Ultrathin Film PtxPd(1-x) Alloy Catalysts for Formic Acid Oxidation Synthesized by Surface Limited Redox Replacement of Underpotentially Deposited H Monolayer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Electrochemical Cell Set-Up
2.2. Electrode Preparation
2.3. Characterization of Au Electrodes before Use
2.4. PtxPd(1-x) Alloy Ultrathin Film Growth:
2.5. Electrochemical Testing and Characterization
2.6. Compositional Analysis
2.7. Formic Acid Oxidation Testing
3. Results and Discussion
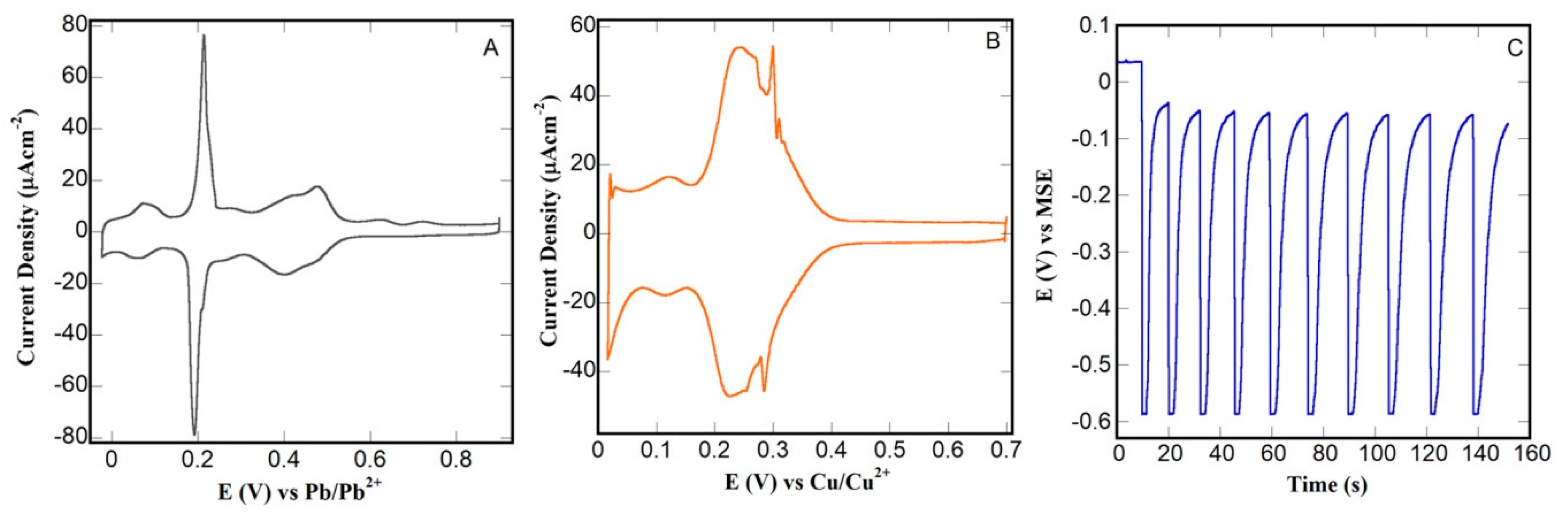
3.1. Catalyst Deposition by SLRR of Hupd
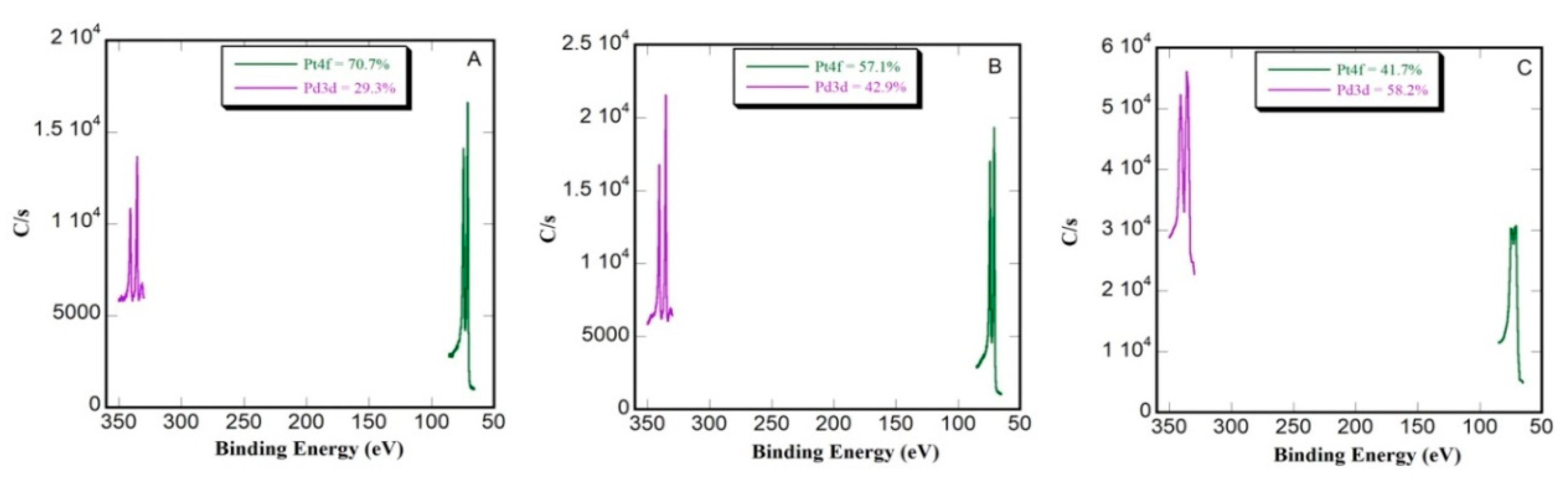
3.2. Compositional Characterization by XPS
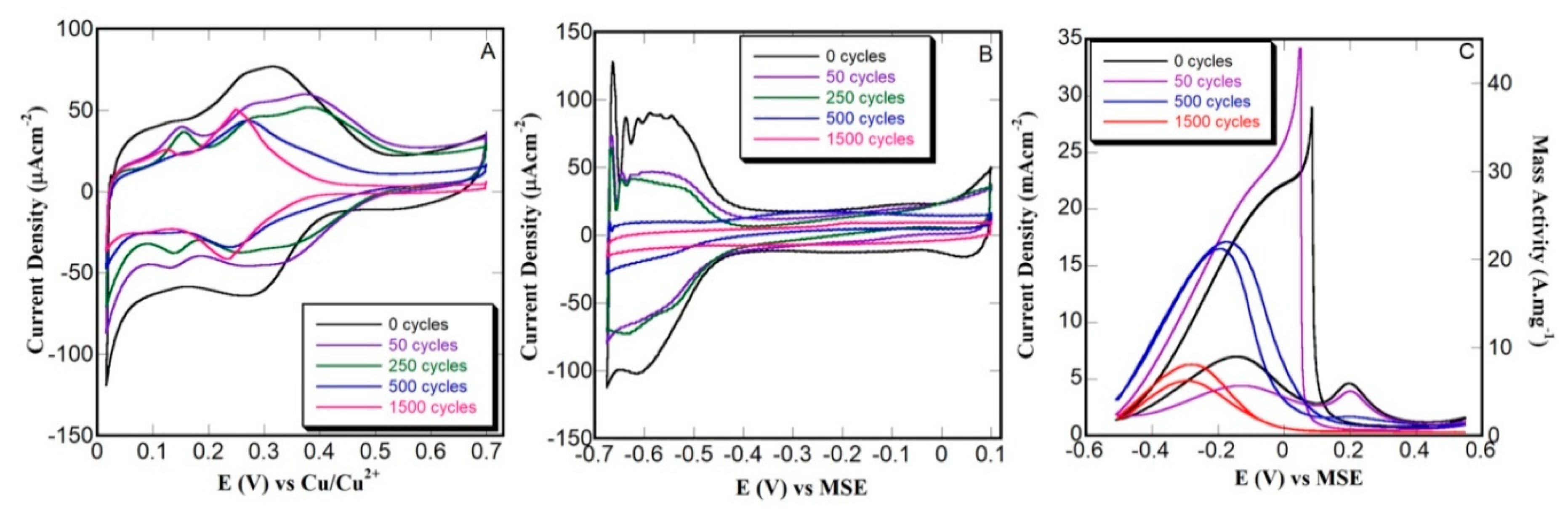
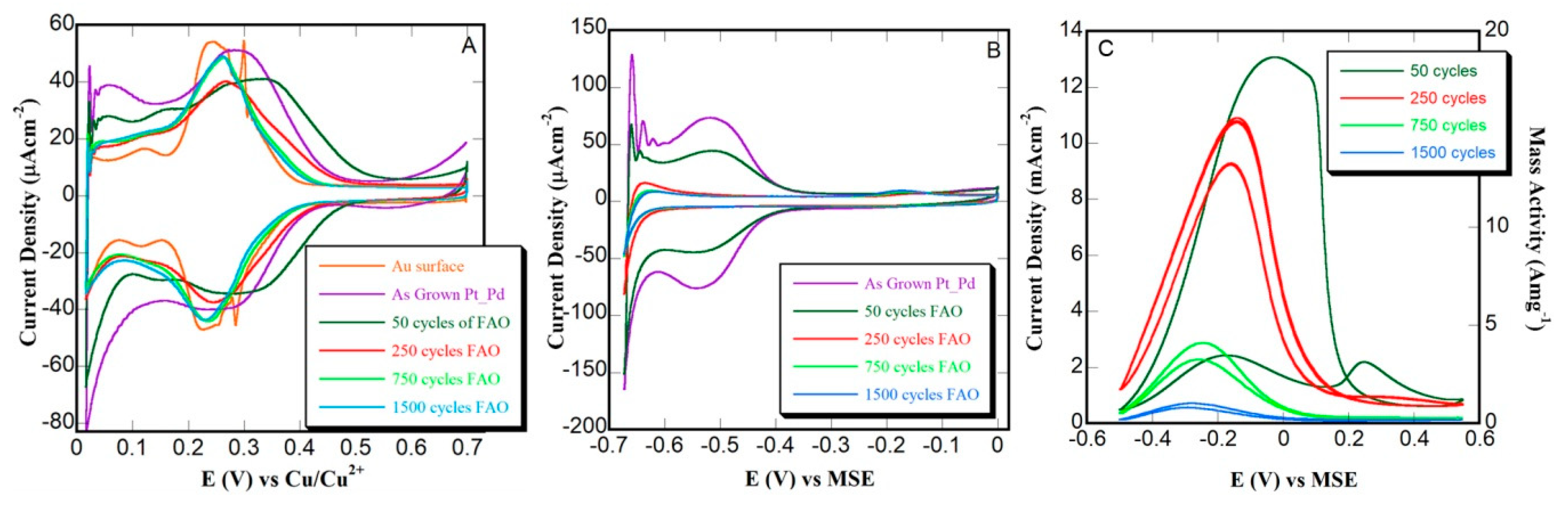
3.3. Thin Film Characterization and Potentiodynamic FAO Testing
3.4. Constant Potential (Potentiostatic) FAO Testing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barbir, F.; Gómez, T. Efficiency and economics of proton exchange membrane (pem) fuel cells. Int. J. Hydrogen Energy 1996, 21, 891–901. [Google Scholar] [CrossRef]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Capon, A.; Parson, R. The oxidation of formic acid at noble metal electrodes: I. Review of previous work. J. Electroanal. Chem. Interfac. Electrochem. 1973, 44, 1–7. [Google Scholar] [CrossRef]
- Wagner, F.T.; Lakshmanan, B.; Mathias, M.F. Electrochemistry and the future of the automobile. J. Phys. Chem. Lett. 2010, 1, 2204–2219. [Google Scholar] [CrossRef]
- Bell, A.T. The impact of nanoscience on heterogeneous catalysis. Science 2003, 299, 1688–1691. [Google Scholar] [CrossRef] [Green Version]
- Wee, J.-H. A feasibility study on direct methanol fuel cells for laptop computers based on a cost comparison with lithium-ion batteries. J. Power Sources 2007, 173, 424–436. [Google Scholar] [CrossRef]
- Winter, M.; Brodd, R.J. What are batteries, fuel cells, and supercapacitors? Chem. Rev. 2004, 104, 4245–4270. [Google Scholar] [CrossRef] [Green Version]
- Ong, B.C.; Kamarudin, S.K.; Basri, S. Direct liquid fuel cells: A review. Int. J. Hydrogen Energy 2017, 42, 10142–10157. [Google Scholar] [CrossRef]
- Fernandez-Pello, C. Micropower generation using combustion: Issues and approaches. Proc. Combust. Inst. 2002, 29, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Kamarudin, S.K.; Achmad, F.; Daud, W.R.W. Overview on the application of direct methanol fuel cell (dmfc) for portable electronic devices. Int. J. Hydrog. Energy 2009, 34, 6902–6916. [Google Scholar] [CrossRef]
- Dillon, R.; Srinivasan, S.; Aricò, A.S.; Antonucci, V. International activities in dmfc r&d: Status of technologies and potential applications. J. Power Sources 2004, 127, 112–126. [Google Scholar]
- Scott, K.; Xing, L. Chapter 3—Direct methanol fuel cells. In Advances in Chemical Engineering; Sundmacher, K., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 41, pp. 145–196. [Google Scholar]
- Weber, M.; Wang, J.-T.; Wasmus, S.; Savinell, R.F. Formic acid oxidation in a polymer electrolyte fuel cell: A real-time mass-spectrometry study. J. Electrochem. Soc. 1996, 143, L158–L160. [Google Scholar] [CrossRef]
- Wang, X.; Hu, J.-M.; Hsing, I.M. Electrochemical investigation of formic acid electro-oxidation and its crossover through a nafion® membrane. J. Electroanal. Chem. 2004, 562, 73–80. [Google Scholar] [CrossRef]
