
Carbon-α-Fe2O3 Composite Active Material for High-Capacity Electrodes with High Mass Loading and Flat Current Collector for Quasi-Symmetric Supercapacitors

 , ,
, , {kind=link}
{kind=link}
{kind=link}
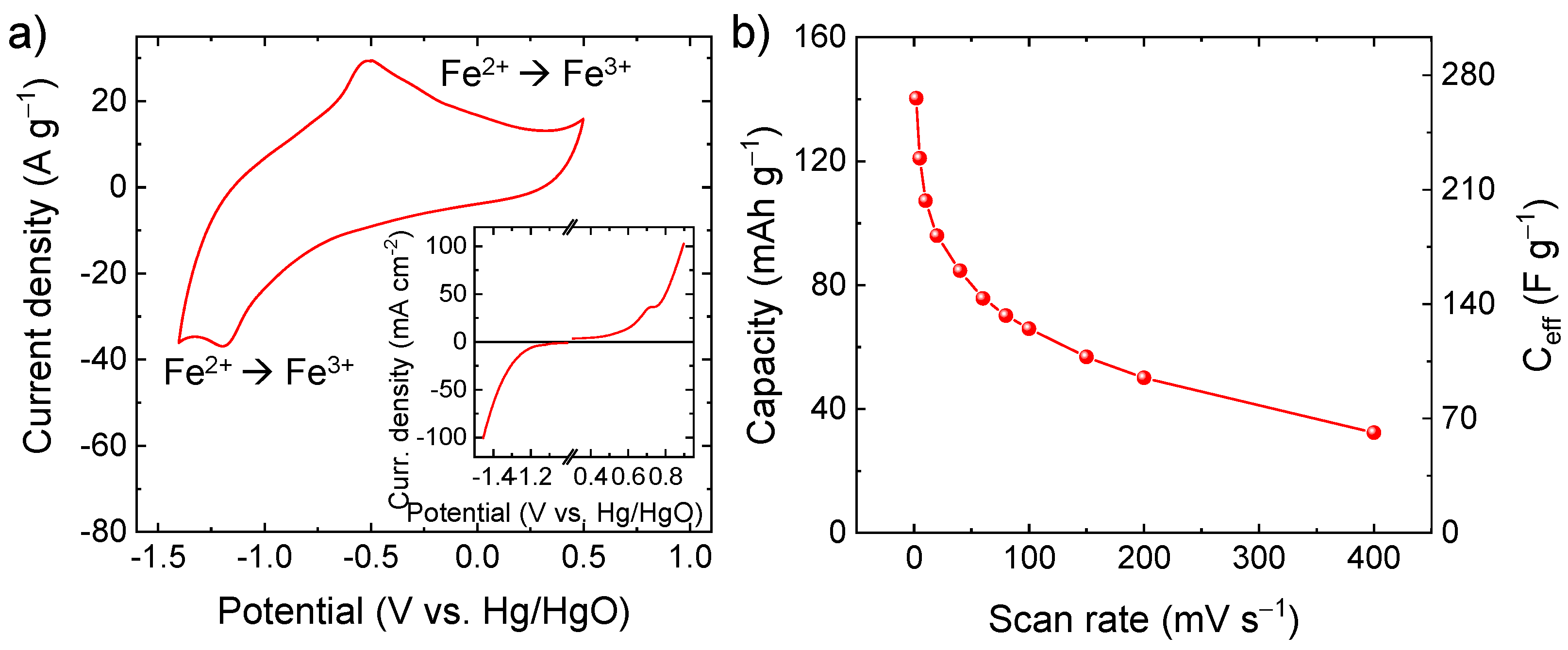
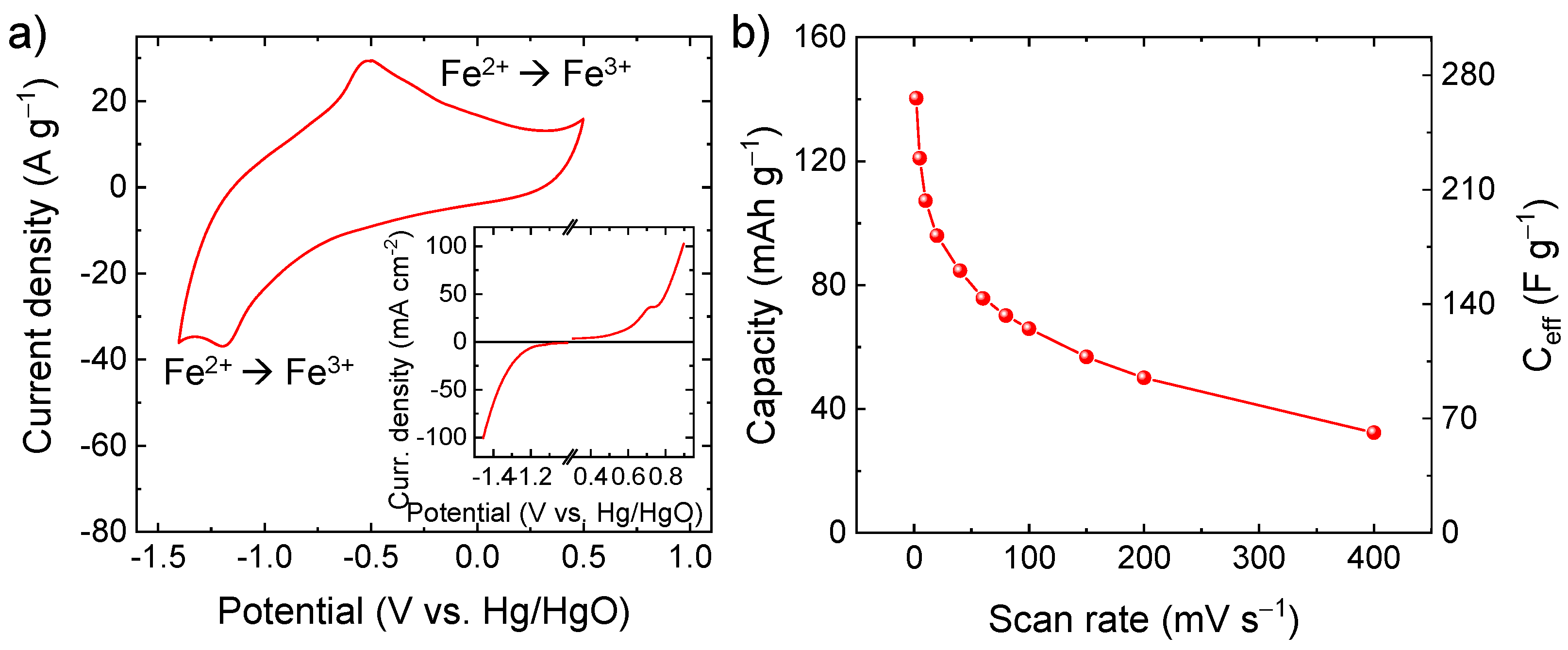{kind=link}
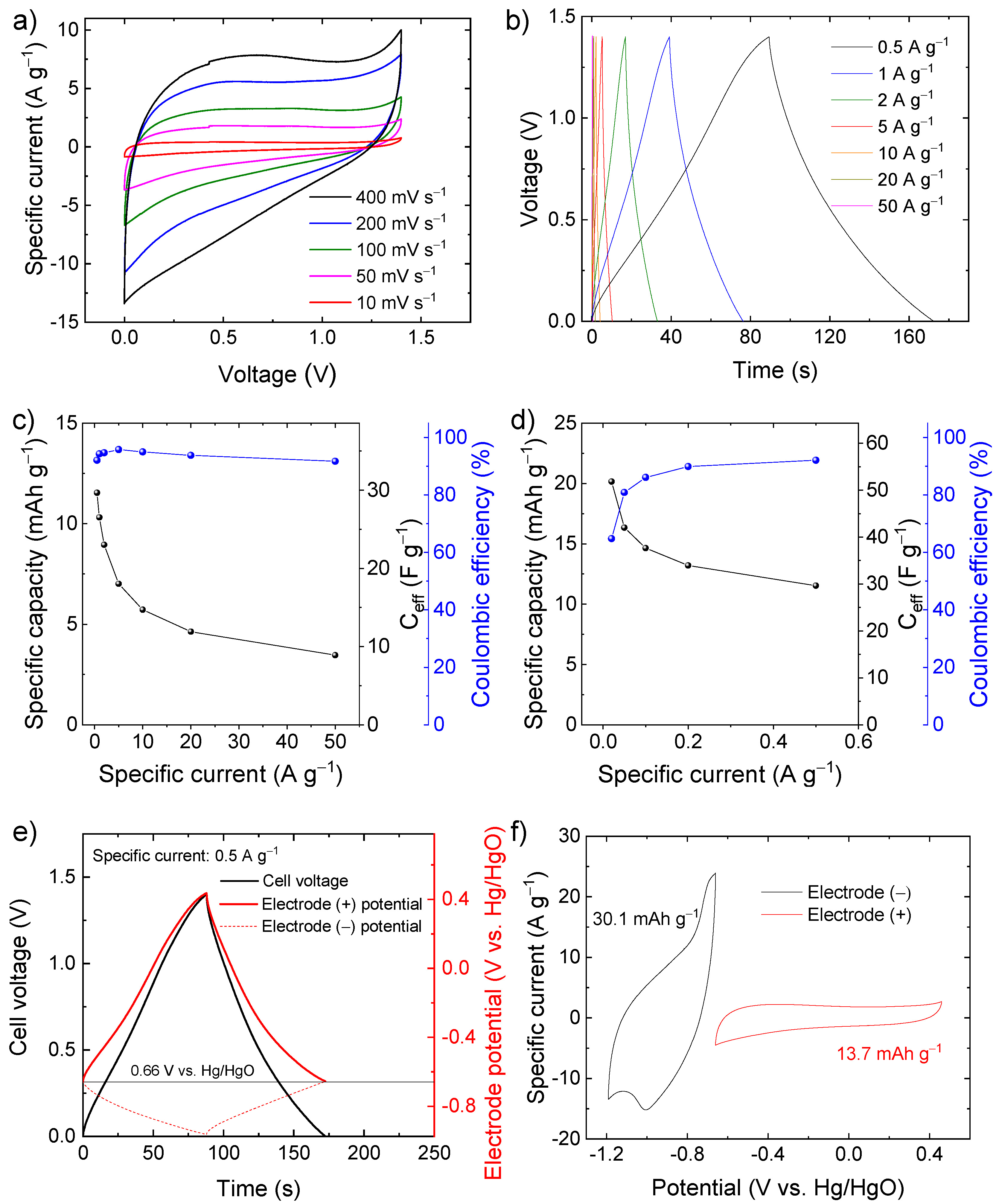
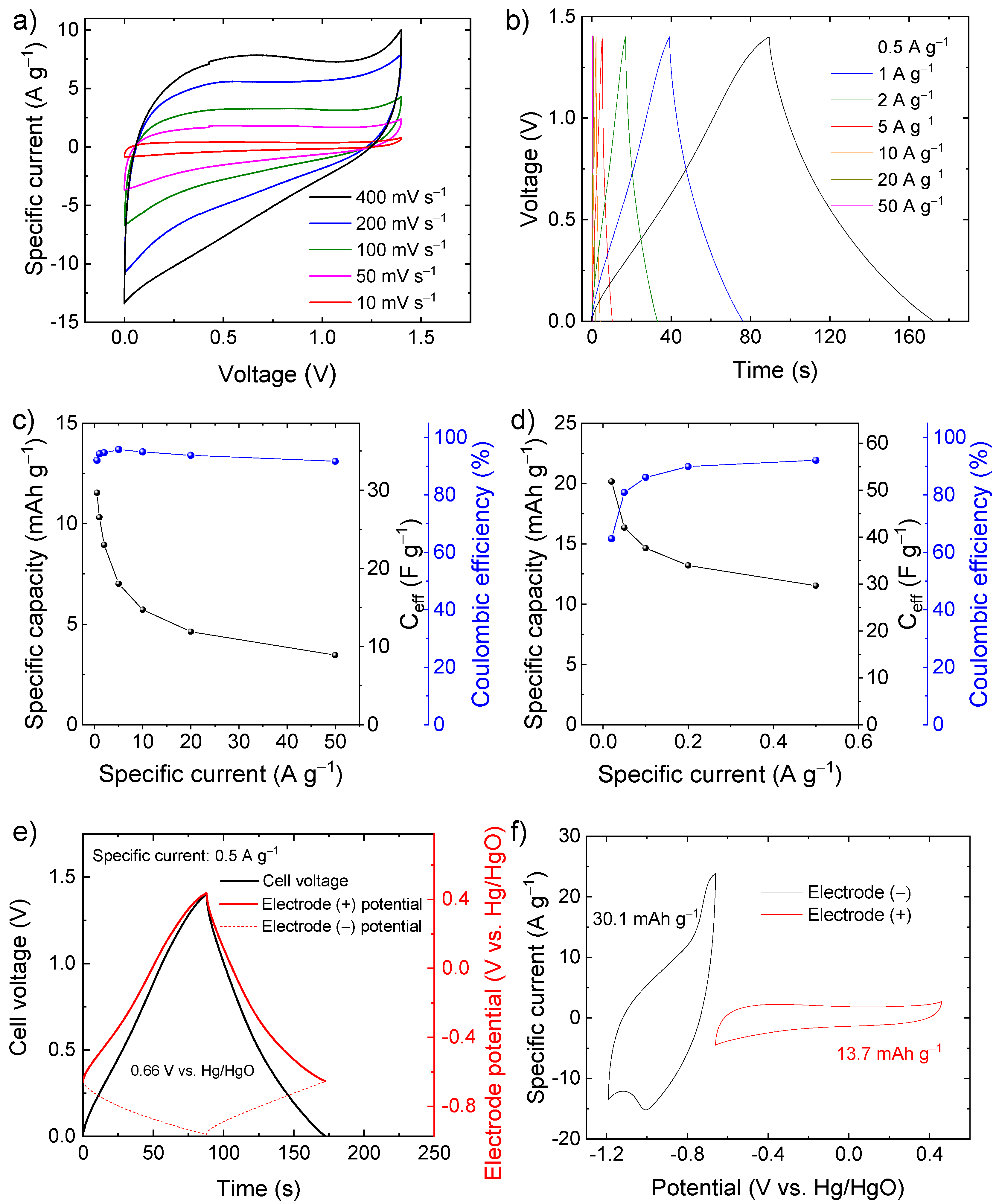{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
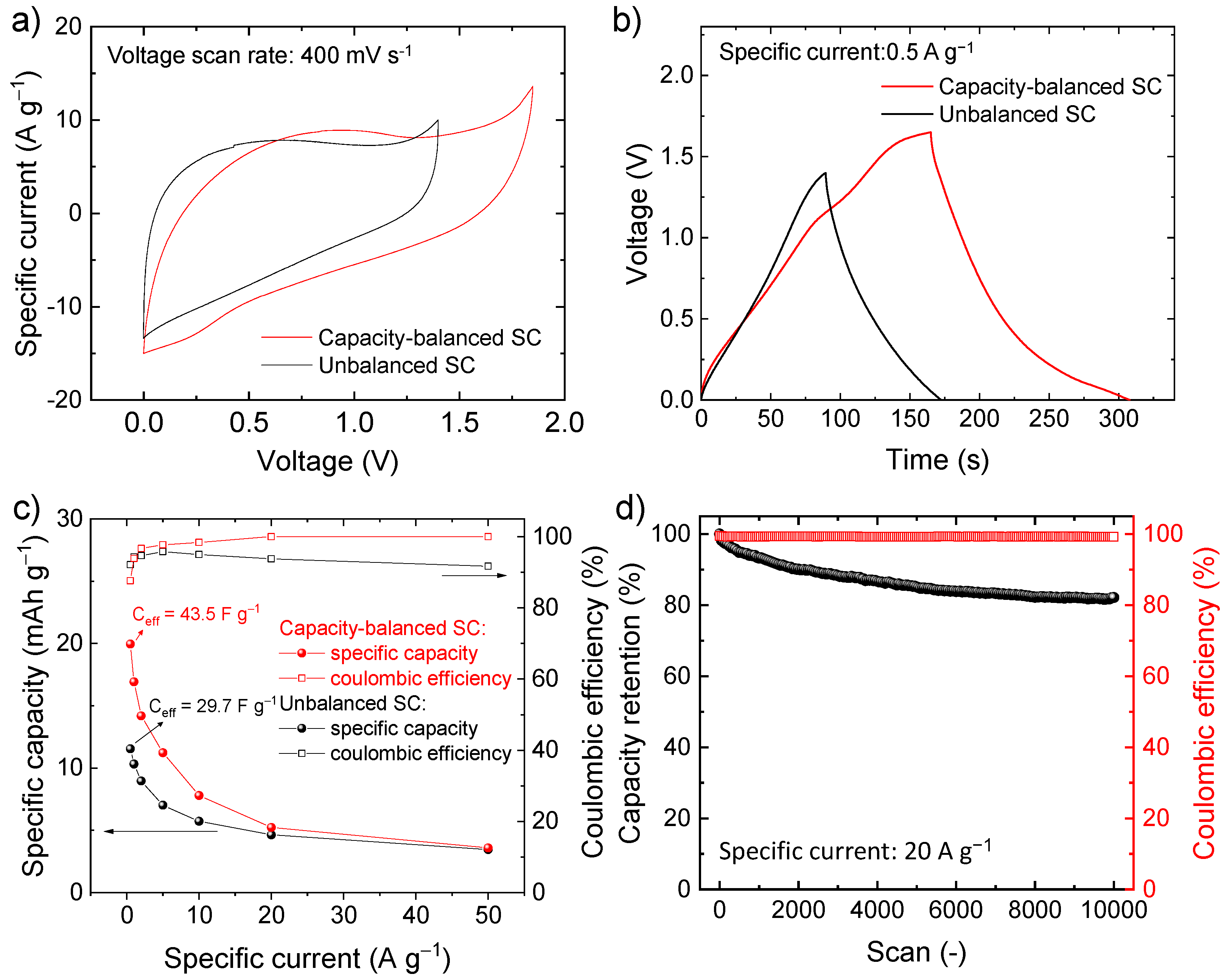
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raza, W.; Ali, F.; Raza, N.; Luo, Y.; Kim, K.H.; Yang, J.; Kumar, S.; Mehmood, A.; Kwon, E.E. Recent Advancements in Supercapacitor Technology. Nano Energy 2018, 52, 441–473. [Google Scholar] [CrossRef]
- Bobba, S.; Carrara, S.; Huisman, J.; Mathieux, F.; Pavel, C. Critical Raw Materials for Strategic Technologies and Sectors in the EU—A Foresight Study; European Commission: Brussels Belgium, 2020; ISBN 9789276153375. [Google Scholar]
- Huang, S.; Zhu, X.; Sarkar, S.; Zhao, Y. Challenges and Opportunities for Supercapacitors. APL Mater. 2019, 7, 100901. [Google Scholar] [CrossRef]
- Zhu, X. Recent Advances of Transition Metal Oxides and Chalcogenides in Pseudo-Capacitors and Hybrid Capacitors: A Review of Structures, Synthetic Strategies, and Mechanism Studies. J. Energy Storage 2022, 49, 104148. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, A.; Zhang, X.; Mu, J.; Che, H.; Wang, Y.; Wang, T.T.; Zhang, Z.; Kou, Z. Fundamentals, Advances and Challenges of Transition Metal Compounds-Based Supercapacitors. Chem. Eng. J. 2021, 412, 128611. [Google Scholar] [CrossRef]
- Bellani, S.; Najafi, L.; Tullii, G.; Ansaldo, A.; Oropesa-Nuñez, R.; Prato, M.; Colombo, M.; Antognazza, M.R.; Bonaccorso, F. ITO Nanoparticles Break Optical Transparency/High-Areal Capacitance Trade-off for Advanced Aqueous Supercapacitors. J. Mater. Chem. A 2017, 5, 25177–25186. [Google Scholar] [CrossRef]
- Fu, L.; Qu, Q.; Holze, R.; Kondratiev, V.V.; Wu, Y. Composites of Metal Oxides and Intrinsically Conducting Polymers as Supercapacitor Electrode Materials: The Best of Both Worlds? J. Mater. Chem. A 2019, 7, 14937–14970. [Google Scholar] [CrossRef]
- Naskar, P.; Maiti, A.; Chakraborty, P.; Kundu, D.; Biswas, B.; Banerjee, A. Chemical Supercapacitors: A Review Focusing on Metallic Compounds and Conducting Polymers. J. Mater. Chem. A 2021, 9, 1970–2017. [Google Scholar] [CrossRef]
