
An Overview of Basis Set Effects for Diatomic Boron Nitride Compounds (B2N(∓,0)): A Quantum Symmetry Breaking


Abstract
:1. Introduction
1.1. BNB Structures
1.2. Quantum Theory of Symmetry Breaking (SB)
2. Materials and Methods
Various Basis Sets
3. Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, J.M.L.; François, J.-P.; Gijbels, R. Ab initio study of boron, nitrogen, and boron–nitrogen clusters. I. Isomers and thermochemistry of B3, B2N, BN2, and N3J. Chem. Phys. 1989, 90, 6469. [Google Scholar] [CrossRef]
- Asmis, K.R.; Taylor, T.R.; Neumark, D.M. Anion photoelectron spectroscopy of B2N−. J. Chem. Phys. 1999, 111, 8838. [Google Scholar] [CrossRef]
- Martin, J.M.L.; François, J.-P.; Gijbels, R. Some cost-effective approximations to CCSD and QCISD. Chem. Phys. Lett. 1990, 172, 354–360. [Google Scholar] [CrossRef]
- Knight, L.B.; Hill, D.W., Jr.; Kirk, T.J.; Arrington, C.A. Laser vaporization generation of B 14NH, B 15NH, B 14ND, B 16O, and B 17O: Electron-spin-resonance investigation in neon matrices under ultracold trapping conditions. J. Phys. Chem. 1992, 96, 5604. [Google Scholar] [CrossRef]
- Martin, J.M.L.; François, J.-P.; Gijbels, R. The structure, stability, and infrared spectrum of B2N, B2N+, B2N−, BO, B2O and B2N2. Chem. Phys. Lett. 1992, 193, 243–250. [Google Scholar] [CrossRef]
- Hassanzadeh, P.; Andrews, L. Pulsed laser-assisted reactions of boron and nitrogen atoms in a condensing nitrogen stream. J. Phys. Chem. 1992, 96, 9177–9182. [Google Scholar] [CrossRef]
- Andrews, L.; Hassanzadeh, P.; Burkholder, T.R.; Martin, J.M.L. Reactions of pulsed laser produced boron and nitrogen atoms in a condensing argon stream. J. Chem. Phys. 1993, 98, 922. [Google Scholar] [CrossRef]
- Thompson, C.A.; Andrews, L. Reactions of B Atoms with NH3 to Produce HBNH, BNBH, and B2N. J. Am. Chem. Soc. 1995, 117, 10125–10126. [Google Scholar] [CrossRef]
- Li, X.; Paldus, J. Real or artifactual symmetry breaking in the BNB radical: A multireference coupled cluster viewpoint. J. Chem. Phys. 2007, 126, 224304. [Google Scholar] [CrossRef]
- Martin, J.M.L.; El-Yazal, J.; François, J.-P.; Gijbels, R. The structure and energetics of B3N2, B2N3, and BN4. Mol. Phys. 1995, 85, 527–537. [Google Scholar] [CrossRef]
- Meloni, G.; Sai Baba, M.; Gingerich, A. Knudsen cell mass spectrometric investigation of the B2N molecule. J. Chem. Phys. 2000, 113, 8995. [Google Scholar] [CrossRef]