- Rice, C.; Ha, S.; Masel, R.I.; Waszczuk, P.; Wieckowski, A.; Barnard, T. Direct formic acid fuel cells. J. Power Sources 2002, 111, 83–89. [Google Scholar] [CrossRef]
- Zhu, Y.; Ha, S.Y.; Masel, R.I. High power density direct formic acid fuel cells. J. Power Sources 2004, 130, 8–14. [Google Scholar] [CrossRef]
- Miesse, C.M.; Jung, W.S.; Jeong, K.-J.; Lee, J.K.; Lee, J.; Han, J.; Yoon, S.P.; Nam, S.W.; Lim, T.-H.; Hong, S.-A. Direct formic acid fuel cell portable power system for the operation of a laptop computer. J. Power Sources 2006, 162, 532–540. [Google Scholar] [CrossRef]
- Capon, A.; Parsons, R. The oxidation of formic acid on noble metal electrodes. Ii. A comparison of the behaviour of pure electrodes. J. Electroanal. Chem. 1973, 44, 239–254. [Google Scholar] [CrossRef]
- Llorca, M.J.; Feliu, J.M.; Aldaz, A.; Clavilier, J. Formic acid oxidation on pdad + pt(100) and pdad + pt(111) electrodes. J. Electroanal. Chem. 1994, 376, 151–160. [Google Scholar] [CrossRef]
- Kita, H.; Lei, H.W. Oxidation of formic acid in acid solution on pt single-crystal electrodes. J. Electroanal. Chem. 1995, 388, 167–177. [Google Scholar] [CrossRef]
- Lu, G.-Q.; Crown, A.; Wieckowski, A. Formic acid decomposition on polycrystalline platinum and palladized platinum electrodes. J. Phys. Chem. B 1999, 103, 9700–9711. [Google Scholar] [CrossRef]
- Parsons, R.; VanderNoot, T. The oxidation of small organic molecules. A survey of recent fuel cell related research. J. Electroanal. Chem. 1988, 257, 9–45. [Google Scholar] [CrossRef]
- Sun, S.G.; Clavilier, J.; Bewick, A. The mechanism of electrocatalytic oxidation of formic acid on pt (100) and pt (111) in sulphuric acid solution: An emirs study. J. Electroanal. Chem. Interfac. Electrochem. 1988, 240, 147–159. [Google Scholar] [CrossRef]
- Xia, X.H. Influence of underpotential deposited lead upon the oxidation of hcooh in hclo[sub 4] at platinum electrodes. J. Electrochem. Soc. 1993, 140, 2559. [Google Scholar] [CrossRef]
- Cuesta, A.; Cabello, G.; Osawa, M.; Gutiérrez, C. Mechanism of the electrocatalytic oxidation of formic acid on metals. ACS Catal. 2012, 2, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Osawa, M.; Komatsu, K.-i.; Samjeské, G.; Uchida, T.; Ikeshoji, T.; Cuesta, A.; Gutiérrez, C. The role of bridge-bonded adsorbed formate in the electrocatalytic oxidation of formic acid on platinum. Angew. Chem. Int. Ed. 2011, 50, 1159–1163. [Google Scholar] [CrossRef]
- Miki, A.; Ye, S.; Osawa, M. Surface-enhanced ir absorption on platinum nanoparticles: An application to real-time monitoring of electrocatalytic reactions. Chem. Commun. 2002, 21, 1500–1501. [Google Scholar] [CrossRef]
- Samjeské, G.; Osawa, M. Current oscillations during formic acid oxidation on a pt electrode: Insight into the mechanism by time-resolved ir spectroscopy. Angew. Chem. Int. Ed. 2005, 44, 5694–5698. [Google Scholar] [CrossRef]
- Samjeské, G.; Miki, A.; Ye, S.; Yamakata, A.; Mukouyama, Y.; Okamoto, H.; Osawa, M. Potential oscillations in galvanostatic electrooxidation of formic acid on platinum: A time-resolved surface-enhanced infrared study. J. Phys. Chem. B 2005, 109, 23509–23516. [Google Scholar] [CrossRef]
- Samjeské, G.; Miki, A.; Ye, S.; Osawa, M. Mechanistic study of electrocatalytic oxidation of formic acid at platinum in acidic solution by time-resolved surface-enhanced infrared absorption spectroscopy. J. Phys. Chem. B 2006, 110, 16559–16566. [Google Scholar] [CrossRef]
