
An Experimental Approach to Assessing the Roles of Magnesium, Calcium, and Carbonate Ratios in Marine Carbonates


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of the Seed Material
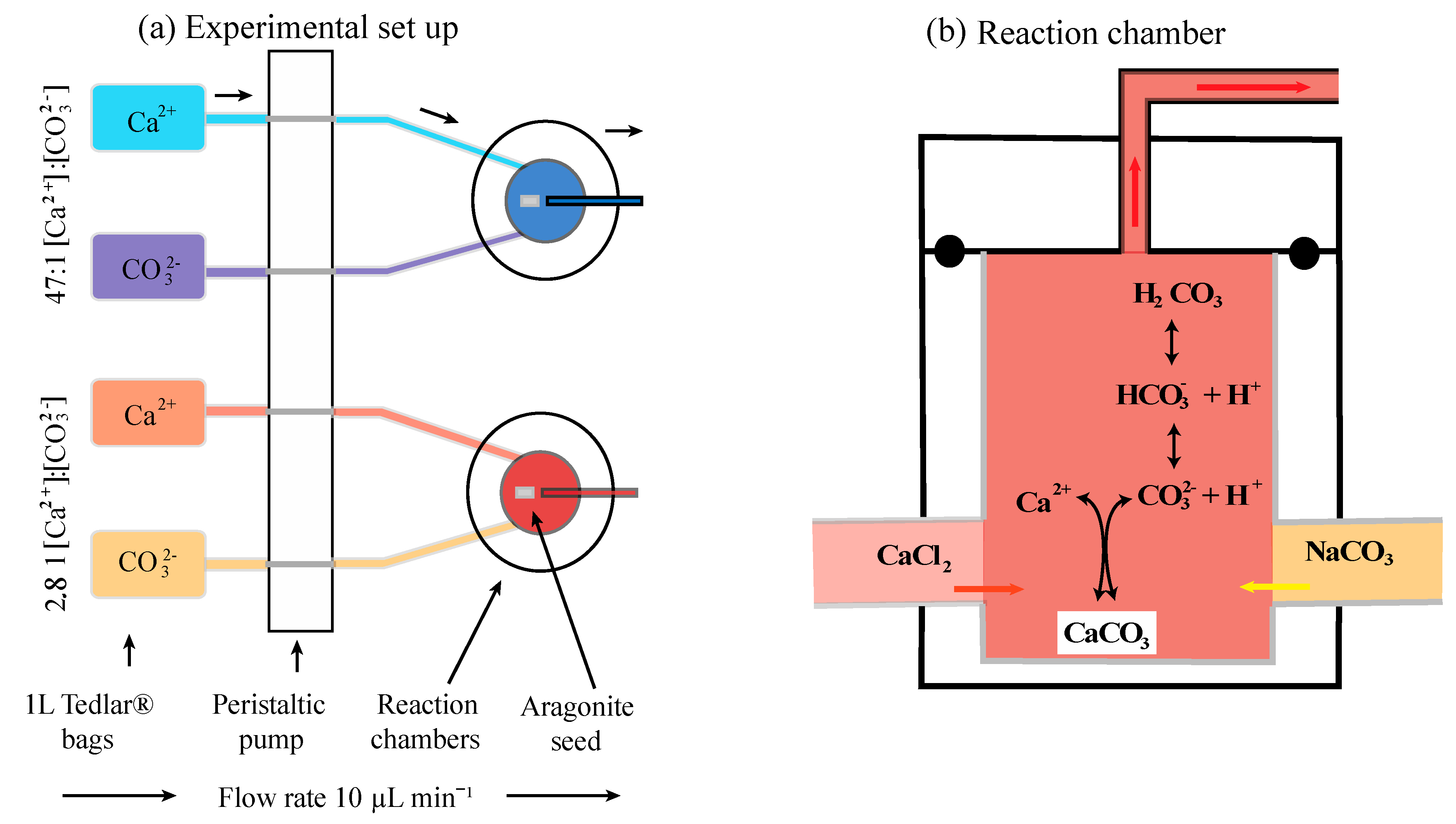
2.2. Experimental Setup
2.3. Preparation of Stock Solutions
3. Results
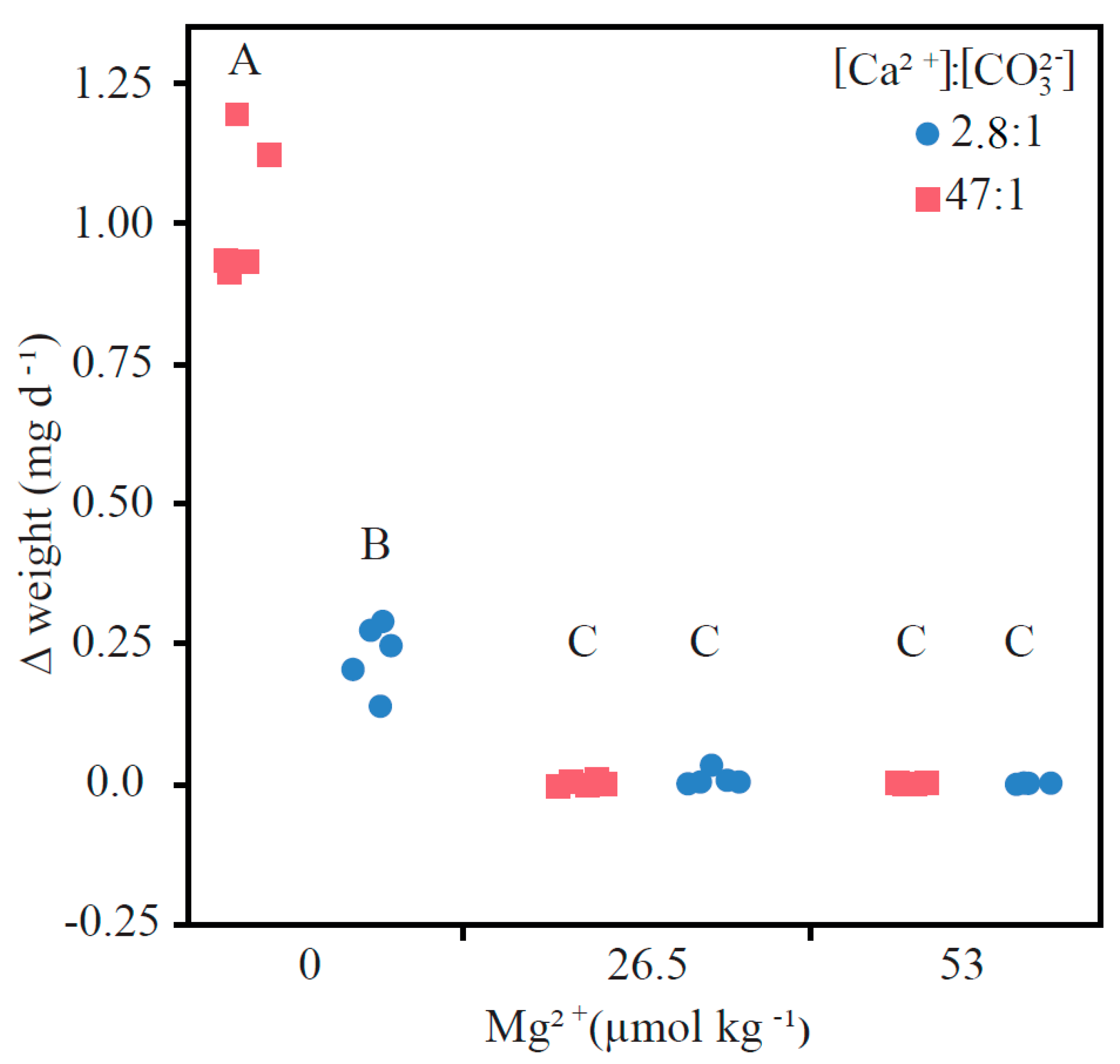
3.1. Precipitation Rates
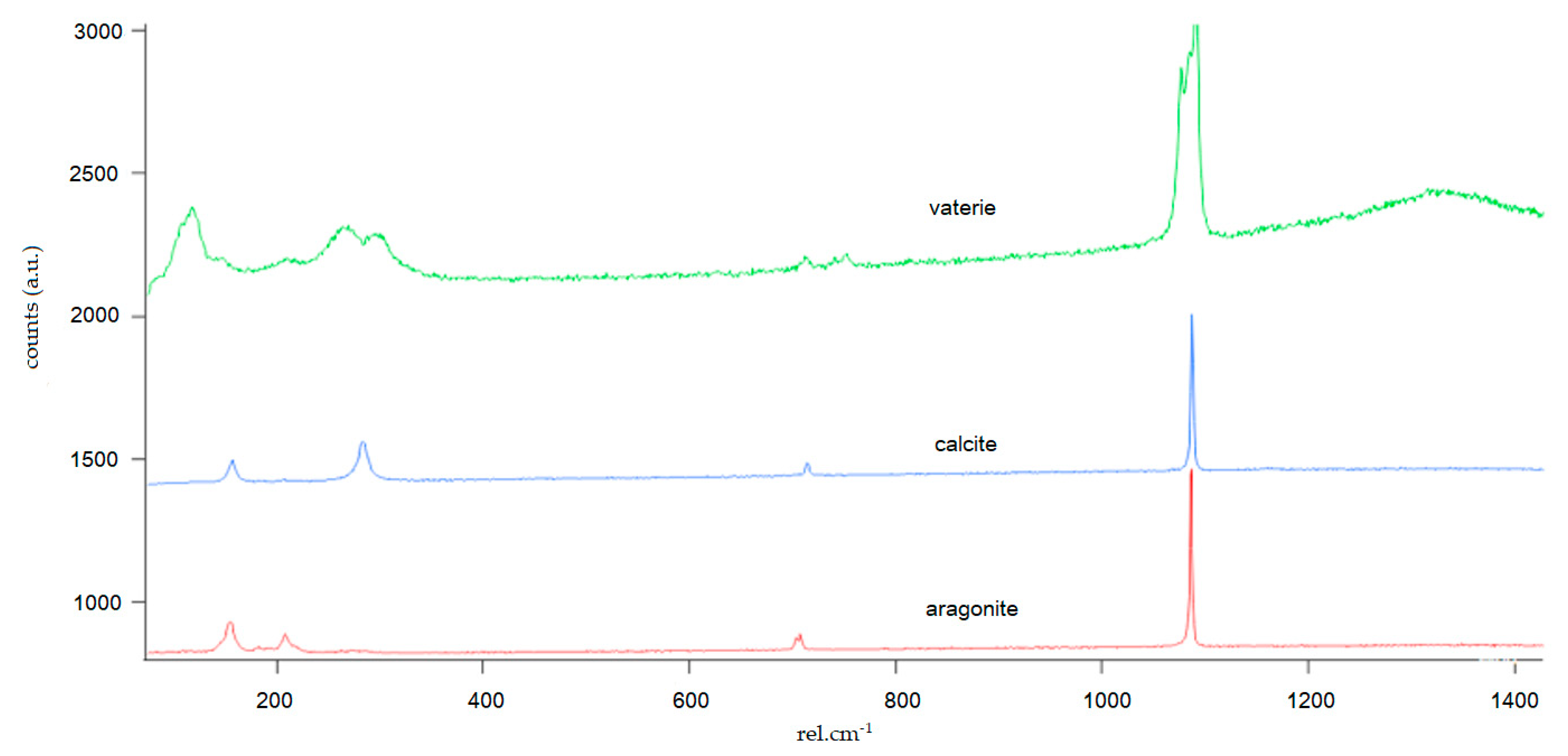
3.2. Mineralogy and Crystal Morphology
4. Discussion
4.1. Comparing Low and High Ca:CO3 Scenarios
4.2. The Connection between Mg and Calcification
4.3. Polynucleation and Spontaneous Nucleation
