
Adult Presentation of X-Linked Hypophosphatemia


Abstract
:1. Introduction
2. Diagnosis of Adult XLH
3. Symptoms of Adult XLH
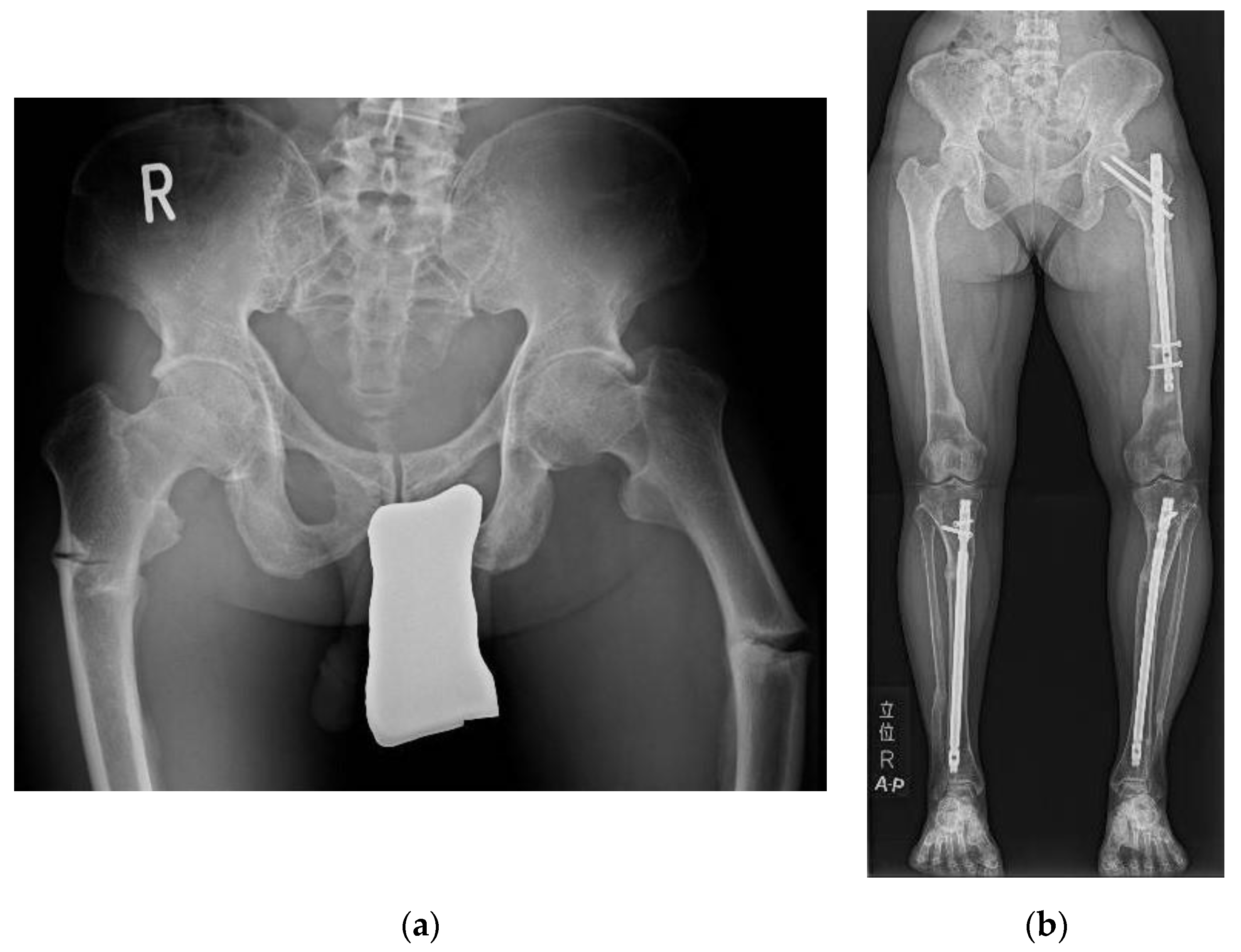
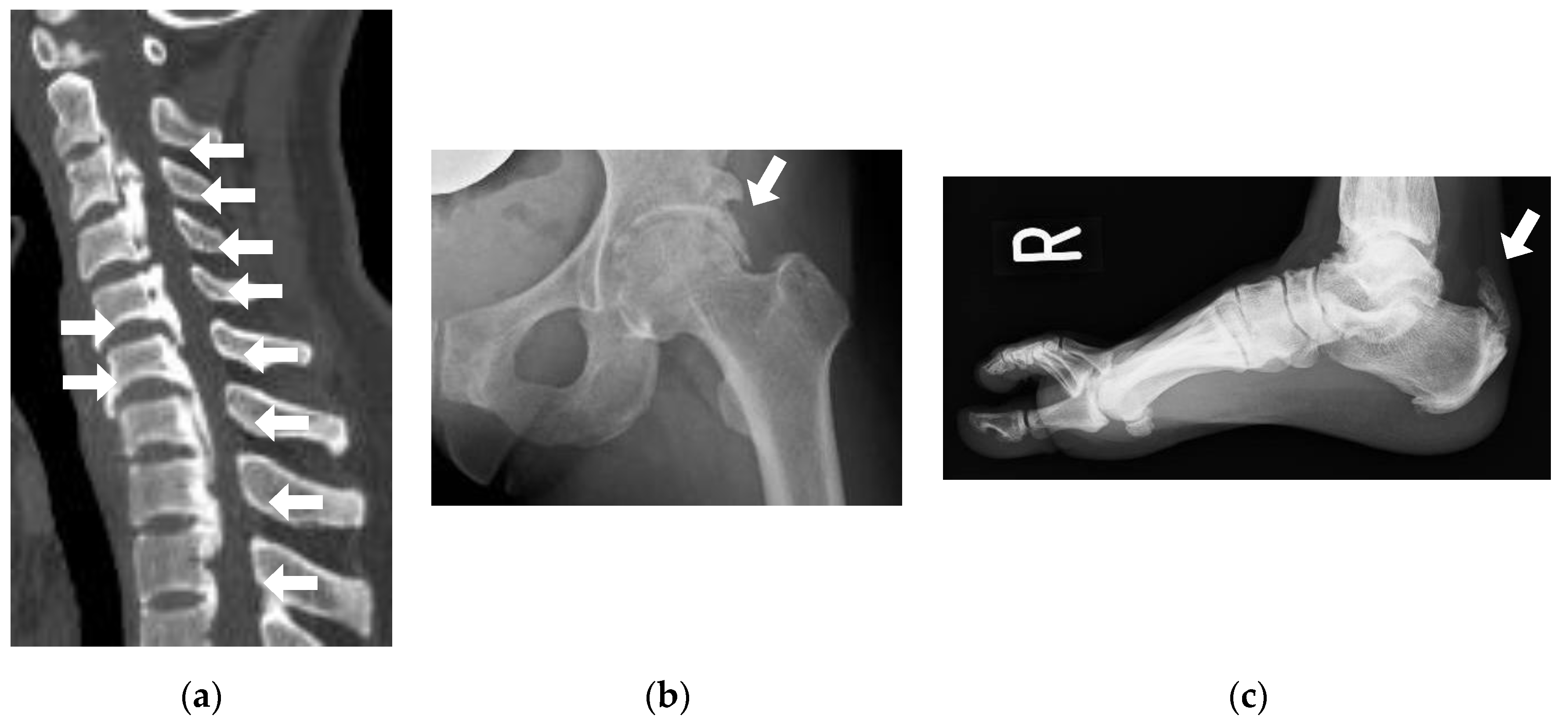
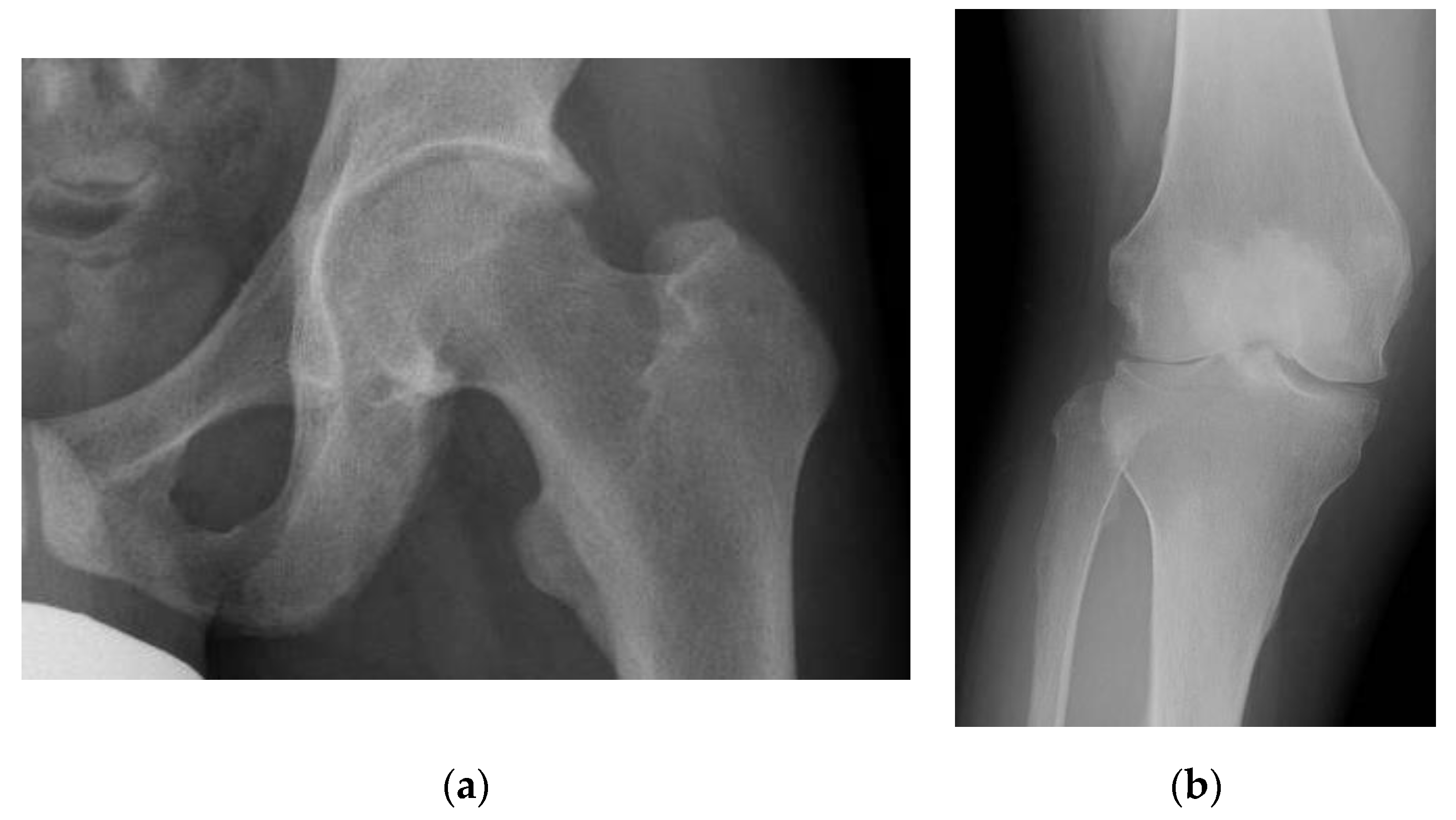
3.1. Pseudofracture and Fracture
3.2. Muscle Weakness
3.3. Dental Health
3.4. Enthesopathy
3.5. Osteoarthritis
3.6. SHPT/THPT
3.7. CKD
3.8. Hypertension and Left Ventricular Hypertrophy
3.9. Hearing Loss
4. QOL of Adult XLH
5. Transition of XLH Patients
6. Treatment of Adult XLH
6.1. Indication and Selection of Treatment
6.2. Conventional Therapy (Active Vitamin D and Phosphate Supplementation)
6.3. Burosumab
7. Remaining Problems and Future Research Topics in Adult XLH Patients
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carpenter, T.O.; Imel, E.A.; Holm, I.A.; Jan de Beur, S.M.; Insogna, K.L. A clinician’s guide to X-linked hypophosphatemia. J. Bone Miner. Res. 2011, 26, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, M.; Roschger, P.; Klaushofer, K.; Veilleux, L.N.; Roughley, P.; Glorieux, F.H.; Rauch, F. Cortical and trabecular bone density in X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 2013, 98, E954–E961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Koga, M.; Kinoshita, Y.; Taniguchi, Y.; Kobayashi, H.; Fukumoto, S.; Nangaku, M.; Makita, N.; Ito, N. Incidence of Complications in 25 Adult Patients with X-linked Hypophosphatemia. J. Clin. Endocrinol. Metab. 2021, 106, e3682–e3692. [Google Scholar] [CrossRef]
- DeLacey, S.; Liu, Z.; Broyles, A.; El-Azab, S.A.; Guandique, C.F.; James, B.C.; Imel, E.A. Hyperparathyroidism and parathyroidectomy in X-linked hypophosphatemia patients. Bone 2019, 127, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Aono, Y.; Yamazaki, Y.; Yasutake, J.; Kawata, T.; Hasegawa, H.; Urakawa, I.; Fujita, T.; Wada, M.; Yamashita, T.; Fukumoto, S.; et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J. Bone Miner. Res. 2009, 24, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Insogna, K.L.; Briot, K.; Imel, E.A.; Kamenický, P.; Ruppe, M.D.; Portale, A.A.; Weber, T.; Pitukcheewanont, P.; Cheong, H.I.; Jan de Beur, S.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults with X-Linked Hypophosphatemia: Week 24 Primary Analysis. J. Bone Miner. Res. 2018, 33, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, T.O.; Whyte, M.P.; Imel, E.A.; Boot, A.M.; Högler, W.; Linglart, A.; Padidela, R.; Van’t Hoff, W.; Mao, M.; Chen, C.Y.; et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N. Engl. J. Med. 2018, 378, 1987–1998. [Google Scholar] [CrossRef] [Green Version]