- Mukouyama, Y.; Kikuchi, M.; Samjeské, G.; Osawa, M.; Okamoto, H. Potential oscillations in galvanostatic electrooxidation of formic acid on platinum: A mathematical modeling and simulation. J. Phys. Chem. B 2006, 110, 11912–11917. [Google Scholar] [CrossRef]
- Chen, Y.X.; Heinen, M.; Jusys, Z.; Behm, R.J. Kinetics and mechanism of the electrooxidation of formic acid—Spectroelectrochemical studies in a flow cell. Angew. Chem. Int. Ed. 2006, 45, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Capon, A.; Parsons, R. The oxidation of formic acid at noble metal electrodes part iii. Intermediates and mechanism on platinum electrodes. J. Electroanal. Chem. Interfac. Electrochem. 1973, 45, 205–231. [Google Scholar] [CrossRef]
- Beden, B.; Bewick, A.; Lamy, C. A comparative study of formic acid adsorption on a platinum electrode by both electrochemical and emirs techniques. J. Electroanal. Chem. Interfac. Electrochem. 1983, 150, 505–511. [Google Scholar] [CrossRef]
- Bin, D.; Yang, B.; Ren, F.; Zhang, K.; Yang, P.; Du, Y. Facile synthesis of pdni nanowire networks supported on reduced graphene oxide with enhanced catalytic performance for formic acid oxidation. J. Mater. Chem. A 2015, 3, 14001–14006. [Google Scholar] [CrossRef]
- Baldauf, M.; Kolb, D.M. Formic acid oxidation on ultrathin pd films on au(hkl) and pt(hkl) electrodes. J. Phys. Chem. B 1996, 100, 11375–11381. [Google Scholar] [CrossRef]
- Clavilier, J.; Parsons, R.; Durand, R.; Lamy, C.; Leger, J.M. Formic acid oxidation on single crystal platinum electrodes. Comparison with polycrystalline platinum. J. Electroanal. Chem. Interfac. Electrochem. 1981, 124, 321–326. [Google Scholar] [CrossRef]
- Lamy, C.; Léger, J. Electrocatalytic oxidation of small organic molecules at platinum single crystals. J. Chim. Phys. 1991, 88, 1649–1671. [Google Scholar] [CrossRef]
- Iwasita, T.; Xia, X.; Herrero, E.; Liess, H.-D. Early stages during the oxidation of hcooh on single-crystal pt electrodes as characterized by infrared spectroscopy. Langmuir 1996, 12, 4260–4265. [Google Scholar] [CrossRef]
- Wasmus, S.; Tryk, D.A.; Vielstich, W. Electrochemical behavior of nitromethane and its influence on the electro-oxidation of formic acid: An on-line ms study. J. Electroanal. Chem. 1994, 377, 205–214. [Google Scholar] [CrossRef]
- Więckowski, A.; Sobkowski, J. Comparative study of adsorption and oxidation of formic acid and methanol on platinized electrodes in acidic solution. J. Electroanal. Chem. Interfac. Electrochem. 1975, 63, 365–377. [Google Scholar] [CrossRef]
- Brummer, S.B. Comparison of adsorbed formic acid and carbon monoxide on platinum electrodes. J. Phys. Chem. 1965, 69, 1363–1365. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Marković, N.; Ross, P.N.; Cairns, E.J. Electro-oxidation of small organic molecules on well-characterized ptru alloys. Electrochim. Acta 1994, 39, 1825–1832. [Google Scholar] [CrossRef]
- Taylor, A.H.; Kirkland, S.; Brummer, S.B. Effect of adsorbed layers on the anodic oxidation of simple organic compounds. Part 4.—Ag adsorption on pt and its effect on hcooh oxidation. Trans. Faraday Soc. 1971, 67, 819–827. [Google Scholar] [CrossRef]
- Motoo, S.; Watanabe, M. Electrocatalysis by sn and ge ad-atoms. J. Electroanal. Chem. Interfac. Electrochem. 1976, 69, 429–431. [Google Scholar] [CrossRef]