- Fleischmann, S.; Mitchell, J.B.; Wang, R.; Zhan, C.; Jiang, D.E.; Presser, V.; Augustyn, V. Pseudocapacitance: From Fundamental Understanding to High Power Energy Storage Materials. Chem. Rev. 2020, 120, 6738–6782. [Google Scholar] [CrossRef]
- Choi, C.; Ashby, D.S.; Butts, D.M.; DeBlock, R.H.; Wei, Q.; Lau, J.; Dunn, B. Achieving High Energy Density and High Power Density with Pseudocapacitive Materials. Nat. Rev. Mater. 2020, 5, 5–19. [Google Scholar] [CrossRef]
- Zhao, J.; Burke, A.F. Electrochemical Capacitors: Materials, Technologies and Performance. Energy Storage Mater. 2021, 36, 31–55. [Google Scholar] [CrossRef]
- Liu, J.; Wang, J.; Xu, C.; Jiang, H.; Li, C.; Zhang, L.; Lin, J.; Shen, Z.X. Advanced Energy Storage Devices: Basic Principles, Analytical Methods, and Rational Materials Design. Adv. Sci. 2018, 5, 1700322. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, S.; Zhang, Y.; Wang, X.; Cummings, P.T.; Wu, J.; Simon, P.; Gogotsi, Y.; Presser, V.; Augustyn, V. Continuous Transition from Double-Layer to Faradaic Charge Storage in Confined Electrolytes. Nat. Energy 2022, 7, 222–228. [Google Scholar] [CrossRef]
- Wei, W.; Cui, X.; Chen, W.; Ivey, D.G. Manganese Oxide -Based Materials as Electrochemical Supercapacitor Electrodes. Chem. Soc. Rev. 2011, 40, 1697–1721. [Google Scholar] [CrossRef] [PubMed]
- An, C.; Zhang, Y.; Guo, H.; Wang, Y. Metal Oxide-Based Supercapacitors: Progress and Prospectives. Nanoscale Adv. 2019, 1, 4644–4658. [Google Scholar] [CrossRef]
- Nithya, V.D.; Arul, N.S. Review on α-Fe2O3 Based Negative Electrode for High Performance Supercapacitors. J. Power Sources 2016, 327, 297–318. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Simon, P. True Performance Metrics in Electrochemical Energy Storage. Science 2012, 335, 167. [Google Scholar] [CrossRef]
- Guan, C.; Liu, J.; Wang, Y.; Mao, L.; Fan, Z.; Shen, Z.; Zhang, H.; Wang, J. Iron Oxide-Decorated Carbon for Supercapacitor Anodes with Ultrahigh Energy Density and Outstanding Cycling Stability. ACS Nano 2015, 9, 5198–5207. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, S.P.; Shao, Z. Intercalation Pseudocapacitance in Electrochemical Energy Storage: Recent Advances in Fundamental Understanding and Materials Development. Mater. Today Adv. 2020, 7, 100072. [Google Scholar] [CrossRef]