- Portale, A.A.; Carpenter, T.O.; Brandi, M.L.; Briot, K.; Cheong, H.I.; Cohen-Solal, M.; Crowley, R.; Jan De Beur, S.; Eastell, R.; Imanishi, Y.; et al. Continued Beneficial Effects of Burosumab in Adults with X-Linked Hypophosphatemia: Results from a 24-Week Treatment Continuation Period after a 24-Week Double-Blind Placebo-Controlled Period. Calcif. Tissue Res. 2019, 105, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Imel, E.A.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Ward, L.M.; Nilsson, O.; Simmons, J.H.; Padidela, R.; Namba, N.; Cheong, H.I.; et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: A randomised, active-controlled, open-label, phase 3 trial. Lancet 2019, 393, 2416–2427. [Google Scholar] [CrossRef]
- Briot, K.; Portale, A.A.; Brandi, M.L.; Carpenter, T.O.; Cheong, H.I.; Cohen-Solal, M.; Crowley, R.K.; Eastell, R.; Imanishi, Y.; Ing, S.; et al. Burosumab treatment in adults with X-linked hypophosphataemia: 96-week patient-reported outcomes and ambulatory function from a randomised phase 3 trial and open-label extension. RMD Open 2021, 7, e001714. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, S.; Ozono, K.; Michigami, T.; Minagawa, M.; Okazaki, R.; Sugimoto, T.; Takeuchi, Y.; Matsumoto, T. Pathogenesis and diagnostic criteria for rickets and osteomalacia—Proposal by an expert panel supported by the Ministry of Health, Labour and Welfare, Japan, the Japanese Society for Bone and Mineral Research, and the Japan Endocrine Society. J. Bone Miner. Metab. 2015, 33, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Kubota, T.; Kitanaka, S.; Fujiwara, I.; Adachi, M.; Takeuchi, Y.; Yamagami, H.; Kimura, T.; Shinoda, T.; Minagawa, M.; et al. Clinical performance of a novel chemiluminescent enzyme immunoassay for FGF23. J. Bone Miner. Metab. 2021, 39, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Hidaka, N.; Koga, M.; Ogawa, N.; Takahashi, S.; Miyazaki, H.; Nangaku, M.; Makita, N.; Ito, N. Performance evaluation of the new chemiluminescent intact FGF23 assay relative to the existing assay system. J. Bone Miner. Metab. 