 | 126.4 | 93.8 | |
| 218 | 146.4 | 79.3 | |
| 219 | 56.7 | 21.5 | |
| Code | MA | 220 | 221 | 222 | 223 | 224 | 225 | 226 | 227 | 228 | 229 | 230 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CC50 | 97.3 | 157.2 | 90.6 | 208.8 | 97.4 | 185.3 | 218.1 | 513.8 | 356.4 | 405.4 | 324.2 | 141.6 |
| IC50 (μM) | 99.8 | 236.3 | 85.2 | 135 | 108 | 131.9 | 134.7 | 116.1 | 158.9 | 136.7 | 218.6 | 4.1 |
| Virus Inhibition | ||||||
|---|---|---|---|---|---|---|
| Polio 1 1 | HSV1 2 | HSV2 3 | VV 4 | VSV 5 | SWN 6 | Rotavirus |
| − | − | ± | − | ± | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trabelsi, Y.; Znati, M.; Ben Jannet, H.; Bouajila, J. Advances in Research on Semi-Synthesis, Biotransformation and Biological Activities of Novel Derivatives from Maslinic Acid. Chemistry 2024, 6, 1146-1188. https://doi.org/10.3390/chemistry6050067
Trabelsi Y, Znati M, Ben Jannet H, Bouajila J. Advances in Research on Semi-Synthesis, Biotransformation and Biological Activities of Novel Derivatives from Maslinic Acid. Chemistry. 2024; 6(5):1146-1188. https://doi.org/10.3390/chemistry6050067
Chicago/Turabian StyleTrabelsi, Yosra, Mansour Znati, Hichem Ben Jannet, and Jalloul Bouajila. 2024. "Advances in Research on Semi-Synthesis, Biotransformation and Biological Activities of Novel Derivatives from Maslinic Acid" Chemistry 6, no. 5: 1146-1188. https://doi.org/10.3390/chemistry6050067
APA StyleTrabelsi, Y., Znati, M., Ben Jannet, H., & Bouajila, J. (2024). Advances in Research on Semi-Synthesis, Biotransformation and Biological Activities of Novel Derivatives from Maslinic Acid. Chemistry, 6(5), 1146-1188. https://doi.org/10.3390/chemistry6050067