- Li, J.; Chen, D.; Wu, Q. α-Fe2O3 Based Carbon Composite as Pure Negative Electrode for Application as Supercapacitor. Eur. J. Inorg. Chem. 2019, 10, 1301–1312. [Google Scholar] [CrossRef]
- Zeng, Y.; Yu, M.; Meng, Y.; Fang, P.; Lu, X.; Tong, Y. Iron-Based Supercapacitor Electrodes: Advances and Challenges. Adv. Energy Mater. 2016, 6, 1601053. [Google Scholar] [CrossRef]
- Lu, X.; Zeng, Y.; Yu, M.; Zhai, T.; Liang, C.; Xie, S.; Balogun, M.-S.; Tong, Y. Oxygen-Deficient Hematite Nanorods as High-Performance and Novel Negative Electrodes for Flexible Asymmetric Supercapacitors. Adv. Mater. 2014, 26, 3148–3155. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Deng, S.; Fan, H.M.; Mhaisalkar, S.; Tan, H.R.; Tok, E.S.; Loh, K.P.; Chin, W.S.; Sow, C.H. α-Fe2O3 Nanotubes-Reduced Graphene Oxide Composites as Synergistic Electrochemical Capacitor Materials. Nanoscale 2012, 4, 2958–2961. [Google Scholar] [CrossRef]
- Del Rio Castillo, A.E.; Pellegrini, V.; Ansaldo, A.; Ricciardella, F.; Sun, H.; Marasco, L.; Buha, J.; Dang, Z.; Gagliani, L.; Lago, E.; et al. High-Yield Production of 2D Crystals by Wet-Jet Milling. Mater. Horizons 2018, 5, 890–904. [Google Scholar] [CrossRef]
- Bellani, S.; Petroni, E.; Del Rio Castillo, A.E.; Curreli, N.; Martín-García, B.; Oropesa-Nuñez, R.; Prato, M.; Bonaccorso, F. Scalable Production of Graphene Inks via Wet-Jet Milling Exfoliation for Screen-Printed Micro-Supercapacitors. Adv. Funct. Mater. 2019, 29, 1807659. [Google Scholar] [CrossRef]
- Bellani, S.; Martín-Garciá, B.; Oropesa-Nuñez, R.; Romano, V.; Najafi, L.; Demirci, C.; Prato, M.; Del Rio Castillo, A.E.; Marasco, L.; Mantero, E.; et al. “Ion Sliding” on Graphene: A Novel Concept to Boost Supercapacitor Performance. Nanoscale Horizons 2019, 4, 1077–1091. [Google Scholar] [CrossRef]
- Garakani, M.A.; Bellani, S.; Pellegrini, V.; Oropesa-Nuñez, R.; Castillo, A.E.D.R.; Abouali, S.; Najafi, L.; Martín-García, B.; Ansaldo, A.; Bondavalli, P.; et al. Scalable Spray-Coated Graphene-Based Electrodes for High-Power Electrochemical Double-Layer Capacitors Operating over a Wide Range of Temperature. Energy Storage Mater. 2021, 34, 1–11. [Google Scholar] [CrossRef]
- Chodankar, N.R.; Pham, H.D.; Nanjundan, A.K.; Fernando, J.F.S.; Jayaramulu, K.; Golberg, D.; Han, Y.K.; Dubal, D.P. True Meaning of Pseudocapacitors and Their Performance Metrics: Asymmetric versus Hybrid Supercapacitors. Small 2020, 16, 2002806. [Google Scholar] [CrossRef] [PubMed]
