
The Autophagy-Lysosomal Machinery Enhances Cytotrophoblast–Syncytiotrophoblast Fusion Process

 , and
, and 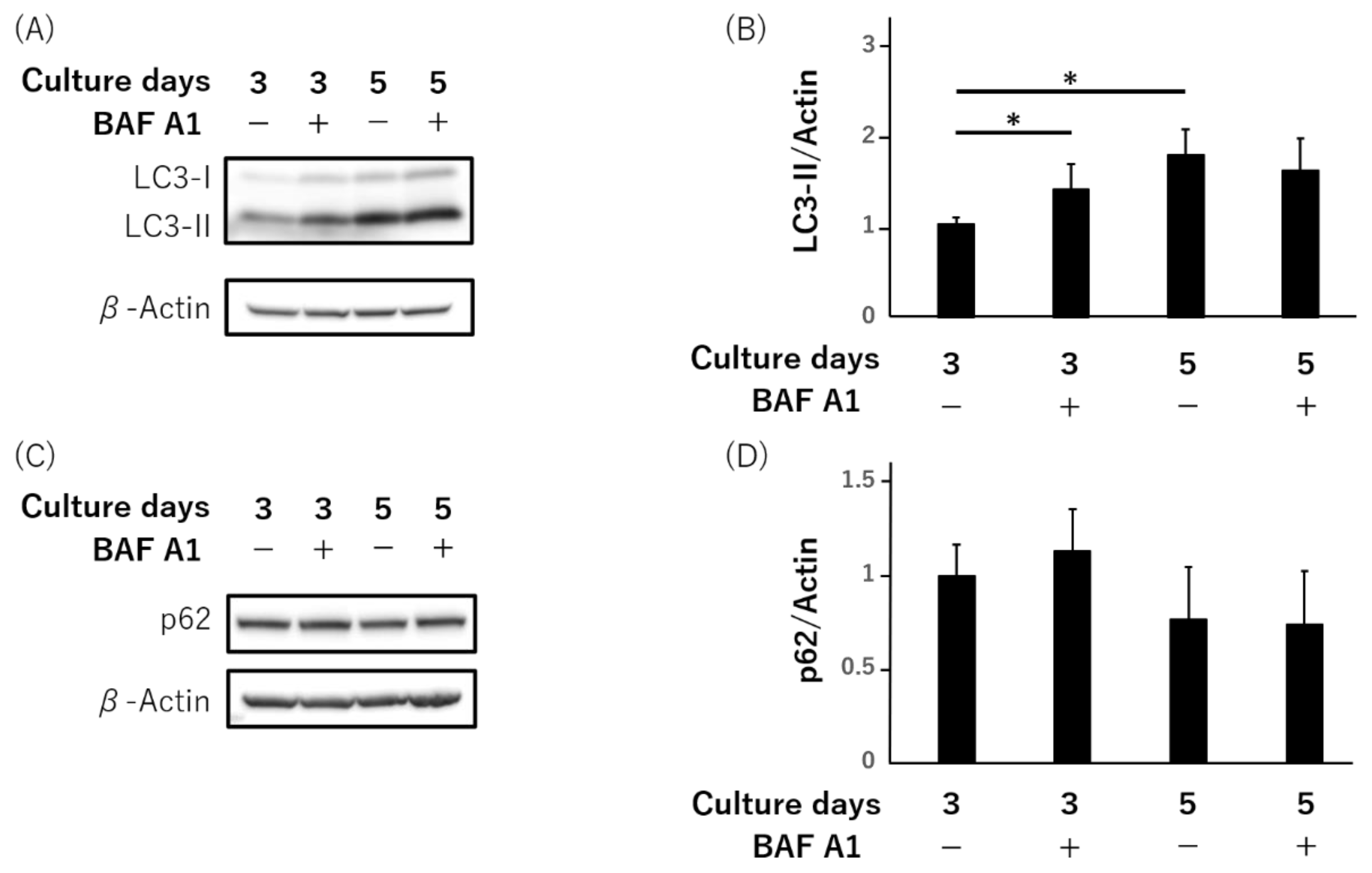{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Western Blotting and Antibodies
2.4. Cell Fusion Index and Immunocytochemistry of Di-8-ANEPPS or LysoTracker DND-99
2.5. Water-Soluble Tetrazolium-1 (WST-1) Assay
2.6. Statistical Analysis
3. Results
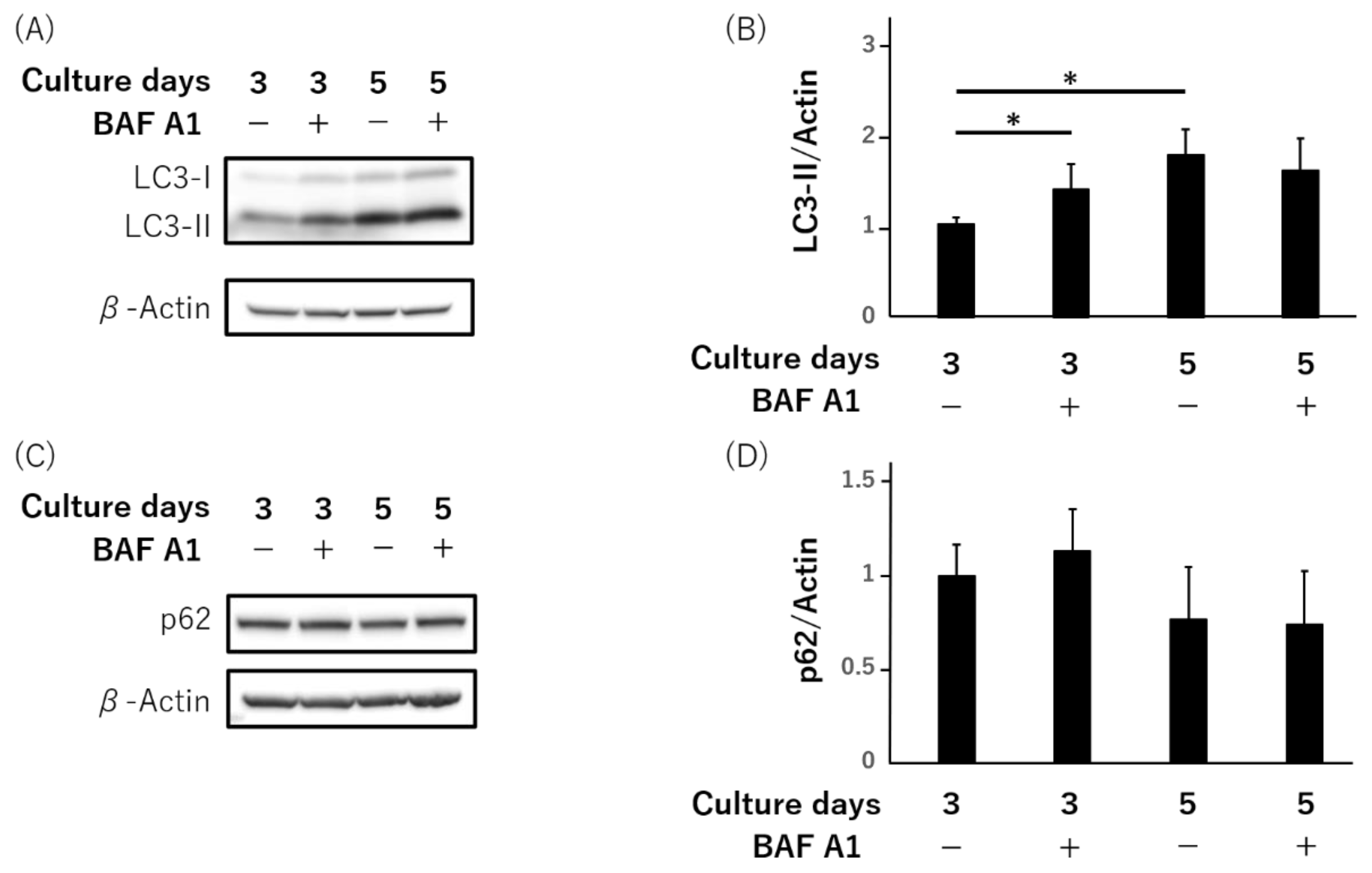
3.1. LC3-II, Not Autophagy-Flux, Is Increased during Syncytialization in Primary Human Trophoblasts
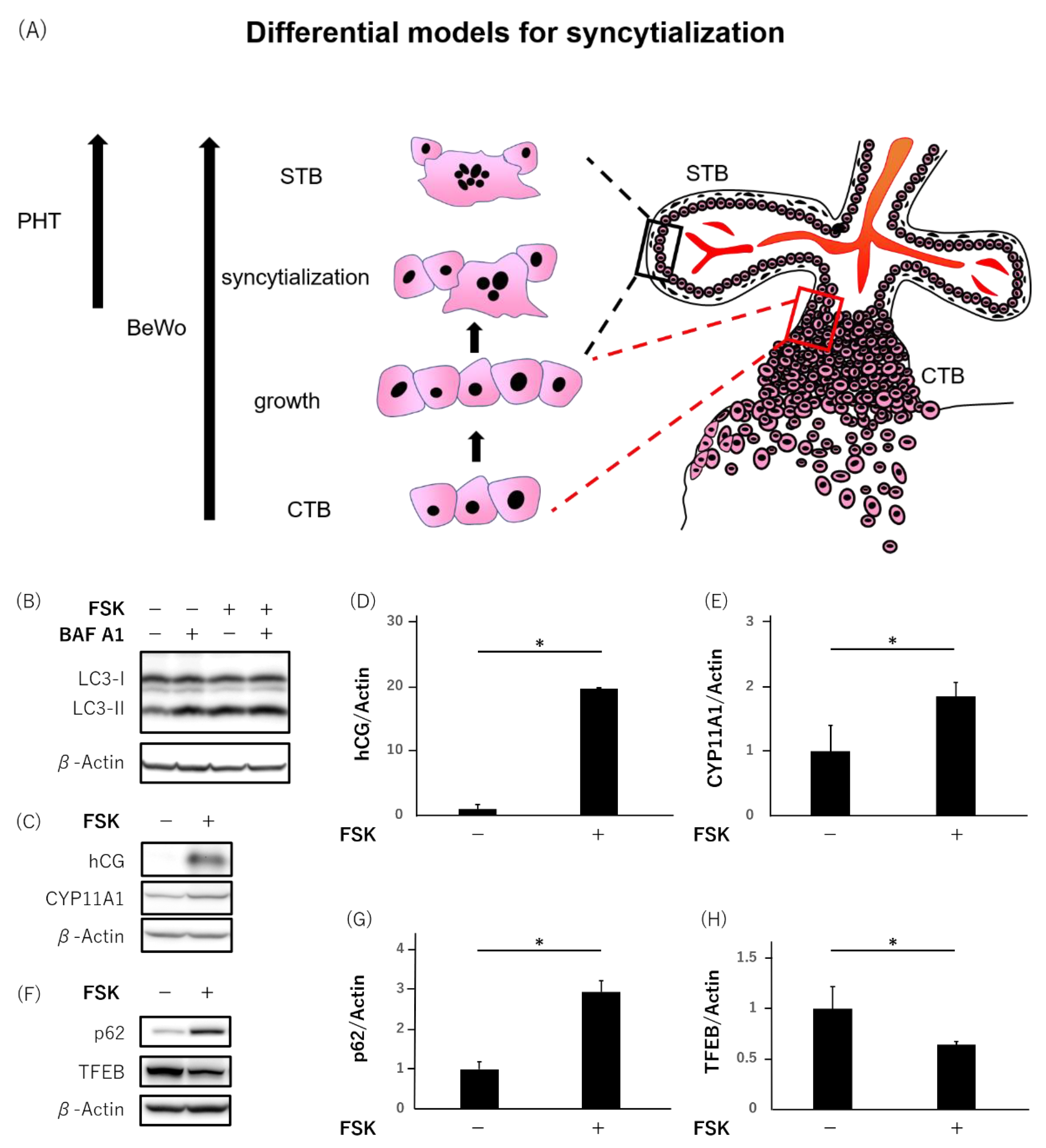
3.2. Downregulation of TFEB during the Syncytialization in BeWo Cells
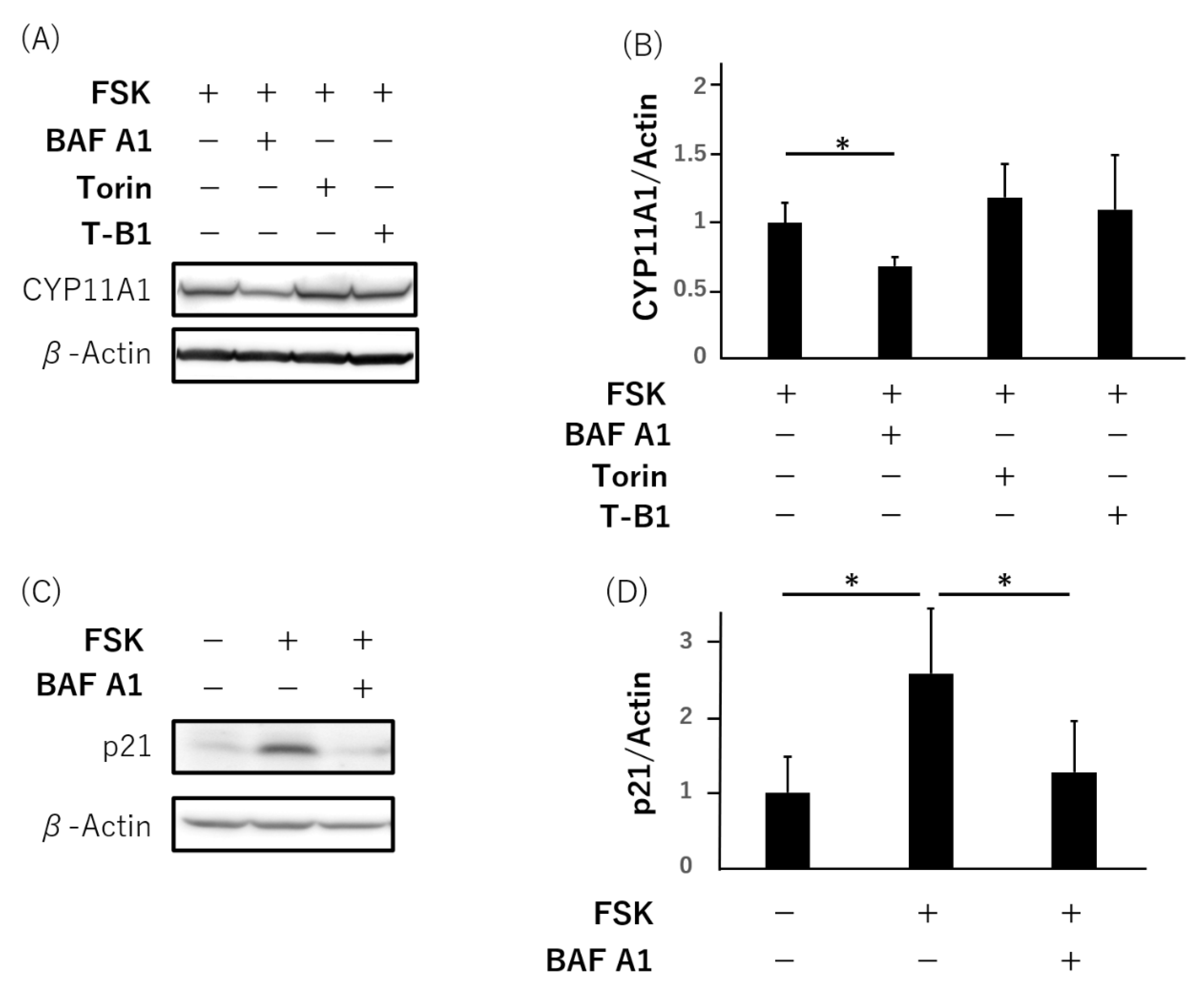
3.3. Failure of Syncytialization Mediated by Autophagy Inhibition in BeWo Cells
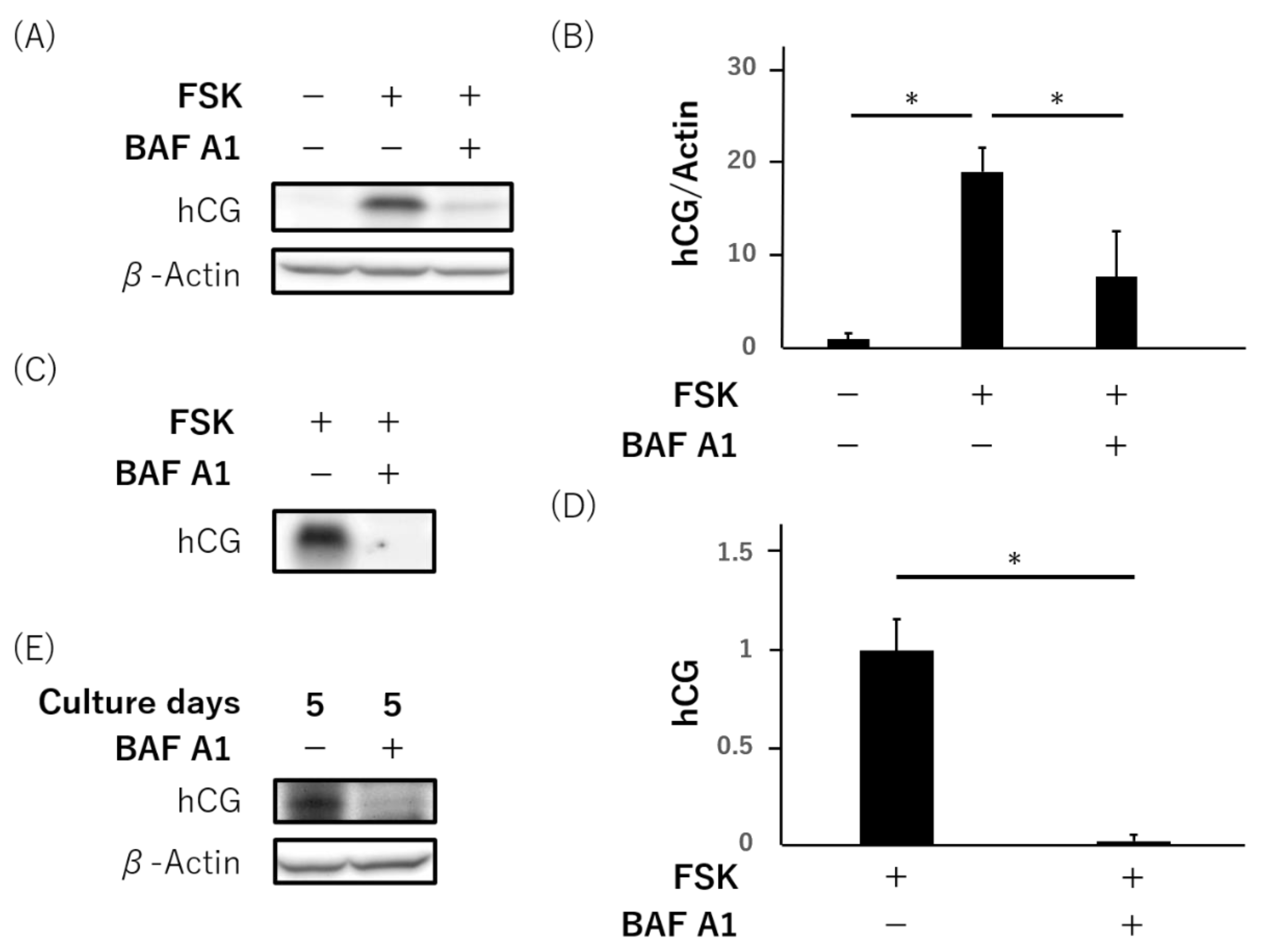
3.4. Failure of hCG Production and Secretion by Bafilomycin A1 in Primary Human Trophoblasts
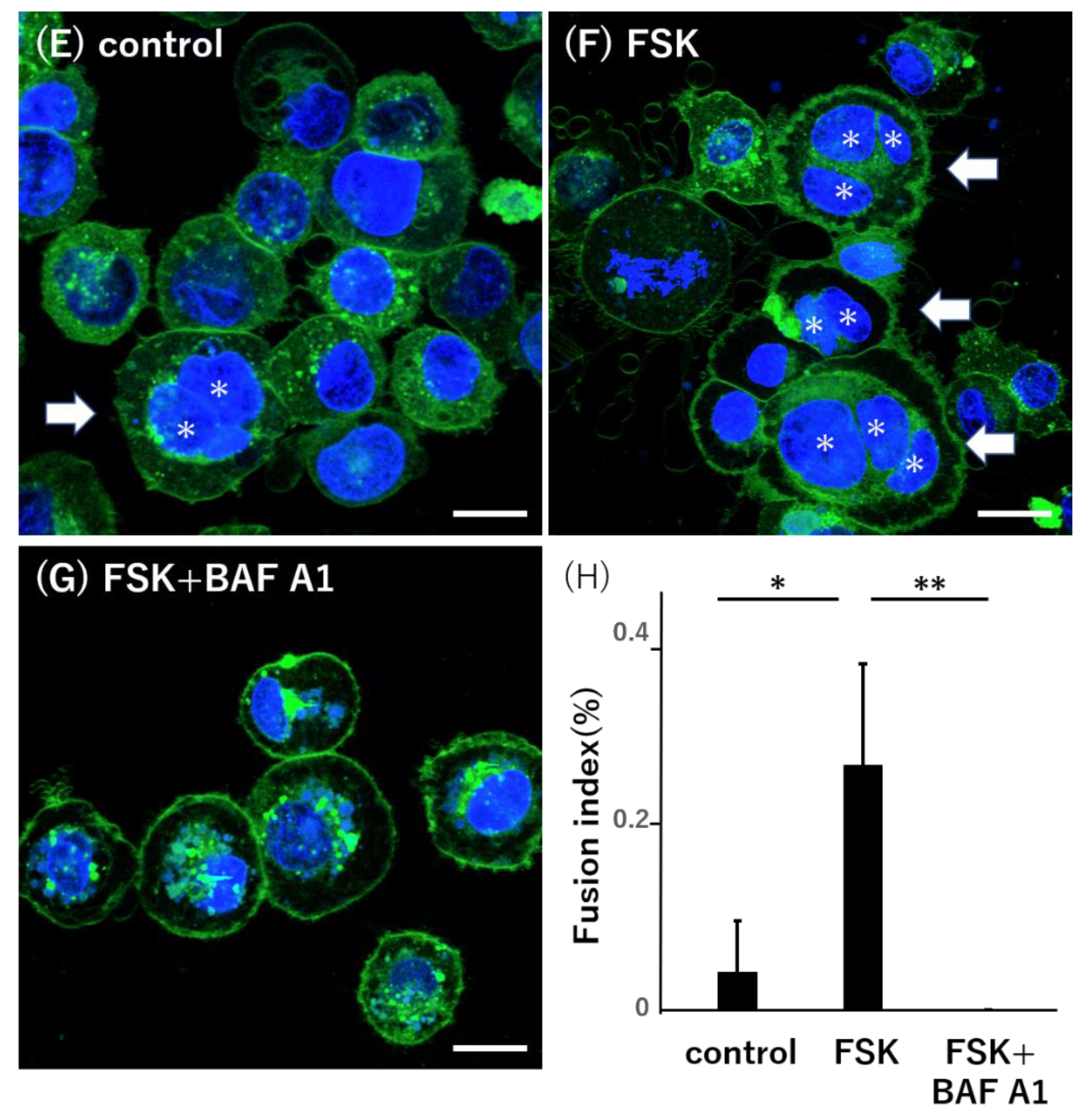
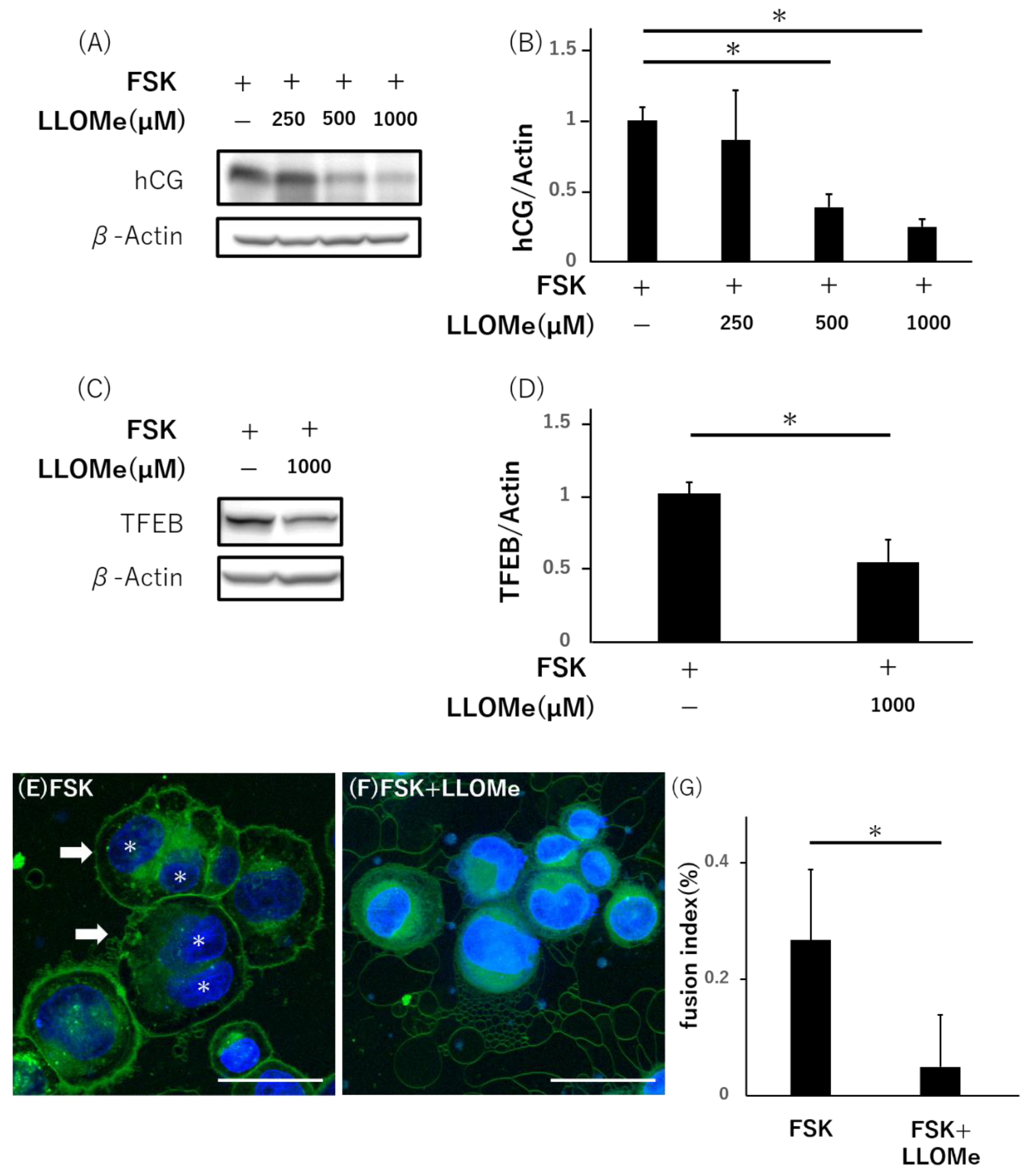
3.5. Lysosomal Impairment Inhibits Syncytialization in BeWo Cells
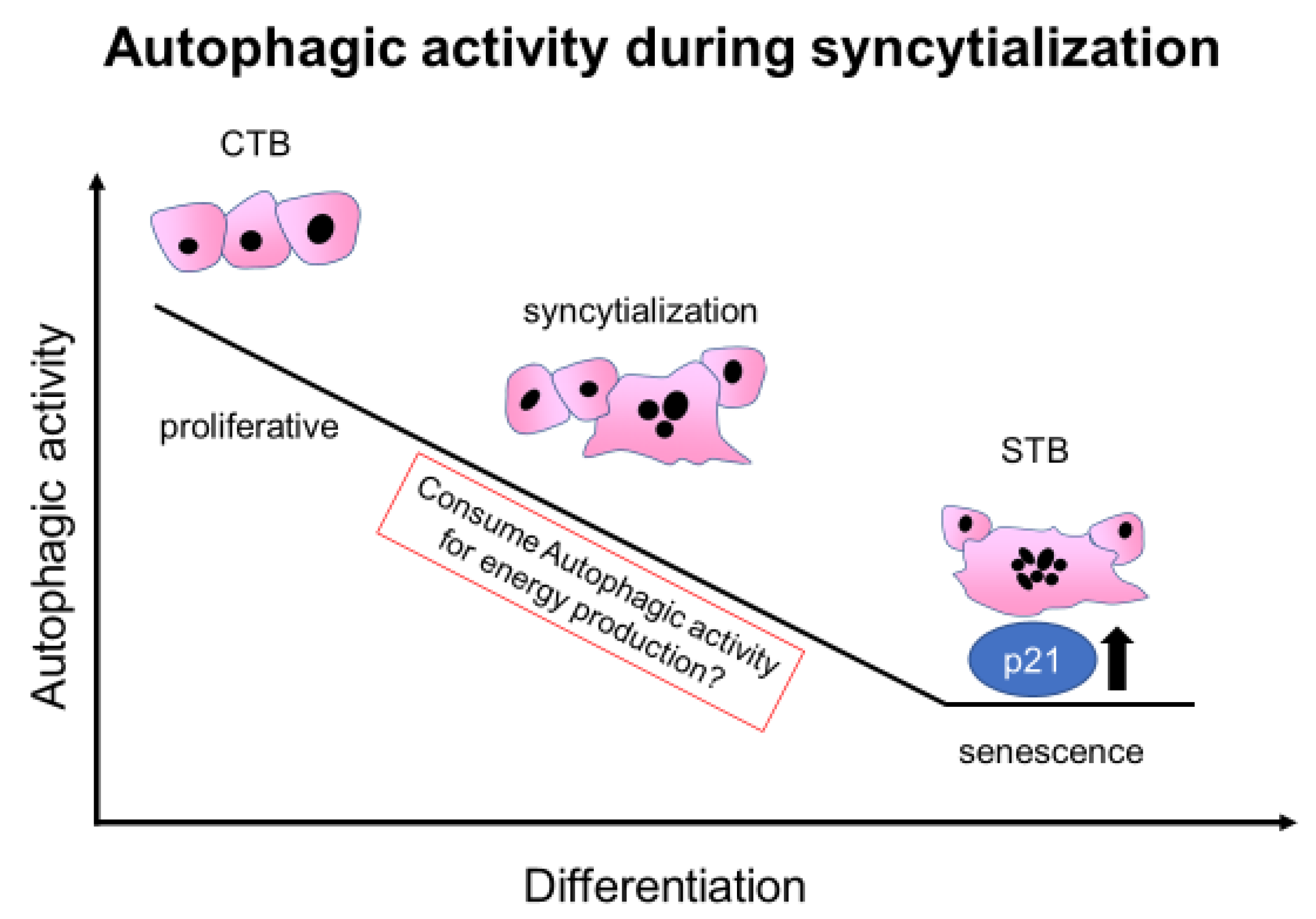
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nakashima, A.; Tsuda, S.; Kusabiraki, T.; Aoki, A.; Ushijima, A.; Shima, T.; Cheng, S.B.; Sharma, S.; Saito, S. Current Understanding of Autophagy in Pregnancy. Int. J. Mol. Sci. 2019, 20, 2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tranquilli, A.L.; Landi, B.; Giannubilo, S.R.; Sibai, B.M. Preeclampsia: No longer solely a pregnancy disease. Pregnancy Hypertens. 