- Graham, W.R.M.; Weltner, W., Jr. B atoms, B2 and H2BO molecules: ESR and optical spectra at 4 °K. J. Chem. Phys. 1976, 65, 1516–1521. [Google Scholar] [CrossRef]
- Monajjemi, M. Non bonded interaction between BnNn (stator) and BN(−,0,+)B (rotor) systems: A quantum rotation in IR region. Chem. Phys. 2013, 425, 29–45. [Google Scholar] [CrossRef]
- Monajjemi, M.; Lee, V.S.; Khaleghian, M.; Honarparvar, B.; Mollaamin, F. Theoretical Description of Electromagnetic Nonbonded Interactions of Radical, Cationic, and Anionic NH2BHNBHNH2 Inside of the B18N18 Nanoring. J. Phys. Chem. C 2010, 114, 15315–15330. [Google Scholar] [CrossRef]
- Monajjemi, M.; Boggs, J.E. A New Generation of BnNn Rings as a Supplement to Boron Nitride Tubes and Cages. J. Phys. Chem. A 2013, 117, 1670–1684. [Google Scholar] [CrossRef] [PubMed]
- Monajjemi, M. Quantum investigation of non-bonded interaction between the B15N15 ring and BH2NBH2 (radical, cation, anion) systems: A nano molecularmotor. Struct. Chem. 2012, 23, 551–580. [Google Scholar] [CrossRef]
- Walsh, A.D. The electronic orbitals, shapes, and spectra of polyatomic molecules. Part I. AH2 molecules. J. Chem. Soc. 1953, 2260–2266. [Google Scholar] [CrossRef]
- Wigner, E.P. Group Theory and Its Application to the Quantum Mechanics of Atomic Spectra; Academic Press: New York, NY, USA, 1959; p. 259. [Google Scholar]
- Löwdin, P.O. Proton tunneling in DNA and its biological implications. Rev. Mod. Phys. 1963, 35, 496. [Google Scholar] [CrossRef]
- Barone, V. Recent Advances in Density Functional Methods; Parts, I., Chong, D.P., Eds.; World Scientific Publishing Co., Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Chopra, N.G.; Luyken, R.J.; Herrey, K.; Crespi, V.H.; Cohen, M.L.; Louie, S.G.; Zettl, A. Boron Nitride Nanotubes. Science 1995, 269, 966–967. [Google Scholar] [CrossRef]
- Blase, X.; Rubio, A.; Louie, S.G.; Cohen, M.L. Stability and Band Gap Constancy of Boron Nitride Nanotubes. Europhys. Lett. (EPL) 1994, 28, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Blase, X.; Charlier, J.C.; de Vita, A.; Car, R. Theory of composite B𝑥C𝑦N𝑧 nanotube heterojunctions. Appl. Phys. Lett. 1997, 70, 197. [Google Scholar] [CrossRef]