4.4. CaCO3 Polymorphs
4.5. Implications for Coral Reef Calcifiers
5. Conclusions
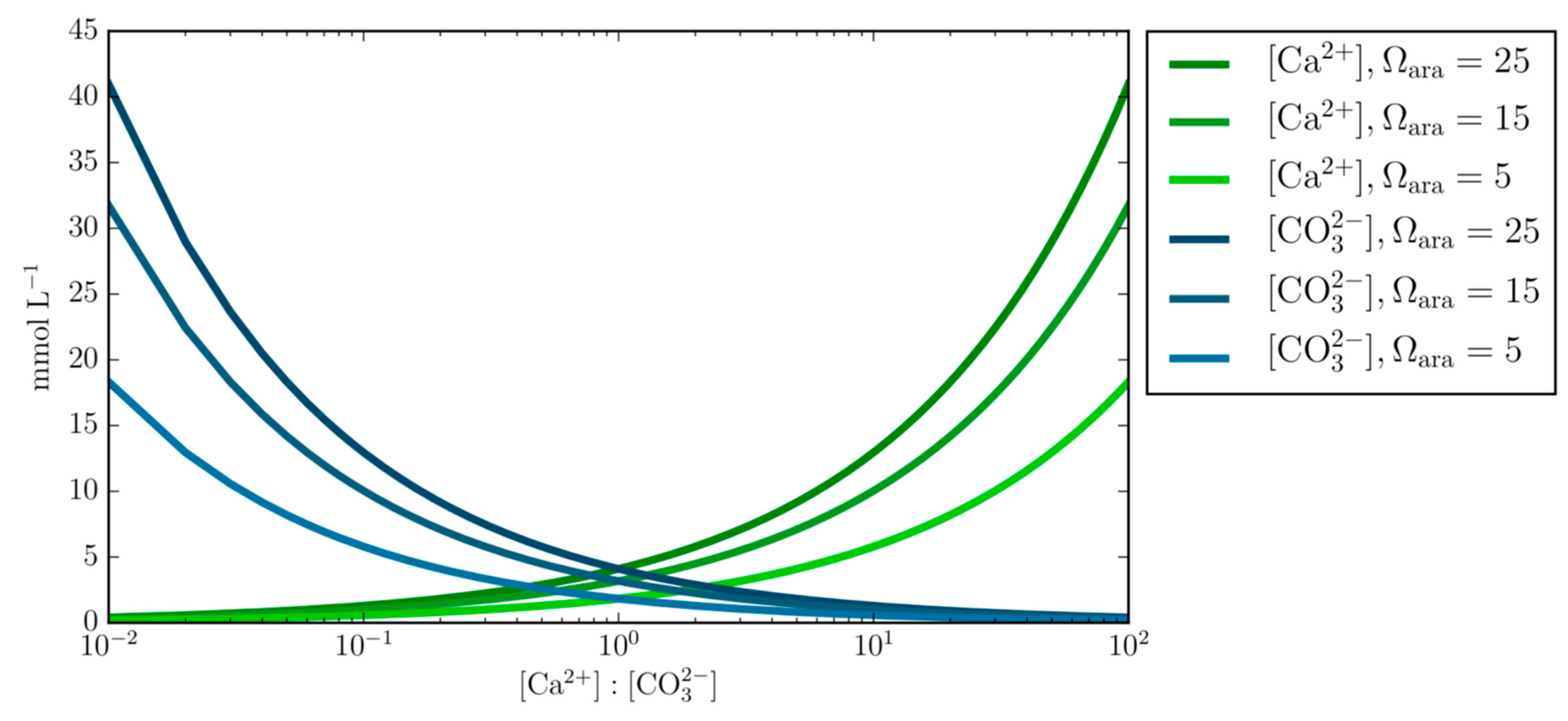
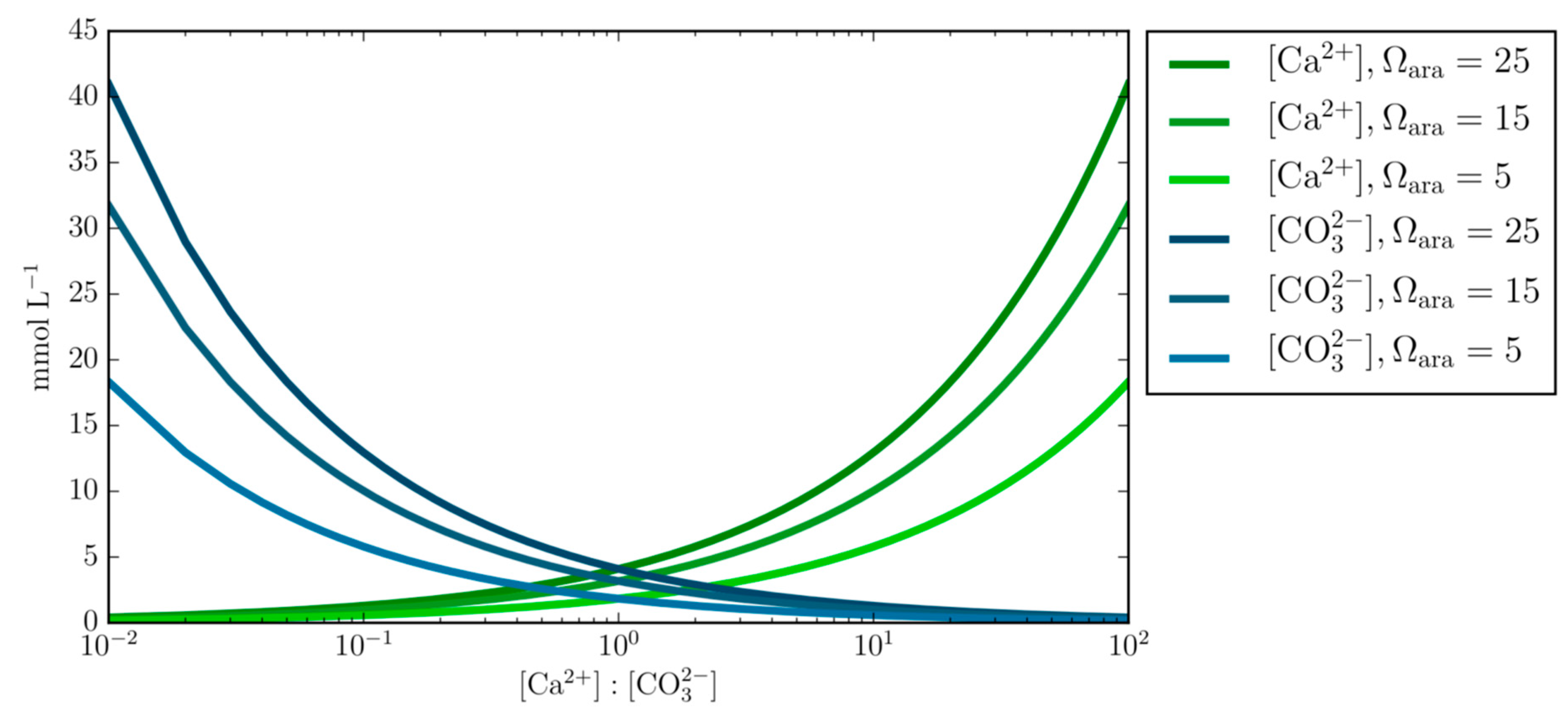
- Varying concentrations of calcium and carbonate ions at fixed Ωara demonstrates the underlining principal that calcium and carbonate ion concentrations can obtain the same Ωara value at different Ca:CO3 stoichiometry and questions the generalized applicability of the empirical equation that prescribes the calcification rate as a function of Ωara alone.
- As shown, calcifying fluid stoichiometry alters the precipitation rate and morphology of CaCO3 at a constant Ω and pH. Therefore, our findings suggest caution should be applied when inferring saturation state from the crystal morphology, particularly if other factors e.g., Mg, temperature, or DIC are not known.
- When comparing a strong proton removal scenario and a DIC concentrating mechanism to a scenario with ambient seawater pH and DIC conditions, calcite precipitation rates were three times greater in the ambient seawater conditions. Implying ambient seawater pH and DIC within the calcifying fluid is sufficient to induce calcification provided homeostasis is maintained.