2012, 2, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2020, 226, S907–S927. [Google Scholar] [CrossRef]
- Dupressoir, A.; Marceau, G.; Vernochet, C.; Benit, L.; Kanellopoulos, C.; Sapin, V.; Heidmann, T. Syncytin-A and syncytin-B, two fusogenic placenta-specific murine envelope genes of retroviral origin conserved in Muridae. Proc. Natl. Acad. Sci. USA 2005, 102, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Dupressoir, A.; Vernochet, C.; Bawa, O.; Harper, F.; Pierron, G.; Opolon, P.; Heidmann, T. Syncytin-A knockout mice demonstrate the critical role in placentation of a fusogenic, endogenous retrovirus-derived, envelope gene. Proc. Natl. Acad. Sci. USA 2009, 106, 12127–12132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupressoir, A.; Vernochet, C.; Harper, F.; Guegan, J.; Dessen, P.; Pierron, G.; Heidmann, T. A pair of co-opted retroviral envelope syncytin genes is required for formation of the two-layered murine placental syncytiotrophoblast. Proc. Natl. Acad. Sci. USA 2011, 108, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langbein, M.; Strick, R.; Strissel, P.L.; Vogt, N.; Parsch, H.; Beckmann, M.W.; Schild, R.L. Impaired cytotrophoblast cell-cell fusion is associated with reduced Syncytin and increased apoptosis in patients with placental dysfunction. Mol. Reprod. Dev. 2008, 75, 175–183. [Google Scholar] [CrossRef]