2022, 40, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Saito, T.; Shimizu, Y.; Hori, M.; Taguchi, M.; Igarashi, T.; Fukumoto, S.; Fujita, T. Mutational analysis of patients with FGF23-related hypophosphatemic rickets. Eur. J. Endocrinol. 2012, 167, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Minisola, S.; Peacock, M.; Fukumoto, S.; Cipriani, C.; Pepe, J.; Tella, S.H.; Collins, M.T. Tumour-induced osteomalacia. Nat. Rev. Dis. Prim. 2017, 3, 17044. [Google Scholar] [CrossRef]
- Shimizu, Y.; Tada, Y.; Yamauchi, M.; Okamoto, T.; Suzuki, H.; Ito, N.; Fukumoto, S.; Sugimoto, T.; Fujita, T. Hypophosphatemia induced by intravenous administration of saccharated ferric oxide: Another form of FGF23-related hypophosphatemia. Bone 2009, 45, 814–816. [Google Scholar] [CrossRef]
- Hidaka, N.; Kato, H.; Koga, M.; Katsura, M.; Oyama, Y.; Kinoshita, Y.; Fukumoto, S.; Makita, N.; Nangaku, M.; Ito, N. Induction of FGF23-related hypophosphatemic osteomalacia by alcohol consumption. Bone Rep. 2021, 15, 101144. [Google Scholar] [CrossRef]
- Insogna, K.L.; Rauch, F.; Kamenický, P.; Ito, N.; Kubota, T.; Nakamura, A.; Zhang, L.; Mealiffe, M.; San Martin, J.; Portale, A.A. Burosumab Improved Histomorphometric Measures of Osteomalacia in Adults with X-Linked Hypophosphatemia: A Phase 3, Single-Arm, International Trial. J. Bone Miner. Res. 2019, 34, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- Veilleux, L.N.; Cheung, M.S.; Glorieux, F.H.; Rauch, F. The muscle-bone relationship in X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 2013, 98, E990–E995. [Google Scholar] [CrossRef] [Green Version]
- Ito, N.; Fukumoto, S.; Takeuchi, Y.; Takeda, S.; Suzuki, H.; Yamashita, T.; Fujita, T. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J. Bone Miner. Metab. 2007, 25, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Beck-Nielsen, S.S.; Brusgaard, K.; Rasmussen, L.M.; Brixen, K.; Brock-Jacobsen, B.; Poulsen, M.R.; Vestergaard, P.; Ralston, S.H.; Albagha, O.M.; Poulsen, S.; et al. Phenotype presentation of hypophosphatemic rickets in adults. Calcif. Tissue Res. 2010, 87, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Liu, R.; White, N.; Alon, U.S.; Cobb, C.M. Periodontal status of patients with hypophosphatemic rickets: A case series. J. Periodontol. 2011, 82, 1530–1535. [Google Scholar] [CrossRef] [PubMed]