- Han, W.; Bando, Y.; Kurashima, K.; Sato, T. Synthesis of boron nitride nanotubes from carbon nanotubes by a substitution reaction. Appl. Phys. Lett. 1998, 73, 3085. [Google Scholar] [CrossRef]
- Walker, T.E.H.; Richards, W.G. Molecular spin-orbit coupling constants. Role of core polarization. J. Chem. Phys. 1970, 52, 1311. [Google Scholar] [CrossRef]
- Koseki, S.; Schmidt, M.W.; Gordon, M.S. MCSCF/6-31G(d,p) calculations of one-electron spin-orbit coupling constants in diatomic molecules. J. Phys. Chem. 1992, 96, 10768–10772. [Google Scholar] [CrossRef]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecule: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Besler, B.H.; Merz, K.M., Jr.; Kollman, P.A. Atomic Charges Derived from Semiempirical Methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Chirlian, L.E.; Francl, M.M. Atomic Charges Derived from Electrostatic Potentials: A Detailed Study. J. Comput. Chem. 1987, 8, 894–905. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining Atom-Centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Najaflou, N.; Monajjemi, M. A NMR Study of Sodium/Potassium Pumping System in the Node of Ranvier Myelin-Sheath. Biointerface Res. Appl. Chem. 2021, 11, 14260–14277. [Google Scholar] [CrossRef]
- Monajjemi, M.; Kandemirli, F.; Sakhaeinia, H.; Mollaamin, F. Biophysical Interface of Anti-Matter for Virtual Living in Real World: A Reality in Chemical Converted in Parallel Worlds. Biointerface Res. Appl. Chem. 2022, 12, 2646–2659. [Google Scholar] [CrossRef]
- Martin, F.; Zipse, H. Charge distribution in the water molecule—A comparison of methods. J. Comput. Chem. 2005, 26, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA; Oxfordshire, UK, 2009. [Google Scholar]
- Monajjemi, M.; Mahdavian, L.; Mollaamin, F.; Khaleghian, M. Interaction of Na, Mg, Al, Si with carbon nanotube (CNT): NMR and IR study. Russ. J. Inorg. Chem. 2009, 54, 1465–1473. [Google Scholar] [CrossRef]
- Bakhshi, K.; Mollaamin, F.; Monajjemi, M. Exchange and correlation effect of hydrogen chemisorption on nano V(100) surface: A DFT study by generalized gradient approximation (GGA). J. Comput. Theor. Nanosci. 2011, 8, 763–768. [Google Scholar] [CrossRef]
- Tahan, A.; Mollaamin, F.; Monajjemi, M. Thermochemistry and NBO analysis of peptide bond: Investigation of basis sets and binding energy. Russ. J. Phys. Chem. A 2009, 83, 587–597. [Google Scholar] [CrossRef]