- Mg exerts a stronger effect on the instability of CaCO3 than Ca:CO3 stoichiometry, in which Mg incorporation locally disturbs the coordination environment in the aragonite structure [87,103]. These differences emphasize the importance of Mg removal from the calcifying fluid. Future studies are recommended to additionally monitor the Mg concentration in the calcifying fluid along with the carbon chemistry.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. | Seed | t0 | te | D | DΔ |
|---|---|---|---|---|---|
| 1a | 1Mg-Free | 0.480 | 30.290 | 32 | 0.932 |
| 1a | 2Mg-Free | 0.657 | 36.500 | 32 | 1.120 |
| 1a | 3Mg-Free | 0.883 | 30.620 | 32 | 0.929 |
| 1a | 4Mg-Free | 1.057 | 30.220 | 32 | 0.911 |
| 1a | 5Mg-Free | 1.202 | 39.370 | 32 | 1.193 |
| 1b | 6Mg-Free | 0.591 | 9.320 | 32 | 0.273 |
| 1b | 7Mg-Free | 0.862 | 5.260 | 32 | 0.137 |
| 1b | 8Mg-Free | 0.983 | 7.480 | 32 | 0.203 |
| 1b | 9Mg-Free | 1.064 | 10.300 | 32 | 0.289 |
| 1b | 10Mg-Free | 1.218 | 9.070 | 32 | 0.245 |
| 2a | 1Mg | 0.212 | 0.310 | 70 | 0.001 |
| 2a | 2Mg | 0.715 | 0.570 | 70 | −0.002 |
| 2a | 3Mg | 0.777 | 0.649 | 70 | −0.002 |
| 2a | 4Mg | 0.924 | 0.718 | 70 | −0.003 |
| 2a | 5Mg | 1.013 | 1.006 | 70 | 0.000 |
| 2b | 6Mg | 0.423 | 0.322 | 70 | −0.001 |
| 2b | 7Mg | 0.766 | 13.003 | 70 | 0.175 |
| 2b | 8Mg | 0.818 | 0.793 | 70 | 0.000 |
| 2b | 9Mg | 0.972 | 0.944 | 70 | 0.000 |
| 2b | 10Mg | 1.336 | 1.381 | 70 | 0.001 |
| 3a | 1Mg + | 0.364 | 0.360 | 38 | 0.000 |
| 3a | 2Mg + | 0.816 | 0.643 | 38 | −0.005 |
| 3a | 3Mg + | 0.947 | 0.902 | 38 | −0.001 |
| 3a | 4Mg + | 1.150 | 1.146 | 38 | 0.000 |
| 3a | 5Mg + | 1.274 | 1.212 | 38 | −0.002 |
| 3b | 6Mg + | 0.668 | 0.534 | 38 | −0.004 |
| 3b | 7Mg + | 0.918 | 0.922 | 38 | 0.000 |
| 3b | 8Mg + | 1.069 | 1.064 | 38 | 0.000 |
| 3b | 9Mg + | 1.220 | 1.237 | 38 | 0.000 |
| 3b | 10Mg + | 1.449 | 1.485 | 38 | 0.001 |
| Exp. | Calcium Stock (mg/5 L) | Carbonate Stock (mg/5 L) | |||
|---|---|---|---|---|---|
| CaCl2 | MgCl2 | NaCl | NaHCO3 | NaCl | |
| 1a | 15.583 | 0.000 | 164.416 | 1.493 | 178.507 |
| 1b | 3.809 | 0.000 | 176.191 | 6.107 | 173.893 |
| 2a | 15.583 | 53.874 | 110.542 | 1.493 | 178.507 |
| 2b | 3.809 | 53.874 | 122.316 | 6.107 | 173.893 |
| 3a | 15.583 | 107.749 | 56.667 | 1.493 | 178.507 |
| 3b | 3.809 | 107.749 | 68.442 | 6.107 | 173.893 |

- Script A1. Below are the calculations used to modify the artificial seawater for the six experiments.
References
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: New York, NY, USA, 1989; p. 336. [Google Scholar]
- Spalding, M.D.; Ravilious, C.; Green, E.P. World Atlas of Coral Reefs; University of California Press: Berkeley, CA, USA, 2001; p. 424. [Google Scholar]
- Knowlton, N.; Brainard, R.E.; Fisher, R.; Moews, M.; Plaisance, L.; Caley, M.J. Coral Reef Biodiversity. In Life in the World’s Oceans: Diversity, Distribution, and Abundance; Wiley online Books; McIntyre, A.D., Ed.; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2010; pp. 65–78. [Google Scholar]
- Nystrom, M.; Folke, C.; Moberg, F. Coral reef disturbance and resilience in a human-dominated environment. Trends Ecol. Evol. 2000, 15, 413–417. [Google Scholar] [CrossRef]
- Teng, H.H.; Dove, P.M.; Orme, C.A.; de Yoreo, J.J. Thermodynamics of calcite growth: Baseline for understanding biomineral formation. Science 1998, 282, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Hoegh-Guldberg, O.; Mumby, P.J.; Hooten, A.J.; Steneck, R.S.; Greenfield, P.; Gomez, E.; Harvell, C.D.; Sale, P.F.; Edwards, A.J.; Caldeira, K.; et al. Coral Reefs Under Rapid Climate Change and Ocean Acidification. Science 2007, 318, 1737–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allemand, D.; Ferrierpages, C.; Furla, P.; Houlbreque, F.; Puverel, S.; Reynaud, S.; Tambutté, E.; Tambutté, S.; Zoccola, D. Biomineralisation in reef-building corals: From molecular mechanisms to environmental control. Comptes Rendus Palevol. 2004, 3, 453–467. [Google Scholar] [CrossRef]
- Tambutté, S.; Holcomb, M.; Ferrier-Pagés, C.; Reynaud, S.; Tambutté, É.; Zoccola, D.; Allemand, D. Coral biomineralization: From the gene to the environment. J. Exp. Mar. Bio Ecol. 2011, 408, 58–78. [Google Scholar] [CrossRef]
- Al-Horani, F.A.; Ferdelman, T.; Al-Moghrabi, S.M.; De Beer, D. Spatial distribution of calcification and photosynthesis in the scleractinian coral Galaxea fascicularis. Coral. Reefs. 2005, 24, 173–180. [Google Scholar] [CrossRef]
- Tambutté, E.; Allemand, D.; Zoccola, D.; Meibom, A.; Lotto, S.; Caminiti, N.; Segonds, N. Observations of the tissue-skeleton interface in the scleractinian coral Stylophora pistillata. Coral. Reefs. 2007, 26, 517–529. [Google Scholar]
- Al-Horani, F.A.; Al-Moghrabi, S.M.; De Beer, D. The mechanism of calcification and its relation to photosynthesis in the scleractinian coral Galaxea fascicularis. Mar. Biol. 2003, 142, 419–426. [Google Scholar] [CrossRef]
- Ries, J.B. A physicochemical framework for interpreting the biological calcification response to CO2 induced ocean acidification. Geochim. Cosmochim. Acta. 2011, 75, 4053–4064. [Google Scholar] [CrossRef]
- Furla, P.; Allemand, D.; Orsenigo, M.N. Involvement of H+ -ATPase and carbonic anhydrase in inorganic carbon uptake for endosymbiont photosynthesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R870–R881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohn, S.; Merico, A. Modelling coral polyp calcification in relation to ocean acidification. Biogeosciences 2012, 9, 4441–4454. [Google Scholar] [CrossRef] [Green Version]