- Hua, Y.; Wang, J.; Yuan, D.L.; Qi, Y.; Tang, Z.; Zhu, X.; Jiang, S.W. A tag SNP in syncytin-2 3-UTR significantly correlates with the risk of severe preeclampsia. Clin. Chim. Acta 2018, 483, 265–270. [Google Scholar] [CrossRef]
- Wang, R.; Yu, R.; Zhu, C.; Lin, H.Y.; Lu, X.; Wang, H. Tubulin detyrosination promotes human trophoblast syncytium formation. J. Mol. Cell. Biol. 2019, 11, 967–978. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Shima, T.; Tsuda, S.; Aoki, A.; Kawaguchi, M.; Yoneda, S.; Yamaki-Ushijima, A.; Cheng, S.B.; Sharma, S.; Saito, S. Disruption of Placental Homeostasis Leads to Preeclampsia. Int. J. Mol. Sci. 2020, 21, 3298. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka-Tatematsu, M.; Nakashima, A.; Fujita, N.; Shima, T.; Yoshimori, T.; Saito, S. Autophagy induced by HIF1alpha overexpression supports trophoblast invasion by supplying cellular energy. PLoS ONE 2013, 8, e76605. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Cheng, S.B.; Ikawa, M.; Yoshimori, T.; Huber, W.J.; Menon, R.; Huang, Z.; Fierce, J.; Padbury, J.F.; Sadovsky, Y.; et al. Evidence for lysosomal biogenesis proteome defect and impaired autophagy in preeclampsia. Autophagy 2020, 16, 1771–1785. [Google Scholar] [CrossRef] [PubMed]