- Noori, A.; El-Kady, M.F.; Rahmanifar, M.S.; Kaner, R.B.; Mousavi, M.F. Towards Establishing Standard Performance Metrics for Batteries, Supercapacitors and Beyond. Chem. Soc. Rev. 2019, 48, 1272–1341. [Google Scholar] [CrossRef]
- Ansaldo, A.; Bondavalli, P.; Bellani, S.; Del Rio Castillo, A.E.; Prato, M.; Pellegrini, V.; Pognon, G.; Bonaccorso, F. High-Power Graphene–Carbon Nanotube Hybrid Supercapacitors. ChemNanoMat 2017, 3, 436–446. [Google Scholar] [CrossRef]
- Laheäär, A.; Przygocki, P.; Abbas, Q.; Béguin, F. Appropriate Methods for Evaluating the Efficiency and Capacitive Behavior of Different Types of Supercapacitors. Electrochem. Commun. 2015, 60, 21–25. [Google Scholar] [CrossRef]
- Xue, J.; Wu, T.; Dai, Y.; Xia, Y. Electrospinning and Electrospun Nanofibers: Methods, Materials, and Applications. Chem. Rev. 2019, 119, 5298–5415. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, Q.; Li, K.; Wu, Y.; Liu, H. Optimization of Stabilization Conditions for Electrospun Polyacrylonitrile Nanofibers. Polym. Degrad. Stab. 2012, 97, 1511–1519. [Google Scholar] [CrossRef]
- Xu, T.; Nguyen, A.; Rosas, N.; Flores, I.; Chen, C.; Gan, J.B.; Hamdan, A.S.; Gan, Y.X. Effect of Pyrolysis Temperature on the Electrical Property and Photosensitivity of a PAN-PMMA Derived Carbon Fiber. ChemEngineering 2019, 3, 86. [Google Scholar] [CrossRef]
- Gao, Z.; Kaneko, T.; Hou, D.; Nakada, M. Kinetics of Thermal Degradation of Poly(Methyl Methacrylate) Studied with the Assistance of the Fractional Conversion at the Maximum Reaction Rate. Polym. Degrad. Stab. 2004, 84, 399–403. [Google Scholar] [CrossRef]
- Holland, B.J.; Hay, J.N. The Kinetics and Mechanisms of the Thermal Degradation of Poly(Methyl Methacrylate) Studied by Thermal Analysis-Fourier Transform Infrared Spectroscopy. Polymer 2001, 42, 4825–4835. [Google Scholar] [CrossRef]
- Peterson, J.D.; Vyazovkin, S.; Wight, C.A. Kinetic Study of Stabilizing Effect of Oxygen on Thermal Degradation of Poly(Methyl Methacrylate). J. Phys. Chem. B 1999, 103, 8087–8092. [Google Scholar] [CrossRef]
- Peng, Y.T.; Lo, C.T. Electrospun Porous Carbon Nanofibers as Lithium Ion Battery Anodes. J. Solid State Electrochem. 2015, 19, 3401–3410. [Google Scholar] [CrossRef]
- Mohapatra, P.; Shaw, S.; Mendivelso-Perez, D.; Bobbitt, J.M.; Silva, T.F.; Naab, F.; Yuan, B.; Tian, X.; Smith, E.A.; Cademartiri, L. Calcination Does Not Remove All Carbon from Colloidal Nanocrystal Assemblies. Nat. Commun. 2017, 8, 2038. [Google Scholar] [CrossRef]