- Biosse Duplan, M.; Coyac, B.R.; Bardet, C.; Zadikian, C.; Rothenbuhler, A.; Kamenicky, P.; Briot, K.; Linglart, A.; Chaussain, C. Phosphate and Vitamin D Prevent Periodontitis in X-Linked Hypophosphatemia. J. Dent. Res. 2016, 96, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.; Olear, E.A.; Insogna, K.L.; Katz, L.; Baker, S.; Kaur, R.; Simpson, C.A.; Sterpka, J.; Dubrow, R.; Zhang, J.H.; et al. Conventional Therapy in Adults with X-Linked Hypophosphatemia: Effects on Enthesopathy and Dental Disease. J. Clin. Endocrinol. Metab. 2015, 100, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Portale, A.A.; Högler, W.; Simmons, J.H.; Gottesman, G.S.; Padidela, R.; Namba, N.; et al. Impact of Burosumab Compared with Conventional Therapy in Younger versus Older Children with X-Linked Hypophosphatemia. J. Clin. Endocrinol. Metab. 2022, dgac296. [Google Scholar] [CrossRef]
- Steele, A.; Gonzalez, R.; Garbalosa, J.C.; Steigbigel, K.; Grgurich, T.; Parisi, E.J.; Feinn, R.S.; Tommasini, S.M.; Macica, C.M. Osteoarthritis, Osteophytes, and Enthesophytes Affect Biomechanical Function in Adults with X-linked Hypophosphatemia. J. Clin. Endocrinol. Metab. 2020, 105, e1798–e1814. [Google Scholar] [CrossRef]
- Karaplis, A.C.; Bai, X.; Falet, J.P.; Macica, C.M. Mineralizing enthesopathy is a common feature of renal phosphate-wasting disorders attributed to FGF23 and is exacerbated by standard therapy in hyp mice. Endocrinology 2012, 153, 5906–5917. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Shimizu, Y.; Hori, M.; Taguchi, M.; Igarashi, T.; Fukumoto, S.; Fujita, T. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone 2011, 49, 913–916. [Google Scholar] [CrossRef]
- Kato, H.; Ansh, A.J.; Lester, E.R.; Kinoshita, Y.; Hidaka, N.; Hoshino, Y.; Koga, M.; Taniguchi, Y.; Uchida, T.; Yamaguchi, H.; et al. Identification of ENPP1 Haploinsufficiency in Patients with Diffuse Idiopathic Skeletal Hyperostosis and Early-Onset Osteoporosis. J. Bone Miner. Res. 2022, 37, 1125–1135. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Kintzinger, K.; Hackbarth, M.E.; Botschen, U.; Nitschke, Y.; Mughal, M.Z.; Baujat, G.; Schnabel, D.; Yuen, E.; Gahl, W.A.; et al. Ectopic Calcification and Hypophosphatemic Rickets: Natural History of ENPP1 and ABCC6 Deficiencies. J. Bone Miner. Res. 2021, 36, 2193–2202. [Google Scholar] [CrossRef]