- Venn, A.A.; Tambutté, E.; Holcomb, M.; Laurent, J.; Allemand, D.; Tambutté, S. Impact of seawater acidification on pH at the tissue–skeleton interface and calcification in reef corals. Proc. Natl. Acad. Sci. USA 2013, 110, 1634–1639. [Google Scholar] [CrossRef] [Green Version]
- Fabricius, K.E.; De’ath, G.; Noonan, S.; Uthicke, S. Ecological effects of ocean acidification and habitat complexity on reef-associated macroinvertebrate communities. Proc. R. Soc. B Biol. Sci. 2014, 81, 2013479. [Google Scholar] [CrossRef] [Green Version]
- Fabricius, K.E.; Langdon, C.; Uthicke, S.; Humphrey, C.; Noonan, S.; De ’ath, G.; Okazaki, R.; Muehllehner, N.; Glas, M.S.; Lough, J.M. Losers and winners in coral reefs acclimatized to elevated carbon dioxide concentrations. Nat. Clim. Chang. 2011, 1, 165–169. [Google Scholar] [CrossRef]
- Muscatine, L.; Tambutte, E.; Allemand, D. Morphology of coral desmocytes, cells that anchor the calicoblastic epithelium to the skeleton. Coral Reefs. 1997, 16, 205–213. [Google Scholar] [CrossRef]
- Mass, T.; Drake, J.L.; Peters, E.C.; Jiang, W.; Falkowski, P.G. Immunolocalization of skeletal matrix proteins in tissue and mineral of the coral Stylophora pistillata. Proc. Natl. Acad. Sci. USA 2014, 111, 12728–12733. [Google Scholar] [CrossRef] [Green Version]
- Cuif, J.-P.; Dauphin, Y. The Environment Recording Unit in coral skeletons—a synthesis of structural and chemical evidences for a biochemically driven, stepping-growth process in fibres. Biogeosciences 2005, 2, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, F. Sedimentary Carbonate Minerals; Springer: Berlin/Heidelberg, Germany, 1973; p. 228. [Google Scholar]
- Lasaga, A.C. Kinetic Theory in the Earth Sciences; Princeton University Press: Princeton, NJ, USA, 2014; p. 822. [Google Scholar]
- Burton, E.A.; Walter, L.M. Relative precipitation rates of aragonite and Mg calcite from seawater: Temperature or carbonate ion control? Geology 1987, 15, 111–114. [Google Scholar] [CrossRef]
- Burton, E.A.; Walter, L.M. The role of pH in phosphate inhibition of calcite and aragonite precipitation rates in seawater. Geochim. Cosmochim. Acta 1990, 54, 797–808. [Google Scholar] [CrossRef]
- Zuddas, P.; Mucci, A. Kinetics of calcite precipitation from seawater: I. A classical chemical kinetics description for strong electrolyte solutions. Geochim. Cosmochim. Acta 1994, 58, 4353–4362. [Google Scholar] [CrossRef]
- Reddy, M.M.; Nancollas, G.H. The crystallization of calcium carbonate. IV. The effect of magnesium, strontium and sulfate ions. J. Cryst. Growth 1976, 35, 33–38. [Google Scholar] [CrossRef]
- Gattuso, J.P.; Frankignoulle, M.; Bourge, I.; Romaine, S.; Buddemeier, R.W. Effect of calcium carbonate saturation of seawater on coral calcification. Glob. Planet Chang. 1998, 18, 37–46. [Google Scholar] [CrossRef]
- McCulloch, M.; Falter, J.; Trotter, J.; Montagna, P. Coral resilience to ocean acidification and global warming through pH up-regulation. Nat. Clim. Chang. 2012, 2, 623–627. [Google Scholar] [CrossRef]
- Hohn, S.; Merico, A. Quantifying the relative importance of transcellular and paracellular ion transports to coral polyp calcification. Front Earth Sci. 2015, 2, 1–11. [Google Scholar] [CrossRef]
- Holtz, L.M.; Wolf-Gladrow, D.; Thoms, S. Simulating the effects of light intensity and carbonate system composition on particulate organic and inorganic carbon production in Emiliania huxleyi. J. Theor. Biol. 2015, 372, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Nehrke, G.; Reichart, G.J.; Van Cappellen, P.; Meile, C.; Bijma, J. Dependence of calcite growth rate and Sr partitioning on solution stoichiometry: Non-Kossel crystal growth. Geochim. Cosmochim. Acta 2007, 71, 2240–2249. [Google Scholar] [CrossRef] [Green Version]
- Hartley, G.; Mucci, A. The influence of PCO2 on the partitioning of magnesium in calcite overgrowth precipitated from artificial seawater at 25° and 1 atm total pressure. Geochim. Cosmochim. Acta 1996, 60, 315–324. [Google Scholar] [CrossRef]
- Ries, J.B. Aragonitic Algae in Calcite Seas: Effect of Seawater Mg/Ca Ratio on Algal Sediment Production. J. Sediment Res. 2006, 76, 515–523. [Google Scholar] [CrossRef]
- Ries, J.B.; Stanley, S.M.; Hardie, L.A. Scleractinian corals produce calcite, and grow more slowly, in artificial Cretaceous seawater. Geology 2006, 34, 525–528. [Google Scholar] [CrossRef]
- Kim, Y.-Y.; Ganesan, K.; Yang, P.; Kulak, A.N.; Borukhin, S.; Pechook, S.; Ribeiro, L.; Kröger, R.; Eichhorn, S.J.; Armes, S.P.; et al. An artificial biomineral formed by incorporation of copolymer micelles in calcite crystals. Nat. Mater. 2011, 10, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Hohn, S.; Reymond, C.E. Coral calcification, mucus, and the origin of skeletal organic molecules. Coral Reefs. 2019, 38, 973–984. [Google Scholar] [CrossRef]
- Cai, W.-J.; Ma, Y.; Hopkinson, B.M.; Grottoli, A.G.; Warner, M.E.; Ding, Q.; Hu, X.; Yuan, X.; Schoepf, V.; Xu, H.; et al. Microelectrode characterization of coral daytime interior pH and carbonate chemistry. Nat. Commun. 2016, 4, 11144. [Google Scholar] [CrossRef] [Green Version]