- Watanabe, M.; Horiuchi, M.; Motoo, S. Electrocatalysis by ad-atoms: Part xxiii. Design of platinum ad-electrodes for formic acid fuel cells with ad-atoms of the ivth and the vth groups. J. Electroanal. Chem. Interfac. Electrochem. 1988, 250, 117–125. [Google Scholar] [CrossRef]
- Herrero, E.; Feliu, J.M.; Aldaz, A. Poison formation reaction from formic acid on pt(100) electrodes modified by irreversibly adsorbed bismuth and antimony. J. Electroanal. Chem. 1994, 368, 101–108. [Google Scholar] [CrossRef]
- Llorca, M.J.; Herrero, E.; Feliu, J.M.; Aldaz, A. Formic acid oxidation on pt(111) electrodes modified by irreversibly adsorbed selenium. J. Electroanal. Chem. 1994, 373, 217–225. [Google Scholar] [CrossRef]
- Kang, S.; Lee, J.; Lee, J.K.; Chung, S.-Y.; Tak, Y. Influence of bi modification of pt anode catalyst in direct formic acid fuel cells. J. Phys. Chem. B 2006, 110, 7270–7274. [Google Scholar] [CrossRef]
- Chen, Q.-S.; Zhou, Z.-Y.; Vidal-Iglesias, F.J.; Solla-Gullón, J.; Feliu, J.M.; Sun, S.-G. Significantly enhancing catalytic activity of tetrahexahedral pt nanocrystals by bi adatom decoration. J. Am. Chem. Soc. 2011, 133, 12930–12933. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Hamnett, A.; Kennedy, B.J.; Manoharan, R.; Weeks, S.A. Methanol oxidation on unsupported and carbon supported pt + ru anodes. J. Electroanal. Chem. Interfac. Electrochem. 1988, 240, 133–145. [Google Scholar] [CrossRef]
- Swathirajan, S.M.; Youssef, M. Electrochemical oxidation of methanol at chemically prepared platinum-ruthenium alloy electrodes. J. Electrochem. Soc. 1991, 138, 1321. [Google Scholar] [CrossRef]
- Herrero, E.; Franaszczuk, K.; Wieckowski, A. Electrochemistry of methanol at low index crystal planes of platinum: An integrated voltammetric and chronoamperometric study. J. Phys. Chem. 1994, 98, 5074–5083. [Google Scholar] [CrossRef]
- Kennedy, B.J.; Hamnett, A. Oxide formation and reactivity for methanol oxidation on platinised carbon anodes. J. Electroanal. Chem. Interfac. Electrochem. 1990, 283, 271–285. [Google Scholar] [CrossRef]
- Lu, Y.; Jiang, Y.; Zhang, R.; Chen, W. Surfactant-free synthesis of cube-like ptru alloy nanoparticles with enhanced electrocatalytic activity toward formic acid oxidation. Sci. Adv. Mater. 2013, 5, 1718–1726. [Google Scholar] [CrossRef]
- Waszczuk, P.; Barnard, T.M.; Rice, C.; Masel, R.I.; Wieckowski, A. A nanoparticle catalyst with superior activity for electrooxidation of formic acid. Electrochem. Commun. 2002, 4, 599–603. [Google Scholar] [CrossRef]
- Qiu, H.J.; Xu, H.T.; Li, X.; Wang, J.Q.; Wang, Y. Core–shell-structured nanoporous ptcu with high cu content and enhanced catalytic performance. J. Mater. Chem. A 2015, 3, 7939–7944. [Google Scholar] [CrossRef]
- Krstajić Pajić, M.N.; Stevanović, S.I.; Radmilović, V.V.; Gavrilović-Wohlmuther, A.; Zabinski, P.; Elezović, N.R.; Radmilović, V.R.; Gojković, S.L.; Jovanović, V.M. Dispersion effect in formic acid oxidation on ptau/c nanocatalyst prepared by water-in-oil microemulsion method. Appl. Catal. B Environ. 2019, 243, 585–593. [Google Scholar] [CrossRef]
- Kong, F.; Du, C.; Ye, J.; Chen, G.; Du, L.; Yin, G. Selective surface engineering of heterogeneous nanostructures: In situ unraveling of the catalytic mechanism on pt–au catalyst. ACS Catal. 2017, 7, 7923–7929. [Google Scholar] [CrossRef]
- Zhang, G.-R.; Zhao, D.; Feng, Y.-Y.; Zhang, B.; Su, D.S.; Liu, G.; Xu, B.-Q. Catalytic pt-on-au nanostructures: Why pt becomes more active on smaller au particles. ACS Nano 2012, 6, 2226–2236. [Google Scholar] [CrossRef]