- Bernhard, E.; Nitschke, Y.; Khursigara, G.; Sabbagh, Y.; Wang, Y.; Rutsch, F. A Reference Range for Plasma Levels of Inorganic Pyrophosphate in Children Using the ATP Sulfurylase Method. J. Clin. Endocrinol. Metab. 2021, 107, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Maulding, N.D.; Kavanagh, D.; Zimmerman, K.; Coppola, G.; Carpenter, T.O.; Jue, N.K.; Braddock, D.T. Genetic pathways disrupted by ENPP1 deficiency provide insight into mechanisms of osteoporosis, osteomalacia, and paradoxical mineralization. Bone 2020, 142, 115656. [Google Scholar] [CrossRef]
- Trombetti, A.; Al-Daghri, N.; Brandi, M.L.; Cannata-Andía, J.B.; Cavalier, E.; Chandran, M.; Chaussain, C.; Cipullo, L.; Cooper, C.; Haffner, D.; et al. Interdisciplinary management of FGF23-related phosphate wasting syndromes: A Consensus Statement on the evaluation, diagnosis and care of patients with X-linked hypophosphataemia. Nat. Rev. Endocrinol. 2022, 18, 366–384. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Prideaux, M.; Wijenayaka, A.R.; Yang, D.; Ormsby, R.T.; Bonewald, L.F.; Atkins, G.J. Sclerostin Directly Stimulates Osteocyte Synthesis of Fibroblast Growth Factor-23. Calcif. Tissue Res. 2021, 109, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Fukumoto, S.; Cheong, H.I.; Michigami, T.; Namba, N.; Ito, N.; Tokunaga, S.; Gibbs, Y.; Ozono, K. Long-term outcomes for Asian patients with X-linked hypophosphataemia: Rationale and design of the SUNFLOWER longitudinal, observational cohort study. BMJ Open 2020, 10, e036367. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Takagi, M.; Takeda, R.; Miyai, K.; Hasegawa, Y. Hypertension is a characteristic complication of X-linked hypophosphatemia. Endocr. J. 2017, 64, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Beck-Nielsen, S.S.; Mughal, Z.; Haffner, D.; Nilsson, O.; Levtchenko, E.; Ariceta, G.; de Lucas Collantes, C.; Schnabel, D.; Jandhyala, R.; Mäkitie, O. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J. Rare Dis. 2019, 14, 58. [Google Scholar] [CrossRef]
- Skrinar, A.; Dvorak-Ewell, M.; Evins, A.; Macica, C.; Linglart, A.; Imel, E.A.; Theodore-Oklota, C.; San Martin, J. The Lifelong Impact of X-Linked Hypophosphatemia: Results from a Burden of Disease Survey. J. Endocr. Soc. 2019, 3, 1321–1334. [Google Scholar] [CrossRef] [Green Version]