- Raybaud, V.; Tambutté, S.; Ferrier-Pagès, C.; Reynaud, S.; Venn, A.A.; Tambutté, É.; Nival, P.; Allemand, D. Computing the carbonate chemistry of the coral calcifying medium and its response to ocean acidification. J. Theor. Biol. 2017, 424, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevilgen, D.S.; Venn, A.A.; Hu, M.Y.; Tambutté, E.; de Beer, D.; Planas-Bielsa, V.; Tambutté, S. Full in vivo characterization of carbonate chemistry at the site of calcification in corals. Sci. Adv. 2019, 5, 7447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf-Gladrow, D.A.; Zeebe, R.E.; Klaas, C.; Körtzinger, A.; Dickson, A.G. Total alkalinity: The explicit conservative expression and its application to biogeochemical processes. Mar. Chem. 2007, 106, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Millero, F.; Huang, F.; Graham, T.; Pierrot, D. The dissociation of carbonic acid in NaCl solutions as a function of concentration and temperature. Geochim. Cosmochim. Acta 2007, 71, 46–55. [Google Scholar] [CrossRef]
- Schleinkofer, N.; Raddatz, J.; Freiwald, A.; David Evans, D.; Beuck, L.; Rüggeberg, A.; Liebetrau, V. Environmental and biological controls on Na/Ca ratios in scleractinian cold-water corals. Biogeosciences 2019, 16, 3565–3582. [Google Scholar] [CrossRef] [Green Version]
- DeCarlo, T.M.; Comeau, S.; Cornwall, C.E.; McCulloch, M.T. Coral resistance to ocean acidification linked to increased calcium at the site of calcification. Proc. R. Soc. B Biol. Sci. 2018, 285, 1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, N.; Cohen, I.; Finch, A.A.; Erez, J.; Tudhope, A.W. Corals concentrate dissolved inorganic carbon to facilitate calcification. Nat. Commun. 2014, 5, 5741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Nooijer, L.J.; Spero, H.J.; Erez, J.; Bijma, J.; Reichart, G.J. Biomineralization in perforate Foraminifera. Earth Sci. Rev. 2014, 135, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Venn, A.A.; Tambutté, E.; Holcomb, M.; Allemand, D.; Tambutté, S. Live tissue imaging shows reef corals elevate pH under their calcifying tissue relative to seawater. PLoS ONE 2011, 6, e20013.65. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable prenucleation calcium carbonate clusters. Science 2009, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihli, J.; Clark, J.N.; Kanwal, N.; Kim, Y.-Y.; Holden, M.A.; Harder, R.J.; Tang, C.C.; Ashbrook, S.E.; Robinson, I.K.; Meldrum, F.C. Visualization of the effect of additives on the nanostructures of individual bio-inspired calcite crystals. Chem. Sci. 2019, 10, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Plummer, L.N.; Busenberg, E. The solubilities of calcite, aragonite and vaterite in CO2-H2O solutions between 0 and 90 °C, and an evaluation of the aqueous model for the system CaCO3-CO2-H2O. Geochim. Cosmochim. Acta 1982, 46, 1011–1040. [Google Scholar] [CrossRef]
- De Visscher, A.; Vanderdeelen, J. Estimation of the Solubility Constant of Calcite, Aragonite, and Vaterite at 25°C Based on Primary Data Using the Pitzer Ion Interaction Approach. Monatshefte fur Chemie 2003, 134, 769–775. [Google Scholar] [CrossRef]
- Zhong, S.; Mucci, A. Calcite precipitation in seawater using a constant addition technique: A new overall reaction kinetic expression. Geochim. Cosmochim. Acta 1993, 57, 1409–1417. [Google Scholar]
- Allison, N.; Finch, A.A. δ11B, Sr, Mg and B in a modern Porites coral: The relationship between calcification site pH and skeletal chemistry. Geochim. Cosmochim. Acta 2010, 74, 1790–1800. [Google Scholar] [CrossRef]
- Opdyke, B.N.; Wilkinson, B.H. Paleolatitude distribution of Phanerozoic marine ooids and cements. Palaeogeogr Palaeoclim Palaeoecol 1990, 78, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Morse, J.W.; Wang, Q.; Tsio, M.Y. Influences of temperature and Mg: Ca ratio on CaCO3 precipitates from seawater. Geology 1997, 25, 85–87. [Google Scholar] [CrossRef]
- Higuchi, T.; Fujimura, H.; Yuyama, I.; Harii, S.; Agostini, S.; Oomori, T. Biotic control of skeletal growth by scleractinian corals in aragonite–calcite seas. PLoS ONE 2014, 9, e91021. [Google Scholar] [CrossRef] [Green Version]
- Boon, M.; Rickard, W.D.; Rohl, A.L.; Jones, F. Stabilization of Aragonite: Role of Mg2+ and Other Impurity Ions. Cryst. Growth Des. 2020, 20, 5006–5017. [Google Scholar] [CrossRef]
- Bracco, J.N.; Grantham, M.C.; Stack, A.G. Calcite Growth Rates as a Function of Aqueous Calcium-to-Carbonate Ratio, Saturation Index, and Inhibitor Concentration: Insight into the Mechanism of Reaction and Poisoning by Strontium. Cryst. Growth Des. 2012, 12, 3540–3548. [Google Scholar] [CrossRef]
- Gebrehiwet, T.A.; Redden, G.D.; Fujita, Y.; Beig, M.S.; Smith, R.W. The Effect of the CO32− to Ca2+ Ion activity ratio on calcite precipitation kinetics and Sr2+ partitioning. Geochem. Trans. 2012, 13, 1–11. [Google Scholar] [CrossRef]
- Gaetani, G.A.; Cohen, A.L. Element partitioning during precipitation of aragonite from seawater: A framework for understanding paleoproxies. Geochim. Cosmochim. Acta 2006, 70, 4617–4634. [Google Scholar] [CrossRef]
- Holcomb, M.; Cohen, A.L.; Gabitov, R.; Hutter, J. Compositional and morphological features of aragonite precipitated experimentally from seawater and biogenically by corals. Geochim. Cosmochim. Acta 2009, 73, 166–179. [Google Scholar] [CrossRef]
- Adkins, J.F.; Boyle, E.A.; Curry, W.B.; Lutringer, A. Stable isotopes in deep-sea corals and a new mechanism for “vital effects”. Geochim. Cosmochim. Acta 2003, 67, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Rollion-Bard, C.; Chaussidon, M.; France-Lanord, C. pH control on oxygen isotopic composition of symbiotic corals. Earth Planet Sci. Lett. 2003, 215, 275–288. [Google Scholar] [CrossRef]