- Lemine, O.M. Microstructural Characterisation of α-Fe2O3 Nanoparticles Using, XRD Line Profiles Analysis, FE-SEM and FT-IR. Superlattices Microstruct. 2009, 45, 576–582. [Google Scholar] [CrossRef]
- Chen, L.; Liu, D.; Yang, P. Preparation of α-Fe2O3/RGO Composites toward Supercapacitor Applications. RSC Adv. 2019, 9, 12793–12800. [Google Scholar] [CrossRef]
- De Faria, D.L.A.; Venâncio Silva, S.; De Oliveira, M.T. Raman Microspectroscopy of Some Iron Oxides and Oxyhydroxides. J. Raman Spectrosc. 1997, 28, 873–878. [Google Scholar] [CrossRef]
- Emons, T.T.; Li, J.; Nazar, L.F. Synthesis and Characterization of Mesoporous Indium Tin Oxide Possessing an Electronically Conductive Framework. J. Am. Chem. Soc. 2002, 124, 8516–8517. [Google Scholar] [CrossRef] [PubMed]
- Bersani, D.; Lottici, P.P.; Montenero, A. Micro-Raman Investigation of Iron Oxide Films and Powders Produced by Sol-Gel Syntheses. J. Raman Spectrosc. 1999, 30, 355–360. [Google Scholar] [CrossRef]
- Cao, H.; Wang, G.; Zhang, L.; Liang, Y.; Zhang, S.; Zhang, X. Shape and Magnetic Properties of Single-Crystalline Hematite (α-Fe2O3) Nanocrystals. ChemPhysChem 2006, 7, 1897–1901. [Google Scholar] [CrossRef]
- Shim, S.H.; Duffy, T.S. Raman Spectroscopy of Fe2O3 to 62 GPa. Am. Mineral. 2002, 87, 318–326. [Google Scholar] [CrossRef]
- Yamashita, T.; Hayes, P. Analysis of XPS Spectra of Fe2+ and Fe3+ Ions in Oxide Materials. Appl. Surf. Sci. 2008, 254, 2441–2449. [Google Scholar] [CrossRef]
- López-Sánchez, J.; Del Campo, A.; Román-Sánchez, S.; de la Fuente, Ó.R.; Carmona, N.; Serrano, A. Large Two-Magnon Raman Hysteresis Observed in a Magnetically Uncompensated Hematite Coating across the Morin Transition. Coatings 2022, 12, 540. [Google Scholar] [CrossRef]
- Owens, F.J.; Orosz, J. Effect of Nanosizing on Lattice and Magnon Modes of Hematite. Solid State Commun. 2006, 138, 95–98. [Google Scholar] [CrossRef]
- Farahmandjou, M.; Soflaee, F. Low Temperature Synthesis of α-Fe2O3 Nano-Rods Using Simple Chemical. J. Nanostruct. 2014, 4, 413–418. [Google Scholar]
- Hao, C.; Shen, Y.; Wang, Z.; Wang, X.; Feng, F.; Ge, C.; Zhao, Y.; Wang, K. Preparation and Characterization of Fe2O3 Nanoparticles by Solid-Phase Method and Its Hydrogen Persoxide Sensing Properties. ACS Sustain. Chem. Eng. 2016, 4, 1069–1077. [Google Scholar] [CrossRef]