- Mollaamin, F.; Monajjemi, M. Harmonic Linear Combination and Normal Mode Analysis of Semiconductor Nanotubes Vibrations. J. Comput. Theor. Nanosci. 2015, 12, 1030–1039. [Google Scholar] [CrossRef]
- Monajjemi, M. Cell membrane causes the lipid bilayers to behave as variable capacitors: A resonance with self-induction of helical proteins. Biophys. Chem. 2015, 207, 114–127. [Google Scholar] [CrossRef]
- Monajjemi, M. Liquid-phase exfoliation (LPE) of graphite towards graphene: An ab initio study. J. Mol. Liq. 2017, 230, 461–472. [Google Scholar] [CrossRef]
- Monajjemi, M.; Bagheri, S.; Moosavi, M.S.; Moradiyeh, N.; Zakeri, M.; Attarikhasraghi, N.; Saghayimarouf, N.; Niyatzadeh, G.; Shekarkhand, M.; Khalilimofrad, M.S.; et al. Symmetry breaking of B2N(−,0,+): An aspect of the electric potential and atomic charges. Molecules 2015, 20, 21636–21657. [Google Scholar] [CrossRef]
- Monajjemi, M.; Mohammadian, N.T. S-NICS: An aromaticity criterion for nano molecules. J. Comput. Theor. Nanosci. 2015, 12, 4895–4914. [Google Scholar] [CrossRef]
- Monajjemi, M.; Mollaamin, F. Ellipticity, PDI and FLU, Evaluation of Aromaticity Indexes During Substituting B &N Atoms in Poly-Annulene. Biointerface Res. Appl. Chem. 2021, 11, 8298–8317. [Google Scholar] [CrossRef]
- Otadi, M.; Panahi Shayegh, Z.; Monajjemi, M. Synthesis and Characterization of Mn doped ZnO Nanoparticles and Degradation of Pyridine in a Batch Reactor Using: Taguchi Experimental Designing & Molecular Mechanic Simulation. Biointerface Res. Appl. Chem. 2021, 11, 12471–12482. [Google Scholar] [CrossRef]
- Mohammad Gholiha, H.; Ghadami, A.; Monajjemi, M.; Ehsani, M. Enhanced Physical and Mechanical Properties of Flake–Shape/Vinyl-ester Nanocomposites Through Surface Modification of Graphene and Glass Flake: A Comparison with Simulated Data. Biointerface Res. Appl. Chem. 2021, 11, 11316–11337. [Google Scholar]
- Monajjemi, M.; Robert, W.J.; Boggs, J.E. NMR contour maps as a new parameter of carboxyl’s OH groups in amino acids recognition: A reason of tRNA–amino acid conjugation. Chem. Phys. 2014, 433, 1–11. [Google Scholar] [CrossRef]
- Bagheri, S.; Monajjemi, M. A Novel Cathodic Combination in Sodium-Ion Battery Based on NaNi0.7Co0.3O2, Na2MnO3, and NaCoO2 Combination: Synthesis and Characterization. Biointerface Res. Appl. Chem. 2021, 11, 11316–11337. [Google Scholar]