- Xu, C.; Wang, R.; Chen, M.; Zhang, Y.; Ding, Y. Dealloying to nanoporous au/pt alloys and their structure sensitive electrocatalytic properties. Phys. Chem. Chem. Phys. (Inc. Faraday Trans.) 2010, 12, 239–246. [Google Scholar] [CrossRef]
- Chen, G.; Li, Y.; Wang, D.; Zheng, L.; You, G.; Zhong, C.-J.; Yang, L.; Cai, F.; Cai, J.; Chen, B.H. Carbon-supported ptau alloy nanoparticle catalysts for enhanced electrocatalytic oxidation of formic acid. J. Power Sources 2011, 196, 8323–8330. [Google Scholar] [CrossRef]
- Bi, X.; Wang, R.; Ding, Y. Boosting the performance of pt electro-catalysts toward formic acid electro-oxidation by depositing sub-monolayer au clusters. Electrochim. Acta 2011, 56, 10039–10043. [Google Scholar] [CrossRef]
- Saipanya, S.; Srisombat, L.; Wongtap, P.; Sarakonsri, T. Characterization and formic acid oxidation studies of ptau nanoparticles. J. Nanosci. Nanotechnol. 2014, 14, 8053–8055. [Google Scholar] [CrossRef] [PubMed]
- Jayashree, R.S.; Spendelow, J.S.; Yeom, J.; Rastogi, C.; Shannon, M.A.; Kenis, P.J.A. Characterization and application of electrodeposited pt, pt/pd, and pd catalyst structures for direct formic acid micro fuel cells. Electrochim. Acta 2005, 50, 4674–4682. [Google Scholar] [CrossRef]
- Arenz, M.; Stamenkovic, V.; Schmidt, T.J.; Wandelt, K.; Ross, P.N.; Markovic, N.M. The electro-oxidation of formic acid on pt–pd single crystal bimetallic surfaces. Phys. Chem. Chem. Phys. 2003, 5, 4242–4251. [Google Scholar] [CrossRef]
- Baena-Moncada, A.M.; Morales, G.M.; Barbero, C.; Planes, G.A.; Florez-Montaño, J.; Pastor, E. Formic acid oxidation over hierarchical porous carbon containing ptpd catalysts. Catalysts 2013, 3, 902–913. [Google Scholar] [CrossRef]
- Xu, H.; Song, P.; Yan, B.; Wang, J.; Wang, C.; Shiraishi, Y.; Yang, P.; Du, Y. Pt islands on 3 d nut-like ptag nanocrystals for efficient formic acid oxidation electrocatalysis. ChemSusChem 2018, 11, 1056–1062. [Google Scholar] [CrossRef]
- Qi, Y.; Gao, J.; Zhang, D.; Liu, C. Comparative theoretical study of formic acid decomposition on ptag(111) and pt(111) surfaces. RSC Adv. 2015, 5, 21170–21177. [Google Scholar] [CrossRef]
- Xie, Y.; Dimitrov, N. Ultralow pt loading nanoporous au-cu-pt thin film as highly active and durable catalyst for formic acid oxidation. Appl. Catal. B Environ. 2020, 263, 118366. [Google Scholar] [CrossRef]
- Wang, M.; He, Y.; Li, R.; Ma, Z.; Zhang, Z.; Wang, X. Electrochemical activated ptaucu alloy nanoparticle catalysts for formic acid, methanol and ethanol electro-oxidation. Electrochim. Acta 2015, 178, 259–269. [Google Scholar] [CrossRef]
- Li, C.; Yuan, Q.; Ni, B.; He, T.; Zhang, S.; Long, Y.; Gu, L.; Wang, X. Dendritic defect-rich palladium–copper–cobalt nanoalloys as robust multifunctional non-platinum electrocatalysts for fuel cells. Nat. Commun. 2018, 9, 3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Ren, F.; Bai, T.; Xu, H.; Du, Y. Facile construction of trimetallic ptauru nanostructures with highly porous features and perpendicular pore channels as enhanced formic acid catalysts. Colloids Surf. A Physicochem. Eng. Asp. 2018, 537, 418–424. [Google Scholar] [CrossRef]
- Ye, W.; Chen, S.; Ye, M.; Ren, C.; Ma, J.; Long, R.; Wang, C.; Yang, J.; Song, L.; Xiong, Y. Pt4pdcu0.4 alloy nanoframes as highly efficient and robust bifunctional electrocatalysts for oxygen reduction reaction and formic acid oxidation. Nano Energy 2017, 39, 532–538. [Google Scholar] [CrossRef]