- Watson, E.B. A conceptual model for near-surface kinetic controls on the trace- element and stable isotope composition of abiogenic calcite crystals. Geochim. Cosmochim. Acta 2004, 68, 1473–1488. [Google Scholar] [CrossRef]
- Boistelle, R.; Astier, J.P. Crystallization mechanisms in solution. J. Cryst. Growth. 1988, 90, 14–30. [Google Scholar] [CrossRef]
- Davey, R.J.; Schroeder, S.L.M.; Ter Horst, J.H. Nucleation of organic crystals—A molecular perspective. Angew Chemie Int. Ed. 2013, 52, 2167–2179. [Google Scholar] [CrossRef]
- Mann, S. Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry; Oxford University Press: Oxford, UK, 2001; p. 210. [Google Scholar]
- Gebauer, D.; Kellermeier, M.; Gale, J.D.; Bergström, L.; Cölfen, H. Pre-nucleation clusters as solute precursors in crystallisation. Chem. Soc. Rev. 2014, 43, 2348–2371. [Google Scholar] [CrossRef] [Green Version]
- Bonucci, E. Role of collagen fibrils in calcification. In Calcification in Biological Systems; Bonucci, E., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 19–39. [Google Scholar]
- Rodriguez-Blanco, J.D.; Shaw, S.; Benning, L.G. The kinetics and mechanisms of amorphous calcium carbonate [ACC] crystallization to calcite, viavaterite. Nanoscale 2011, 3, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Wray, J.; Daniels, F. Precipitation of calcite and aragonite. J. Am. Chem. Soc. 1957, 79, 2031–2034. [Google Scholar] [CrossRef]
- Ogino, T.; Suzuki, T.; Sawada, K. The formation and transformation mechanism of calcium carbonate in water. Geochemica Cosmochem Acta. 1987, 51, 2757–2767. [Google Scholar] [CrossRef]
- Sun, W.; Jayaraman, S.; Chen, W.; Persson, K.A.; Ceder, G. Nucleation of metastable aragonite CaCO3 in seawater. Proc Natl. Acad. Sci. USA 2015, 112, 3199–3204. [Google Scholar] [CrossRef] [Green Version]
- Cyronak, T.; Schulz, K.G.; Jokiel, P.L. The Omega myth: What really drives lower calcification rates in an acidifying ocean. ICES J. Mar. Sci. 2016, 73, 558–562. [Google Scholar] [CrossRef] [Green Version]
- Allemand, D.; Tambutte, E.; Girard, J.; Jaubert, J.; Tambutté, E.; Girard, J.; Jaubert, J. Organic matrix synthesis in the scleractinian coral Stylophora pistillata: Role in biomineralization and poten- tial target of the organotin tributyltin. J. Exp. Biol. 1998, 201, 2001–2009. [Google Scholar] [PubMed]
- Watanabe, T.; Fukuda, I.; China, K.; Isa, Y. Molecular analyses of protein components of the organic matrix in the exoskeleton of two scleractinian coral species. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2003, 136, 767–774. [Google Scholar] [CrossRef]
- Helman, Y.; Natale, F.; Sherrell, R.M.; LaVigne, M.; Starovoytov, V.; Gorbunov, M.Y.; Falkowski, P.G. Extracellular matrix production and calcium carbonate precipitation by coral cells in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mass, T.; Drake, J.L.; Haramaty, L.; Kim, J.D.; Zelzion, E.; Bhattacharya, D.; Falkowski, P.G. Cloning and characterization of four novel coral acid-rich proteins that precipitate carbonates in vitro. Curr. Biol. 2003, 23, 1126–1131. [Google Scholar] [CrossRef] [Green Version]
- Kretsinger, R.H. Calcium-Binding Proteins. Ann. Rev. Biochem. 1976, 45, 239–266. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. Intracellular Calcium Homeostasis. Ann. Rev. Biochem. 1987, 56, 395–433. [Google Scholar] [CrossRef]
- DeCarlo, T.M.; Ren, H.; Farfan, G.A. The Origin and Role of Organic Matrix in Coral Calcification: Insights from Comparing Coral Skeleton and Abiogenic Aragonite. Front Mar. Sci. 2018, 5, 170. [Google Scholar] [CrossRef] [Green Version]
- Marin, F.; Smith, M.; Isa, Y.; Muyzer, G.; Westbroek, P. Skeletal matrices, muci, and the origin of invertebrate calcification. Proc. Natl. Acad. Sci. USA 1996, 93, 1554–1559. [Google Scholar] [CrossRef] [Green Version]
- Westbroek, P.; Marin, F. A marriage of bone and nacre. Nature 1998, 392, 861–862. [Google Scholar] [CrossRef] [PubMed]
- Kawano, J.; Sakuma, H.; Nagai, T. Incorporation of Mg2+ in surface Ca2+ sites of aragonite: An ab initio study. Prog. Earth Planet Sci. 2015, 2, 7. [Google Scholar] [CrossRef]
- Cohen, A.L.; McConnaughey, T.A. Geochemical Perspectives on Coral Mineralization. Rev. Mineral. Geochem. 1990, 4, 151–187. [Google Scholar]
- DeCarlo, T.M.; D’Olivo, J.P.; Foster, T.; Holcomb, M.; Becker, T.; McCulloch, M.T. Coral calcifying fluid aragonite saturation states derived from Raman spectroscopy. Biogeosciences 2017, 14, 5253–5269. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, M.; Trotter, J.; Montagna, P.; Falter, J.; Dunbar, R.; Freiwald, A.; Försterra, G.; López Correa, M.; Maier, C.; Rüggeberg, A.; et al. Resilience of cold-water scleractinian corals to ocean acidification: Boron isotopic systematics of pH and saturation state up-regulation. Geochim. Cosmochim. Acta. 2012, 87, 21–34. [Google Scholar] [CrossRef]
- Farfan, G.A.; Cordes, E.E.; Waller, R.G.; DeCarlo, T.M.; Hansel, C.M. Mineralogy of Deep-Sea Coral Aragonites as a Function of Aragonite Saturation State. Front. Mar. Sci. 2018, 5, 473. [Google Scholar] [CrossRef] [Green Version]
- Georgiou, L.; Falter, J.; Trotter, J.; Kline, D.I.; Holcomb, M.; Dove, S.G.; Hoegh-Guldberg, O.; Mcculloch, M. pH homeostasis during coral calcification in a free ocean CO2 enrichment [FOCE] experiment, Heron Island reef flat, Great Barrier Reef. Proc. Natl. Acad. Sci. USA 2015, 112, 13219–13224. [Google Scholar] [CrossRef] [Green Version]