- Aoki, A.; Nakashima, A.; Kusabiraki, T.; Ono, Y.; Yoshino, O.; Muto, M.; Kumasawa, K.; Yoshimori, T.; Ikawa, M.; Saito, S. Trophoblast-Specific Conditional Atg7 Knockout Mice Develop Gestational Hypertension. Am. J. Pathol. 2018, 188, 2474–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralimanoharan, S.; Gao, X.; Weintraub, S.; Myatt, L.; Maloyan, A. Sexual dimorphism in activation of placental autophagy in obese women with evidence for fetal programming from a placenta-specific mouse model. Autophagy 2016, 12, 752–769. [Google Scholar] [CrossRef] [Green Version]
- Kalkat, M.; Garcia, J.; Ebrahimi, J.; Melland-Smith, M.; Todros, T.; Post, M.; Caniggia, I. Placental autophagy regulation by the BOK-MCL1 rheostat. Autophagy 2013, 9, 2140–2153. [Google Scholar] [CrossRef] [Green Version]
- Melland-Smith, M.; Ermini, L.; Chauvin, S.; Craig-Barnes, H.; Tagliaferro, A.; Todros, T.; Post, M.; Caniggia, I. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy 2015, 11, 653–669. [Google Scholar] [CrossRef]
- Motomura, K.; Okada, N.; Morita, H.; Hara, M.; Tamari, M.; Orimo, K.; Matsuda, G.; Imadome, K.I.; Matsuda, A.; Nagamatsu, T.; et al. A Rho-associated coiled-coil containing kinases (ROCK) inhibitor, Y-27632, enhances adhesion, viability and differentiation of human term placenta-derived trophoblasts in vitro. PLoS ONE 2017, 12, e0177994. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Higashisaka, K.; Kusabiraki, T.; Aoki, A.; Ushijima, A.; Ono, Y.; Tsuda, S.; Shima, T.; Yoshino, O.; Nagano, K.; et al. Autophagy is a new protective mechanism against the cytotoxicity of platinum nanoparticles in human trophoblasts. Sci. Rep. 2019, 9, 5478. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H. A simple and robust fluorescent labeling method to quantify trophoblast fusion. Placenta 2019, 77, 16–18. [Google Scholar] [CrossRef]
- Nakashima, A.; Cheng, S.B.; Kusabiraki, T.; Motomura, K.; Aoki, A.; Ushijima, A.; Ono, Y.; Tsuda, S.; Shima, T.; Yoshino, O.; et al. Endoplasmic reticulum stress disrupts lysosomal homeostasis and induces blockade of autophagic flux in human trophoblasts. Sci. Rep. 2019, 9, 11466. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Wang, R.; Zhu, C.; Wang, H.; Lin, H.Y.; Gu, Y.; Cross, J.C.; Wang, H. Fine-Tuned and Cell-Cycle-Restricted Expression of Fusogenic Protein Syncytin-2 Maintains Functional Placental Syncytia. Cell Rep. 2017, 21, 1150–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastida-Ruiz, D.; Yart, L.; Wuillemin, C.; Ribaux, P.; Morris, N.; Epiney, M.; Martinez de Tejada, B.; Cohen, M. The fine-tuning of endoplasmic reticulum stress response and autophagy activation during trophoblast syncytialization. Cell Death Dis. 2019, 10, 651. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Macones, C.; Mysorekar, I.U. ATG16L1 governs placental infection risk and preterm birth in mice and women. JCI Insight 2016, 1, e86654. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Namkoong, S.; Lee, K.I.; Lee, J.I.; Park, R.; Lee, E.J.; Jang, I.S.; Park, J. The integral membrane protein ITM2A, a transcriptional target of PKA-CREB, regulates autophagic flux via interaction with the vacuolar ATPase. Autophagy 2015, 11, 756–768. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; He, G.; Chen, M.; Bao, C.; Chen, Y.; Liu, G.; Zhou, M.; Li, S.; Xu, W.; Liu, X. Abnormal CYP11A1 gene expression induces excessive autophagy, contributing to the pathogenesis of preeclampsia. Oncotarget 2017, 8, 89824–89836. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Cao, G.; Chen, D.; Liu, J.; Yu, B.; Liu, M.; Li, Y.X.; Cao, B.; Sadovsky, Y.; Wang, Y.L. Placental trophoblast syncytialization potentiates macropinocytosis via mTOR signaling to adapt to reduced amino acid supply. Proc. Natl. Acad. Sci. USA 2021, 118, e2017092118. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Shima, T.; Tsuda, S.; Aoki, A.; Kawaguchi, M.; Furuta, A.; Yasuda, I.; Yoneda, S.; Yamaki-Ushijima, A.; Cheng, S.B.; et al. Aggrephagy Deficiency in the Placenta: A New Pathogenesis of Preeclampsia. Int. J. Mol. Sci. 2021, 22, 2432. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Shigeyama, S.; Minami, S.; Shima, T.; Akayama, S.; Matsuda, T.; Esposito, A.; Napolitano, G.; Kuma, A.; Namba-Hamano, T.; et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat. Cell Biol. 2020, 22, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Freitag, N.; Tirado-Gonzalez, I.; Barrientos, G.; Powell, K.L.; Boehm-Sturm, P.; Koch, S.P.; Hecher, K.; Staff, A.C.; Arck, P.C.; Diemert, A.; et al. Galectin-3 deficiency in pregnancy increases the risk of fetal growth restriction (FGR) via placental insufficiency. Cell Death Dis. 2020, 11, 560. [Google Scholar] [CrossRef]
- Higuchi, S.; Miyamoto, T.; Kobara, H.; Yamada, S.; Asaka, R.; Kikuchi, N.; Kashima, H.; Ohira, S.; Shiozawa, T. Trophoblast type-specific expression of senescence markers in the human placenta. Placenta 2019, 85, 56–62. [Google Scholar] [CrossRef]
- Duan, L.; Schimmelmann, M.; Wu, Y.; Reisch, B.; Faas, M.; Kimmig, R.; Winterhager, E.; Koninger, A.; Gellhaus, A. CCN3 Signaling Is Differently Regulated in Placental Diseases Preeclampsia and Abnormally Invasive Placenta. Front. Endocrinol. (Lausanne) 2020, 11, 597549. [Google Scholar] [CrossRef]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I.; Bishop, A.J.; Motoyama, N.; Cao, L.; Finkel, T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furuta, A.; Shima, T.; Kawaguchi, M.; Yamaki-Ushijima, A.; Yasuda, I.; Tsuda, S.; Yoneda, S.; Higashisaka, K.; Cheng, S.