- Jiang, K.; Cai, W.-B. Carbon supported pd-pt-cu nanocatalysts for formic acid electrooxidation: Synthetic screening and componential functions. Appl. Catal. B Environ. 2014, 147, 185–192. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Y.; Shi, R.; Wang, Z. Trimetallic ptpdcu nanowires as an electrocatalyst for methanol and formic acid oxidation. New J. Chem. 2018, 42, 19083–19089. [Google Scholar] [CrossRef]
- Luo, S.; Chen, W.; Cheng, Y.; Song, X.; Wu, Q.; Li, L.; Wu, X.; Wu, T.; Li, M.; Yang, Q.; et al. Trimetallic synergy in intermetallic ptsnbi nanoplates boosts formic acid oxidation. Adv. Mater. 2019, 31, 1903683. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Munoz, F.; Noborikawa, J.; Haan, J.; Scudiero, L.; Ha, S. Carbon supported pd-based bimetallic and trimetallic catalyst for formic acid electrochemical oxidation. Appl. Catal. B Environ. 2016, 180, 758–765. [Google Scholar] [CrossRef]
- Al Amri, Z.; Mercer, M.P.; Vasiljevic, N. Surface limited redox replacement deposition of platinum ultrathin films on gold: Thickness and structure dependent activity towards the carbon monoxide and formic acid oxidation reactions. Electrochim. Acta 2016, 210, 520–529. [Google Scholar] [CrossRef]
- Adzic, R.R.; Zhang, J.; Sasaki, K.; Vukmirovic, M.B.; Shao, M.; Wang, J.X.; Nilekar, A.U.; Mavrikakis, M.; Valerio, J.A.; Uribe, F. Platinum monolayer fuel cell electrocatalysts. Top. Catal. 2007, 46, 249–262. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T.M.; Bandarenka, A.S. Tailoring the catalytic activity of electrodes with monolayer amounts of foreign metals. Chem. Soc. Rev. 2013, 42, 5210–5230. [Google Scholar] [CrossRef]
- Porter, N.S.; Wu, H.; Quan, Z.; Fang, J. Shape-control and electrocatalytic activity-enhancement of pt-based bimetallic nanocrystals. Acc. Chem. Res. 2013, 46, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Koper, M.T.M. Structure sensitivity and nanoscale effects in electrocatalysis. Nanoscale 2011, 3, 2054–2073. [Google Scholar] [CrossRef] [PubMed]
- Kibler, L.A.; El-Aziz, A.M.; Hoyer, R.; Kolb, D.M. Tuning reaction rates by lateral strain in a palladium monolayer. Angew. Chem. Int. Ed. 2005, 44, 2080–2084. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Nørskov, J.K.; Mavrikakis, M. Electronic structure and catalysis on metal surfaces. Annu. Rev. Phys. Chem. 2002, 53, 319–348. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Tong, Y. A coverage-dependent study of pt spontaneously deposited onto au and ru surfaces: Direct experimental evidence of the ensemble effect for methanol electro-oxidation on pt. J. Phys. Chem. B 2005, 109, 17775–17780. [Google Scholar] [CrossRef] [PubMed]
- Strbac, S.; Petrovic, S.; Vasilic, R.; Kovac, J.; Zalar, A.; Rakocevic, Z. Carbon monoxide oxidation on au(111) surface decorated by spontaneously deposited pt. Electrochim. Acta 2007, 53, 998–1005. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Papoutsis, A.; Stoychev, D.; Milchev, A. Electroless deposition of pt on ti—Catalytic activity for the hydrogen evolution reaction. J. Electroanal. Chem. 2000, 486, 48–55. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Stoychev, D.; Lazarov, V.; Papoutsis, A.; Milchev, A. Electroless deposition of pt on ti: Part ii. Catalytic activity for oxygen reduction. J. Electroanal. Chem. 2001, 511, 20–30. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Armyanov, S.; Valova, E.; Hubin, A.; Steenhaut, O.; Pavlidou, E.; Kokkinidis, G.; Sotiropoulos, S. Methanol oxidation at pt−cu, pt−ni, and pt−co electrode coatings prepared by a galvanic replacement process. J. Phys. Chem. C 2010, 114, 5217–5223. [Google Scholar] [CrossRef]