- Monajjemi, M. Metal-doped graphene layers composed with boron nitride–graphene as an insulator: A nanocapacitor. J. Mol. Modeling 2014, 20, 2507. [Google Scholar] [CrossRef] [PubMed]
- Monajjemi, M.; Najafpour, J.; Mollaamin, F. (3,3)4 Armchair carbon nanotube in connection with PNP and NPN junctions: Ab Initio and DFT-based studies. Fuller. Nanotub. Carbon Nanostruct. 2013, 21, 213–232. [Google Scholar] [CrossRef]
- Monajjemi, M.; Jafari Azan, M.; Mollaamin, F. Density functional theory study on B30N20 nanocage in structural properties and thermochemical outlook. Fuller. Nanotub. Carbon Nanostruct. 2013, 21, 503–515. [Google Scholar] [CrossRef]
- Monajjemi, M.; Baie, M.T.; Mollaamin, F. Interaction between threonine and cadmium cation in [Cd(Thr)] (n = 1–3) complexes: Density functional calculations. Russ. Chem. Bull. 2010, 59, 886–889. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Check, C.E.; Gilbert, T.M. Progressive Systematic Underestimation of Reaction Energies by the B3LYP Model as the Number of C−C Bonds Increases: Why Organic Chemists Should Use Multiple DFT Models for Calculations Involving Polycarbon Hydrocarbons. J. Org. Chem. 2005, 70, 9828–9834. [Google Scholar] [CrossRef]
- Grimme, S. Seemingly Simple Stereoelectronic Effects in Alkane Isomers and the Implications for Kohn–Sham Density Functional Theory. Angew. Chem. Int. Ed. 2006, 45, 4460–4464. [Google Scholar] [CrossRef] [PubMed]
- Wodrich, M.D.; Corminboeuf, C.; Schleyer, P.V.R. Systematic Errors in Computed Alkane Energies Using B3LYP and Other Popular DFT Functionals. Org. Lett. 2006, 8, 3631–3634. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.R.; Fokin, A.A.; Pascal, R.A., Jr.; de Meijere, A. Many density functional theory approaches fail to give reliable large hydrocarbon isomer energy differences. Org. Lett. 2006, 8, 3635–3638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A Density Functional That Accounts for Medium-Range Correlation Energies in Organic Chemistry. Org. Lett. 2006, 8, 5753–5755. [Google Scholar] [CrossRef] [PubMed]
- Gwaltney, S.R.; Head-Gordon, M. Calculating the equilibrium structure of the BNB molecule: Real s. artifactual symmetry breaking. Phys. Chem. Chem. Phys. 2001, 3, 4495–4500. [Google Scholar] [CrossRef]