- Seefried, L.; Smyth, M.; Keen, R.; Harvengt, P. Burden of disease associated with X-linked hypophosphataemia in adults: A systematic literature review. Osteoporos. Int. 2020, 32, 7–22. [Google Scholar] [CrossRef]
- Ito, N.; Kang, H.G.; Nishida, Y.; Evins, A.; Skrinar, A.; Cheong, H.I. Burden of disease of X-linked hypophosphatemia in Japanese and Korean patients: A cross-sectional survey. Endocr. J. 2022, 69, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Dahir, K.; Dhaliwal, R.; Simmons, J.; Imel, E.A.; Gottesman, G.S.; Mahan, J.D.; Prakasam, G.; Hoch, A.I.; Ramesan, P.; Díaz-González de Ferris, M. Health Care Transition from Pediatric- to Adult-Focused Care in X-linked Hypophosphatemia: Expert Consensus. J. Clin. Endocrinol. Metab. 2021, 107, 599–613. [Google Scholar] [CrossRef]
- Horn, A.; Wright, J.; Bockenhauer, D.; Van’t Hoff, W.; Eastwood, D.M. The orthopaedic management of lower limb deformity in hypophosphataemic rickets. J. Child. Orthop. 2017, 11, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Bettinelli, A.; Bianchi, M.L.; Mazzucchi, E.; Gandolini, G.; Appiani, A.C. Acute effects of calcitriol and phosphate salts on mineral metabolism in children with hypophosphatemic rickets. J. Pediatr. 1991, 118, 372–376. [Google Scholar] [CrossRef]
- Peacock, M.; Bolognese, M.A.; Borofsky, M.; Scumpia, S.; Sterling, L.R.; Cheng, S.; Shoback, D. Cinacalcet treatment of primary hyperparathyroidism: Biochemical and bone densitometric outcomes in a five-year study. J. Clin. Endocrinol. Metab. 2009, 94, 4860–4867. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Observation | Conventional Therapy | Burosumab |
|---|---|---|
| BAP (≤upper limit of the reference range) | BAP (>upper limit of reference range) with eGFR ≥ 45 mL/min/1.73 m2 | BAP (>upper limit of the reference range) with conventional treatment |
| Stature (≥−1.0 SD) | Short stature (−2.0 ≤ −1.0 SD) with eGFR ≥ 45 mL/min/1.73 m2 | Short stature (<−2.0 SD) |
| Mechanical axis of the leg within zone 1 [43] | Deviation of the mechanical axis of the leg into zone 2 [43] with eGFR ≥ 45 L/min/1.73 m2 | Deviation of the mechanical axis of the leg into zone 3 or greater [43] or history of corrective surgery for leg deformity |