- Fayette, M.; Liu, Y.; Bertrand, D.; Nutariya, J.; Vasiljevic, N.; Dimitrov, N. From au to pt via surface limited redox replacement of pb upd in one-cell configuration. Langmuir 2011, 27, 5650–5658. [Google Scholar] [CrossRef]
- Mitchell, C.; Fayette, M.; Dimitrov, N. Homo- and hetero-epitaxial deposition of au by surface limited redox replacement of pb underpotentially deposited layer in one-cell configuration. Electrochim. Acta 2012, 85, 450–458. [Google Scholar] [CrossRef]
- Fayette, M.; Nutariya, J.; Vasiljevic, N.; Dimitrov, N. A study of pt dissolution during formic acid oxidation. Acs Catal. 2013, 3, 1709–1718. [Google Scholar] [CrossRef]
- Brankovic, S.R.; Wang, J.X.; Adzic, R.R. Metal monolayer deposition by replacement of metal adlayers on electrode surfaces. Surf. Sci. 2001, 474, L173–L176. [Google Scholar] [CrossRef]
- Nutariya, J.; Fayette, M.; Dimitrov, N.; Vasiljevic, N. Growth of pt by surface limited redox replacement of underpotentially deposited hydrogen. Electrochim. Acta 2013, 112, 813–823. [Google Scholar] [CrossRef]
- Achari, I.; Ambrozik, S.; Dimitrov, N. Electrochemical atomic layer deposition of pd ultrathin films by surface limited redox replacement of underpotentially deposited h in a single cell. J. Phys. Chem. C 2017, 121, 4404–4411. [Google Scholar] [CrossRef]
- Herrero, E.; Clavilier, J.; Feliu, J.M.; Aldaz, A. Influence of the geometry of the hanging meniscus contact on the hydrogen oxidation reaction on a pt(111) electrode in sulphuric acid. J. Electroanal. Chem. 1996, 410, 125–127. [Google Scholar] [CrossRef]
- Strmcnik, D.; Tripkovic, D.; van der Vliet, D.; Stamenkovic, V.; Marković, N.M. Adsorption of hydrogen on pt(111) and pt(100) surfaces and its role in the hor. Electrochem. Commun. 2008, 10, 1602–1605. [Google Scholar] [CrossRef]
- Xia, J.; Achari, I.; Ambrozik, S.; Dimitrov, N. Synthesis, characterization, and testing of pt-npg catalysts developed by de-alloying of electrodeposited cuxau(1−x) thin films. Mater. Res. Bull. 2017, 85, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Achari, I.; Ambrozik, S.; Dimitrov, N. Electrochemical atomic layer deposition by surface limited redox replacement of pd thin films in one-cell configuration using cu upd layers: Interrupting mass-transport limited growth. J. Electrochem. Soc. 2018, 165, J3074–J3082. [Google Scholar] [CrossRef]
- Hernández, J.; Solla-Gullón, J.; Herrero, E.; Aldaz, A.; Feliu, J.M. Characterization of the surface structure of gold nanoparticles and nanorods using structure sensitive reactions. J. Phys. Chem. B 2005, 109, 12651–12654. [Google Scholar] [CrossRef]
- Dimitrov, N. Recent advances in the growth of metals, alloys, and multilayers by surface limited redox replacement (slrr) based approaches. Electrochim. Acta 2016, 209, 599–622. [Google Scholar] [CrossRef] [Green Version]
- Vasilic, R.; Viyannalage, L.T.; Dimitrov, N. Epitaxial growth of Ag on Au(111) by galvanic displacement of Pb and Tl monolayers. J. Electrochem. Soc. 2006, 153, C648–C655. [Google Scholar] [CrossRef]