- Ding, H.; Morse, M.D.; Apetrei, C.; Chacaga, L.; Maier, J.P. Resonant two-photon ionization spectroscopy of BNB. J. Chem. Phys. 2006, 125, 194315. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, W.; Bersuker, I.B.; Boggs, J.E. Symmetry breaking in the ground state of BNB: A high level multireference study. J. Chem. Phys. 2009, 130, 184305. [Google Scholar] [CrossRef]
- Al-Saidi, W.A. Ground state structure of BNB using fixed-node diffusion Monte Carlo. Chem. Phys. Lett. 2012, 543, 41–44. [Google Scholar] [CrossRef]
- Kalemos, A. Symmetry breaking in a nutshell: The odyssey of a pseudo problem in molecular physics. J. Chem. Phys. 2013, 138, 224302. [Google Scholar] [CrossRef]
- Kalemos, A.; Dunning, T.H.; Mavridis, A. On symmetry breaking in BNB: Real or artifactual? J. Chem. Phys. 2004, 120, 4. [Google Scholar] [CrossRef]
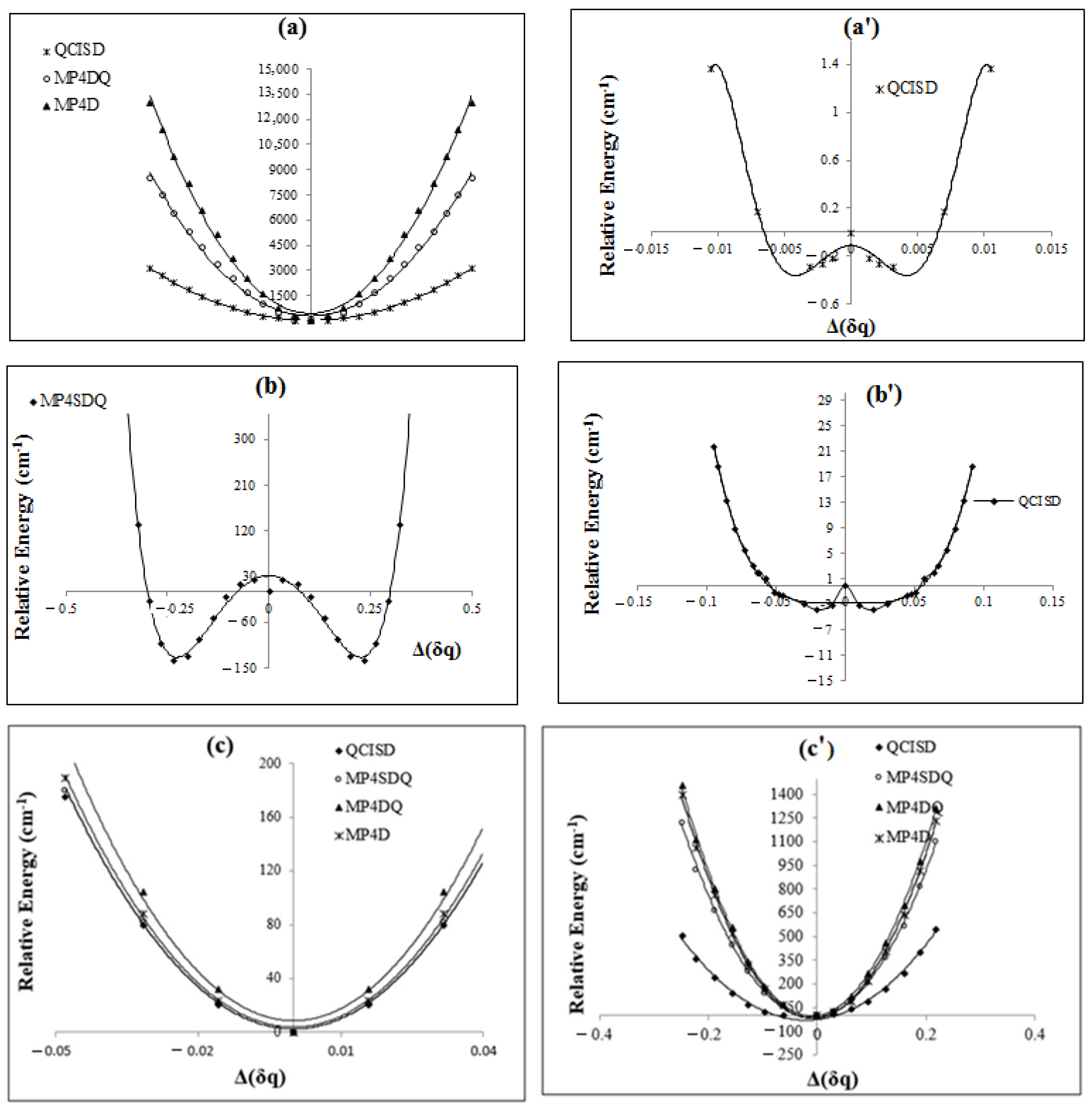
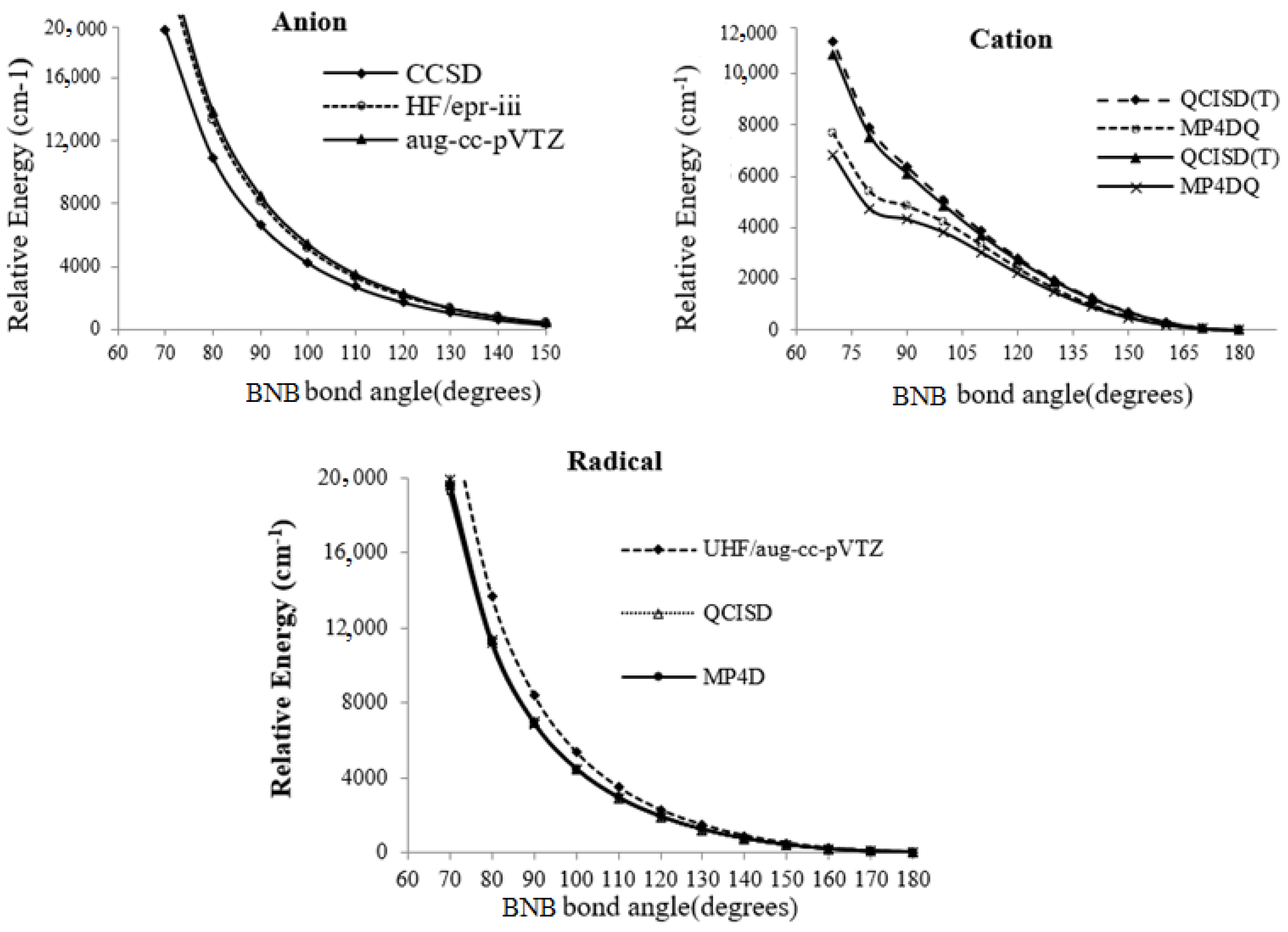


{kind=link}
{kind=link}
{kind=link}
| Basis Sets | Contraction Level | Contraction Scheme | N |
|---|---|---|---|
| 6-311+G(3df) | Original | (12s6p3d1f)/[5s4p3d1f] | 39 |
| uC-6-311+G(3df) | Core-uncontracted | (12s6p3d1f)/[10s4p3d1f] | 44 |
| u-6-311+G(3df) | Fully uncontracted | (12s6p3d1f)/[12s6p3d1f] | 52 |
| aug-cc-pVTZ | Original | (11s6p3d2f)/[5s4p3d2f] | 46 |
| uC-aug-cc-pVTZ | Core-uncontracted | (11s6p3d2f)/[11s4p3d2f] | 52 |
| u-aug-cc-pVTZ | Fully uncontracted | (11s6p3d2f)/[11s6p3d2f] | 58 |
| aug-cc-pVQZ | Original | (13s7p4d3f2g)/[6s5p4d3f2g] | 80 |
| uC-aug-cc-pVQZ | Core-uncontracted | (13s7p4d3f2g)/[13s5p4d3f2g] | 87 |
| u-aug-cc-pVQZ | Fully uncontracted | (13s7p4d3f2g)/[13s7p4d3f2g] | 93 |
| aug-cc-pV5Z | Original | (15s9p5d4f3g2h)/[7s6p5d4f3g2h] | 127 |
| uC-aug-cc-pV5Z | Core-uncontracted | (15s9p5d4f3g2h)/[15s6p5d4f3g2h] | 135 |
| u-aug-cc-pV5Z | Fully uncontracted | (15s9p5d4f3g2h)/[15s9p5d4f3g2h] | 144 |
| aug-cc-pV6Z | Original | (17s11p6d5f4g3h2i)/[8s7p6d5f4g3h2i] | 189 |
| uC-aug-cc-pV6Z | Core-uncontracted | (17s11p6d5f4g3h2i)/[17s7p6d5f4g3h2i] | 198 |
| u-aug-cc-pV6Z | Fully uncontracted | (17s11p6d5f4g3h2i)/[17s11p6d5f4g3h2i] | 210 |
| aug-cc-pCVTZ | Original | (13s8p4d2f)/[7s6p4d2f] | 59 |
| uC-aug-cc-pCVTZ | Core-uncontracted | (13s8p4d2f)/[13s6p4d2f] | 65 |
| u-aug-cc-pCVTZ | Fully uncontracted | (13s8p4d2f)/[13s8p4d2f] | 71 |
| aug-cc-pCVQZ | Original | (16s10p6d4f2g)/[9s8p6d4f2g] | 109 |