| No history of pseudofracture/bone pain/fracture in weight-bearing bone or odontitis/tooth abscesses | Pseudofracture/bone pain in weight-bearing bone or odontitis/tooth abscess once with eGFR ≥ 45 mL/min/1.73 m2 | Pseudofracture/bone pain in weight-bearing bone or odontitis/tooth abscesses more than two times or more than once with conventional treatment or once requiring impending surgery |
| No response or trivial response to burosumab | Fracture in weight-bearing bone | |
| Severe adverse event with burosumab | Uncontrolled severe SHPT with conventional treatment (e.g., peak intact PTH ≥ twice the upper limit of the reference range); | |
| THPT; | ||
| Patients with symptoms described in the “conventional therapy” and eGFR < 45 mL/min/1.73 m2; | ||
| Severe adverse event with conventional therapy |
| Priority | Event | Action |
|---|---|---|
| Initiation of treatment | Start with active vitamin D (0.50 μg b.i.d. for calcitriol or 1.0 μg s.i.d. for alfacalcidol) After 1 to 2 weeks, start phosphate supplementation (800 mg q.i.d.) | |
| Range of dosage | Calcitriol: 0.50 to 0.75 μg b.i.d., alfacalcidol: 0.75 to 1.5 μg s.i.d. Phosphate supplementation: 750 to 1600 mg q.i.d. | |
| Initial phase | Adjust phosphate supplementation for a period of time by 100 mg Goal: peak phosphate 1 within lower 50% of the reference range; laboratory test: every one to four weeks | |
| Maintenance phase (after 12 to 18 months) | Adjust phosphate supplementation for a period of time by 100 mg Goal: BAP within the reference range Laboratory test: every 3 months Uncontrolled BAP: change the treatment to burosumab | |
| High | Severe SHPT (peak intact PTH ≥ 2 × upper limit of the reference range) | Immediately decrease phosphate supplementation by 100 mg or more Increase active vitamin D (0.25 μg daily for calcitriol and 0.25 to 0.5 μg daily for alfacalcidol) After 1 to 2 weeks, increase phosphate supplementation Uncontrolled severe SHPT: change the treatment to burosumab |
| High | Development of THPT (hyperparathyroidism with the value of calcium ≥ the middle of the reference range) | Immediately quit conventional therapy Initiate allosteric modulation of calcium sensing receptor Try to prevent dehydration Conduct parathyroidectomy for all recognizable glands, otherwise continue allosteric modulation of the calcium sensing receptor in patients with contraindication for surgery Change the treatment to burosumab afterward |