- Viyannalage, L.T.; Vasilic, R.; Dimitrov, N. Epitaxial growth of Cu on Au(111) and Ag(111) by surface limited redox replacement - An electrochemical and STM study. J. Phys. Chem. C 2007, 111, 4036–4041. [Google Scholar] [CrossRef]
- Adzic, R.R.; Wang, J.; Vitus, C.M.; Ocko, B.M. The electrodeposition of pb monolayers on low index pt surfaces: An x-ray scattering and scanning tunneling microscopy study. Surf. Sci. 1993, 293, L876–L883. [Google Scholar] [CrossRef]
- Basenbacher, F.; Nielsen, L.P.; Sprunger, P.T.; King, D.A.; Woodruff, D.P. The Chemical Physics of Solid Surfaces; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Aoki, N.; Inoue, H.; Yoshiura, R.; Hasegawa, Y.; Miyazaki, S.; Suzuki, A.; Daimon, H.; Doi, T.; Inaba, M.; Higashi, K.; et al. Electrochemical properties and single cell performance of pd core-pt shell structured catalyst synthesized by a simple direct displacement reaction. J. Electrochem. Soc. 2020, 167, 044513. [Google Scholar] [CrossRef]
- Chen, G.; Liao, M.; Yu, B.; Li, Y.; Wang, D.; You, G.; Zhong, C.-J.; Chen, B.H. Pt decorated pdau/c nanocatalysts with ultralow pt loading for formic acid electrooxidation. Int. J. Hydrogen Energy 2012, 37, 9959–9966. [Google Scholar] [CrossRef]
- Gu, D.; Dey, S.K.; Majhi, P. Effective work function of pt, pd, and re on atomic layer deposited hfo2. Appl. Phys. Lett. 2006, 89, 082907. [Google Scholar] [CrossRef]
- Jagannathan, K.; Benson, D.M.; Robinson, D.B.; Stickney, J.L. Hydrogen sorption kinetics on bare and platinum-modified palladium nanofilms, grown by electrochemical atomic layer deposition (e-ald). J. Electrochem. Soc. 2016, 163, D3047–D3052. [Google Scholar] [CrossRef]
- Ambrozik, S.; Dimitrov, N. The deposition of pt via electroless surface limited redox replacement. Electrochim. Acta 2015, 169, 248–255. [Google Scholar] [CrossRef]
- Ruban, A.; Hammer, B.; Stoltze, P.; Skriver, H.L.; Nørskov, J.K. Surface electronic structure and reactivity of transition and noble metals1communication presented at the first francqui colloquium, brussels, 19–20 february 1996.1. J. Mol. Catal. A Chem. 1997, 115, 421–429. [Google Scholar] [CrossRef]
- Adams, B.D.; Asmussen, R.M.; Ostrom, C.K.; Chen, A. Synthesis and comparative study of nanoporous palladium-based bimetallic catalysts for formic acid oxidation. J. Phys. Chem. C 2014, 118, 29903–29910. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achari, I.; Dimitrov, N. Ultrathin Film PtxPd(1-x) Alloy Catalysts for Formic Acid Oxidation Synthesized by Surface Limited Redox Replacement of Underpotentially Deposited H Monolayer. Electrochem 2020, 1, 4-19. https://doi.org/10.3390/electrochem1010002
Achari I, Dimitrov N. Ultrathin Film PtxPd(1-x) Alloy Catalysts for Formic Acid Oxidation Synthesized by Surface Limited Redox Replacement of Underpotentially Deposited H Monolayer. Electrochem. 2020; 1(1):4-19. https://doi.org/10.3390/electrochem1010002
Chicago/Turabian StyleAchari, Innocent, and Nikolay Dimitrov. 2020. "Ultrathin Film PtxPd(1-x) Alloy Catalysts for Formic Acid Oxidation Synthesized by Surface Limited Redox Replacement of Underpotentially Deposited H Monolayer" Electrochem 1, no. 1: 4-19. https://doi.org/10.3390/electrochem1010002
APA StyleAchari, I., & Dimitrov, N. (2020). Ultrathin Film PtxPd(1-x) Alloy Catalysts for Formic Acid Oxidation Synthesized by Surface Limited Redox Replacement of Underpotentially Deposited H Monolayer. Electrochem, 1(1), 4-19. https://doi.org/10.3390/electrochem1010002