- Fard, G.C.; Mirjalili, M.; Najafi, F. Preparation of Nano-Cellulose/ α-Fe2O3 Hybrid Nanofiber for the Cationic Dyes Removal: Optimization Characterization, Kinetic, Isotherm and Error Analysis. Bulg. Chem. Commun. 2018, 50, 251–261. [Google Scholar]
- Han, T.; Wei, Y.; Jin, X.; Jiu, H.; Zhang, L.; Sun, Y.; Tian, J.; Shang, R.; Hang, D.; Zhao, R. Hydrothermal Self-Assembly of α-Fe2O3 Nanorings@graphene Aerogel Composites for Enhanced Li Storage Performance. J. Mater. Sci. 2019, 54, 7119–7130. [Google Scholar] [CrossRef]
- Mills, P.; Sullivan, J.L. A Study of the Core Level Electrons in Iron and Its Three Oxides by Means of X-Ray Photoelectron Spectroscopy. J. Phys. D Appl. Phys. 1983, 16, 723. [Google Scholar] [CrossRef]
- Mansour, A.N.; Brizzolara, R.A. Characterization of the Surface of γ-Fe2O3 Powder by XPS. Surf. Sci. Spectra 2021, 4, 351. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Puthirath Balan, A.; Radhakrishnan, S.; Woellner, C.F.; Sinha, S.K.; Deng, L.; Reyes, C.D.L.; Rao, B.M.; Paulose, M.; Neupane, R.; Apte, A.; et al. Exfoliation of a Non-van Der Waals Material from Iron Ore Hematite. Nat. Nanotechnol. 2018, 13, 602–609. [Google Scholar] [CrossRef]
- Mohapatra, J.; Ramos, A.; Elkins, J.; Beatty, J.; Xing, M.; Singh, D.; La Plante, E.C.; Ping Liu, J. Ferromagnetism in 2D α-Fe2O3 Nanosheets. Appl. Phys. Lett. 2021, 118, 183102. [Google Scholar] [CrossRef]
- Tao, Y.; Ding, C.; Tan, D.; Yu, F.; Wang, F. Aqueous Dual-Ion Battery Based on a Hematite Anode with Exposed {1 0 4} Facets. ChemSusChem 2018, 11, 4269–4274. [Google Scholar] [CrossRef]
- Kim, H.S.; Piao, Y.; Kang, S.H.; Hyeon, T.; Sung, Y.E. Uniform Hematite Nanocapsules Based on an Anode Material for Lithium Ion Batteries. Electrochem. Commun. 2010, 12, 382–385. [Google Scholar] [CrossRef]
- Modafferi, V.; Triolo, C.; Fiore, M.; Palella, A.; Spadaro, L.; Pianta, N.; Ruffo, R.; Patanè, S.; Santangelo, S.; Musolino, M.G. Effect of Hematite Doping with Aliovalent Impurities on the Electrochemical Performance of α-Fe2O3@rGO-Based Anodes in Sodium-Ion Batteries. Nanomaterials 2020, 10, 1588. [Google Scholar] [CrossRef]