| uC-aug-cc-pCVQZ | Core-uncontracted | (16s10p6d4f2g)/[16s8p6d4f2g] | 116 |
| u-aug-cc-pCVQZ | Fully uncontracted | (16s10p6d4f2g)/[16s10p6d4f2g] | 122 |
| State Number of Electron | Isolated BNB | All Electron Configuration (Total Energy of | Electron Configuration Total Energy of | ||
|---|---|---|---|---|---|
( | |||||
(17e) | |||||
( (17e) | 1.3176 d 1.3176 d | ||||
( | |||||
(17e) | |||||
(17e) | |||||
(18e) | |||||
(18e) | |||||
(18e) | |||||
(18e) | [C] = | = | |||
(16e) | |||||
(16e) | = | ||||
(16e) |
| State of BNB | BN Bonds | force
from ECP | Charges from ESP Fitting | |
|---|---|---|---|---|
| Radical | ||||
Anion | ||||
Cation |
| State (* Ne) | Hybrids Coefficient& | Atomic Occupancies | |
|---|---|---|---|
(* 18e) | ** 0.107 ** 0.021 | ||
(* 18e) | ** 0.107 ** 0.064 | ||
(* 16e) | ** 0.119 ** 0.028 |
| Basis Set and Method | Orbital | |
|---|---|---|
| QCISD/EPR-III | ROHFSA | 7.34 |
| TD/EPR-III//QCISD/EPR-III | ROHFSB | 6.98 |
| TD/EPR-III//QCISD/EPR-III | ROHFSA | 7.12 |
| MP4D/EPR-II//QCISD/EPR-II | ROHFSB | 5.99 |
| QCISD/EPR-II | QRHF(+) | 6.42 |
| CASSCF(10,12)AUG-cc-pvqz | ROHFSA | 4.33 |
| CASSCF(11,12)/AUG-cc-pvqz | QRHF(−) | 4.45 |
| MP4SDQ/EPR-II//QCISD/EPR-II | QRHF(+) | 6.66 |
| MP4SDQ/EPR-III//QCISD/EPR-III | ROHFSB | 7.45 |
| m062x/EPR-II | ROHFSB | 8.33 |
| MP4D/EPR-III//QCISD/EPR-III | ROHFSB | 6.88 |
| MP4D/EPR-II//QCISD/EPR-II | ROHFSB | 7.21 |
| CBS4O | ROHFSA | 7.93 |
| CASSCF(11,12)/UHF | ROHFSB | 6.83 |
| TD/EPR-III//QCISD (T)/EPR-III | QRHF(−) | 7.19 |
| TD/EPR-II | QRHF(+) | 7.55 |
| QCISD(T)/EPR-III | ROHFSA | 7.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monajjemi, M.; Mollaamin, F.; Samiei Soofi, N. An Overview of Basis Set Effects for Diatomic Boron Nitride Compounds (B2N(∓,0)): A Quantum Symmetry Breaking. Quantum Rep. 2022, 4, 338-350. https://doi.org/10.3390/quantum4030024
Monajjemi M, Mollaamin F, Samiei Soofi N. An Overview of Basis Set Effects for Diatomic Boron Nitride Compounds (B2N(∓,0)): A Quantum Symmetry Breaking. Quantum Reports. 2022; 4(3):338-350. https://doi.org/10.3390/quantum4030024
Chicago/Turabian StyleMonajjemi, Majid, Fatemeh Mollaamin, and Neda Samiei Soofi. 2022. "An Overview of Basis Set Effects for Diatomic Boron Nitride Compounds (B2N(∓,0)): A Quantum Symmetry Breaking" Quantum Reports 4, no. 3: 338-350. https://doi.org/10.3390/quantum4030024