-B.; Matsumoto, K.; et al. The Autophagy-Lysosomal Machinery Enhances Cytotrophoblast–Syncytiotrophoblast Fusion Process. Reprod. Med. 2022, 3, 112-126. https://doi.org/10.3390/reprodmed3020010
Furuta A, Shima T, Kawaguchi M, Yamaki-Ushijima A, Yasuda I, Tsuda S, Yoneda S, Higashisaka K, Cheng S-B, Matsumoto K, et al. The Autophagy-Lysosomal Machinery Enhances Cytotrophoblast–Syncytiotrophoblast Fusion Process. Reproductive Medicine. 2022; 3(2):112-126. https://doi.org/10.3390/reprodmed3020010
Chicago/Turabian StyleFuruta, Atsushi, Tomoko Shima, Mihoko Kawaguchi, Akemi Yamaki-Ushijima, Ippei Yasuda, Sayaka Tsuda, Satoshi Yoneda, Kazuma Higashisaka, Shi-Bin Cheng, Kenji Matsumoto, and et al. 2022. "The Autophagy-Lysosomal Machinery Enhances Cytotrophoblast–Syncytiotrophoblast Fusion Process" Reproductive Medicine 3, no. 2: 112-126. https://doi.org/10.3390/reprodmed3020010
APA StyleFuruta, A., Shima, T., Kawaguchi, M., Yamaki-Ushijima, A., Yasuda, I., Tsuda, S., Yoneda, S., Higashisaka, K., Cheng, S.-B., Matsumoto, K., Tsutsumi, Y., Sharma, S., Saito, S., & Nakashima, A. (2022). The Autophagy-Lysosomal Machinery Enhances Cytotrophoblast–Syncytiotrophoblast Fusion Process. Reproductive Medicine, 3(2), 112-126. https://doi.org/10.3390/reprodmed3020010