- Xia, G.; Gao, Q.; Sun, D.; Yu, X. Porous Carbon Nanofibers Encapsulated with Peapod-Like Hematite Nanoparticles for High-Rate and Long-Life Battery Anodes. Small 2017, 13, 1701561. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, Y.; Wei, X.; Liu, C. Construction of Conductive Network in Hematite for Lithium, Sodium and Potassium Storage. J. Alloys Compd. 2021, 889, 161766. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Y.; Meng, D.; Qin, X.; Diao, G. Hydrothermal Synthesis of Hematite Nanoparticles and Their Electrochemical Properties. J. Phys. Chem. C 2012, 116, 16276–16285. [Google Scholar] [CrossRef]
- Chun, S.E.; Whitacre, J.F. Investigating the Role of Electrolyte Acidity on Hydrogen Uptake in Mesoporous Activated Carbons. J. Power Sources 2013, 242, 137–140. [Google Scholar] [CrossRef]
- Binitha, G.; Soumya, M.S.; Madhavan, A.A.; Praveen, P.; Balakrishnan, A.; Subramanian, K.R.V.; Reddy, M.V.; Nair, S.V.; Nair, A.S.; Sivakumar, N. Electrospun α-Fe2O3 Nanostructures for Supercapacitor Applications. J. Mater. Chem. A 2013, 1, 11698–11704. [Google Scholar] [CrossRef]
- Kulal, P.M.; Dubal, D.P.; Lokhande, C.D.; Fulari, V.J. Chemical Synthesis of Fe2O3 Thin Films for Supercapacitor Application. J. Alloys Compd. 2011, 509, 2567–2571. [Google Scholar] [CrossRef]
- Xie, K.; Li, J.; Lai, Y.; Lu, W.; Zhang, Z.; Liu, Y.; Zhou, L.; Huang, H. Highly Ordered Iron Oxide Nanotube Arrays as Electrodes for Electrochemical Energy Storage. Electrochem. Commun. 2011, 13, 657–660. [Google Scholar] [CrossRef]
- Sethuraman, B.; Purushothaman, K.K.; Muralidharan, G. Synthesis of Mesh-like Fe2O3/C Nanocomposite via Greener Route for High Performance Supercapacitors. RSC Adv. 2013, 4, 4631–4637. [Google Scholar] [CrossRef]
- Owusu, K.A.; Qu, L.; Li, J.; Wang, Z.; Zhao, K.; Yang, C.; Hercule, K.M.; Lin, C.; Shi, C.; Wei, Q.; et al. Low-Crystalline Iron Oxide Hydroxide Nanoparticle Anode for High-Performance Supercapacitors. Nat. Commun. 2017, 8, 14264. [Google Scholar] [CrossRef]
- Tang, Q.; Wang, W.; Wang, G. The Perfect Matching between the Low-Cost Fe2O3 Nanowire Anode and the NiO Nanoflake Cathode Significantly Enhances the Energy Density of Asymmetric Supercapacitors. J. Mater. Chem. A 2015, 3, 6662–6670. [Google Scholar] [CrossRef]
- Lu, X.F.; Chen, X.Y.; Zhou, W.; Tong, Y.X.; Li, G.R. α-Fe2O3@PANI Core-Shell Nanowire Arrays as Negative Electrodes for Asymmetric Supercapacitors. ACS Appl. Mater. Interfaces 2015, 7, 14843–14850. [Google Scholar] [CrossRef]