- Mitsuguchi, T.; Matsumoto, E.; Abe, O.; Uchida, T.; Isdale, P.J. Mg/Ca Thermometry in Coral Skeletons. Science. 1996, 274, 961–963. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, G.A.; Cohen, A.L.; Wang, Z.; Crusius, J. A biomineralization approach to developing climate proxies. Geochim. Cosmochim. Acta 2011, 75, 1920–1932. [Google Scholar] [CrossRef]
- Wei, G.; Sun, M.; Li, X.; Nie, B. Mg/Ca, Sr/Ca and U/Ca ratios of a porites coral from Sanya Bay, Hainan Island, South China Sea and their relationships to sea surface temperature. Palaeogeogr Palaeoclimatol Palaeoecol. 2000, 162, 59–74. [Google Scholar] [CrossRef]
- Marchitto, T.M.; Bryan, S.P.; Doss, W.; McCulloch, M.T.; Montagna, P. A simple biomineralization model to explain Li, Mg, and Sr incorporation into aragonitic foraminifera and corals. Earth Planet Sci. Lett. 2018, 481, 20–29. [Google Scholar] [CrossRef] [Green Version]
- De Choudens-Sánchez, V.; González, L.A. Calcite and aragonite precipitation under controlled instantaneous supersaturation: Elucidating the role of CaCO3 saturation state and Mg/Ca ratio on calcium carbonate polymorphism. J. Sediment. Res. 2009, 79, 363–376. [Google Scholar] [CrossRef]
- Falini, G.; Fermani, S.; Goffredo, S. Seminars in Cell & Developmental Biology Coral biomineralization: A focus on intra-skeletal organic matrix and calcification. Semin. Cell Dev. Biol. 2015, 46, 17–26. [Google Scholar] [PubMed]
- Politi, Y.; Arad, T.; Klein, E.; Weiner, S.; Addadi, L. Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase. Science 2004, 306, 1161–1164. [Google Scholar] [CrossRef]
- Mass, T.; Giuffre, A.J.; Sun, C.-Y.; Stifler, C.A.; Frazier, M.J.; Neder, M.; Tamura, N.; Stan, C.V.; Marcus, M.A.; Gilbert, P.U.P.A. Amorphous calcium carbonate particles form coral skeletons. Proc. Natl. Acad. Sci. USA 2017, 114, E7670–E7678. [Google Scholar] [CrossRef] [Green Version]
- Checa, A.G.; Macías-Sánchez, E.; Rodríguez-Navarro, A.B.; Sánchez-Navas, A.; Lagos, N.A. Origin of the biphase nature and surface roughness of biogenic calcite secreted by the giant barnacle Austromegabalanus psittacus. Sci. Rep. 2020, 10, 16784. [Google Scholar] [CrossRef]
- Politi, Y.; Metzler, R.A.; Abrecht, M.; Gilbert, B.; Wilt, F.H.; Sagi, I.; Addadi, L.; Weiner, S.; Gilbert, P.U.P.A. Transformation mechanism of amorphous calcium carbonate into calcite in sea urchin larval spicule. Proc. Natl. Acad. Sci. USA 2008, 105, 17362–17366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, M.C.; Diaz-Pulido, G.; Harvey, A.S.; Adey, W. Coralline algal calcification: A morphological and process-based understanding. PLoS ONE 2019, 14, e0221396. [Google Scholar] [CrossRef] [Green Version]
- Jacob, D.E.; Wirth, R.; Agbaje, O.B.A.; Branson, O.; Eggins, S.M. Planktic foraminifera form their shells via metastable carbonate phases. Nat. Commun. 2017, 8, 1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, S.; Cornwall, C.E.; DeCarlo, T.M.; Doo, S.S.; Carpenter, R.C.; McCulloch, M.T. Resistance to ocean acidification in coral reef taxa is not gained by acclimatization. Nat. Clim. Chang. 2019, 9, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Guillermic, M.; Cameron, L.P.; De Corte, I.; Misra, S.; Bijma, J.; De Beer, D.; Reymond, C.E.; Westphal, H.; Ries, J.B.; Robert, A.; et al. Thermal stress reduces pocilloporid coral resilience to ocean acidification by impairing control over calcifying fluid chemistry. Sci. Adv. 2021, 7, eaba9958. [Google Scholar] [CrossRef]
- Soldati, A.L.; Jacob, D.; Glatzel, P.; Swarbrick, J.C.; Geck, J. Element substitution by living organisms: The case of manganese in mollusc shell aragonite. Sci. Rep. 2016, 6, 22514. [Google Scholar] [CrossRef] [PubMed] [Green Version]



| Exp. | Ca2+:CO32− | Mg2+:Ca2+ | Ca2+ | CO32− | Mg2+ | Ωara | pH | DIC | TA |
|---|---|---|---|---|---|---|---|---|---|
| mol:mol | mol:mol | mM | µM | mM | µM | µM | |||
| 1a | 47 | 0 | 10.6 | 226 | 0 | 10 | 8.7 | 1777 | 2440 |
| 1b | 2.8 | 0 | 2.6 | 926 | 0 | 10 | 8.7 | 7270 | 9885 |
| 2a | 47 | 2.5 | 10.6 | 226 | 26.5 | 10 | 8.7 | 1777 | 2440 |
| 2b | 2.8 | 10.2 | 2.6 | 926 | 26.5 | 10 | 8.7 | 7270 | 9885 |
| 3a | 47 | 5 | 10.6 | 226 | 53 | 10 | 8.7 | 1777 | 2440 |
| 3b | 2.8 | 20.4 | 2.6 | 926 | 53 | 10 | 8.7 | 7270 | 9885 |
| Source | DF | SS | MS | F-Ratio | p > F |
|---|---|---|---|---|---|
| Model | 5 | 4.142 | 0.828 | 240.390 | <0.001 |
| Mg2+ | 2 | 2.592 | 375.980 | <0.001 | |
| Ca2+:CO32− | 1 | 0.510 | 148.143 | <0.001 | |
| Mg2+ * Ca2+:CO32− | 2 | 1.040 | 150.924 | <0.001 | |
| Error | 24 | 0.083 | 0.003 | ||
| Total Error | 29 | 4.224 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reymond, C.E.; Hohn, S. An Experimental Approach to Assessing the Roles of Magnesium, Calcium, and Carbonate Ratios in Marine Carbonates. Oceans 2021, 2, 193-214. https://doi.org/10.3390/oceans2010012
Reymond CE, Hohn S. An Experimental Approach to Assessing the Roles of Magnesium, Calcium, and Carbonate Ratios in Marine Carbonates. Oceans. 2021; 2(1):193-214. https://doi.org/10.3390/oceans2010012
Chicago/Turabian StyleReymond, Claire E., and Sönke Hohn. 2021. "An Experimental Approach to Assessing the Roles of Magnesium, Calcium, and Carbonate Ratios in Marine Carbonates" Oceans 2, no. 1: 193-214. https://doi.org/10.3390/oceans2010012
APA StyleReymond, C. E., & Hohn, S. (2021). An Experimental Approach to Assessing the Roles of Magnesium, Calcium, and Carbonate Ratios in Marine Carbonates. Oceans, 2(1), 193-214. https://doi.org/10.3390/oceans2010012