| High | Overadjustment of peak phosphate within upper 25% of the reference range or over | Immediately decrease phosphate supplementation by 100 mg or more |
| High | Progression of CKD to eGFR < 45 mL/min/1.73 m2 | Decrease conventional therapy Change the treatment to burosumab in patients who still need phosphate supplementation (uncontrolled BAP with active vitamin D). |
| Priority | Event | Action |
|---|---|---|
| Initiation of treatment | 1.0 mg/kg body weight (up to 90 mg) subcutaneous injection every four weeks Response to burosumab should be confirmed by peak phosphate 1; Concomitant use of active vitamin D and phosphate supplementation is not recommended or is prohibited unless another medical condition requires it | |
| Trough phosphate 2 within the higher half of the reference range | Decrease burosumab dose by 0.2 to 0.3 mg/kg Subsequently, fine tune burosumab dose by 0.1 mg/kg to target trough phosphate within lower half of the reference range | |
| High | Trough phosphate over the reference range | Suspend burosumab until phosphate values fall below the normal range Restarted burosumab at approximately half the initial starting dose |
| No or trivial response at peak phosphate | Rule out vitamin D deficiency by measuring 25OHD Severe SHPT: continue burosumab 3 to 6 times and confirm improvement in peak phosphate Continuing no response or trivial response after problems above are ruled out or addressed; change treatment to conventional therapy | |
| High | THPT | Initiate allosteric modulation of the calcium sensing receptor Try to prevent dehydration Conduct parathyroidectomy for all recognizable glands, otherwise continue allosteric modulation of the calcium sensing receptor in patients with contraindication for surgery |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, N. Adult Presentation of X-Linked Hypophosphatemia. Endocrines 2022, 3, 375-390. https://doi.org/10.3390/endocrines3030030
Ito N. Adult Presentation of X-Linked Hypophosphatemia. Endocrines. 2022; 3(3):375-390. https://doi.org/10.3390/endocrines3030030
Chicago/Turabian StyleIto, Nobuaki. 2022. "Adult Presentation of X-Linked Hypophosphatemia" Endocrines 3, no. 3: 375-390. https://doi.org/10.3390/endocrines3030030
APA StyleIto, N. (2022). Adult Presentation of X-Linked Hypophosphatemia. Endocrines, 3(3), 375-390. https://doi.org/10.3390/endocrines3030030