- Dai, Z.; Peng, C.; Chae, J.H.; Ng, K.C.; Chen, G.Z. Cell Voltage versus Electrode Potential Range in Aqueous Supercapacitors. Sci. Rep. 2015, 5, 9854. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, W.; Gao, C.; Zhou, J.; Li, X.; Xie, E. An Overview of Carbon Materials for Flexible Electrochemical Capacitors. Nanoscale 2013, 5, 8799–8820. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, Y.; Li, Y.; Liu, J. Construction of High-Capacitance 3D CoO@Polypyrrole Nanowire Array Electrode for Aqueous Asymmetric Supercapacitor. Nano Lett. 2013, 13, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Shanmugavani, A.; Selvan, R.K. Microwave Assisted Reflux Synthesis of NiCo2O4/NiO Composite: Fabrication of High Performance Asymmetric Supercapacitor with Fe2O3. Electrochim. Acta 2016, 189, 283–294. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najafi, M.; Bellani, S.; Galli, V.; Zappia, M.I.; Bagheri, A.; Safarpour, M.; Beydaghi, H.; Eredia, M.; Pasquale, L.; Carzino, R.; et al. Carbon-α-Fe2O3 Composite Active Material for High-Capacity Electrodes with High Mass Loading and Flat Current Collector for Quasi-Symmetric Supercapacitors. Electrochem 2022, 3, 463-478. https://doi.org/10.3390/electrochem3030032
Najafi M, Bellani S, Galli V, Zappia MI, Bagheri A, Safarpour M, Beydaghi H, Eredia M, Pasquale L, Carzino R, et al. Carbon-α-Fe2O3 Composite Active Material for High-Capacity Electrodes with High Mass Loading and Flat Current Collector for Quasi-Symmetric Supercapacitors. Electrochem. 2022; 3(3):463-478. https://doi.org/10.3390/electrochem3030032
Chicago/Turabian StyleNajafi, Maedeh, Sebastiano Bellani, Valerio Galli, Marilena Isabella Zappia, Ahmad Bagheri, Milad Safarpour, Hossein Beydaghi, Matilde Eredia, Lea Pasquale, Riccardo Carzino, and et al. 2022. "Carbon-α-Fe2O3 Composite Active Material for High-Capacity Electrodes with High Mass Loading and Flat Current Collector for Quasi-Symmetric Supercapacitors" Electrochem 3, no. 3: 463-478. https://doi.org/10.3390/electrochem3030032
APA StyleNajafi, M., Bellani, S., Galli, V., Zappia, M. I., Bagheri, A., Safarpour, M., Beydaghi, H., Eredia, M., Pasquale, L., Carzino, R., Lauciello, S., Panda, J.-K., Brescia, R., Gabatel, L., Pellegrini, V., & Bonaccorso, F. (2022). Carbon-α-Fe2O3 Composite Active Material for High-Capacity Electrodes with High Mass Loading and Flat Current Collector for Quasi-Symmetric Supercapacitors. Electrochem, 3(3), 463-478. https://doi.org/10.3390/electrochem3030032