
Recent Developments in Stereoselective Reactions of Sulfonium Ylides

Abstract
:1. Introduction
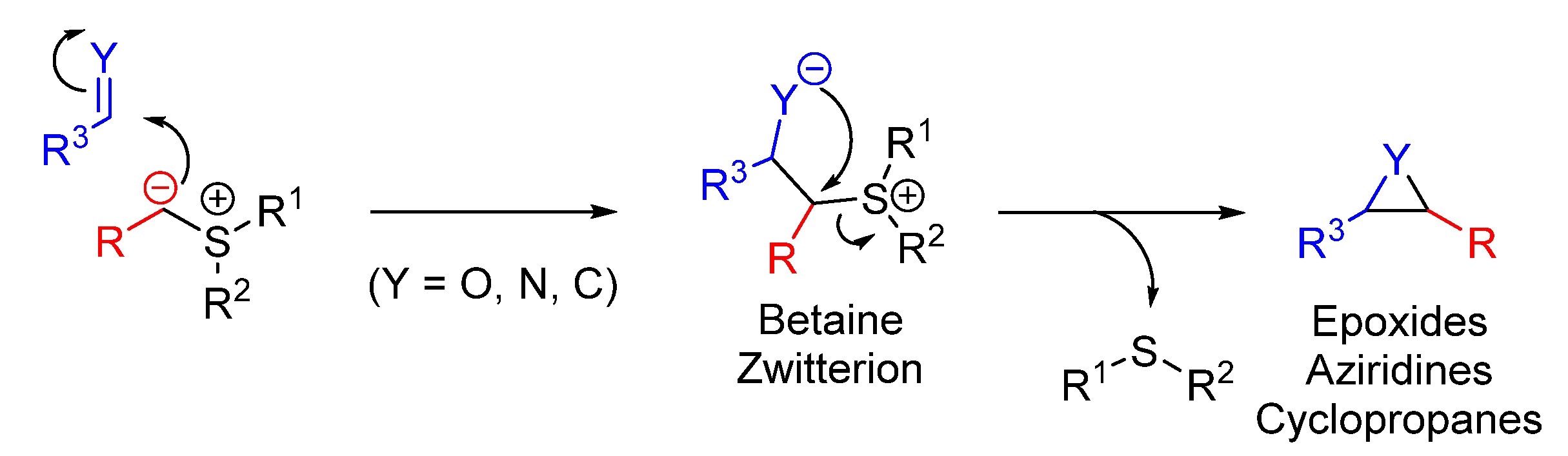
2. The Special Characteristics of Sulfur Ylides
3. Synthesis of Sulfonium Ylides
Synthesis of Sulfonium Salts and Ylides
4. Reactions of Sulfonium Ylides
4.1. Epoxidation
4.1.1. Stoichiometric Sulfonium Ylide-Mediated Epoxidation
4.1.2. Stoichiometric Allyl Sulfonium Ylide-Mediated Epoxidation
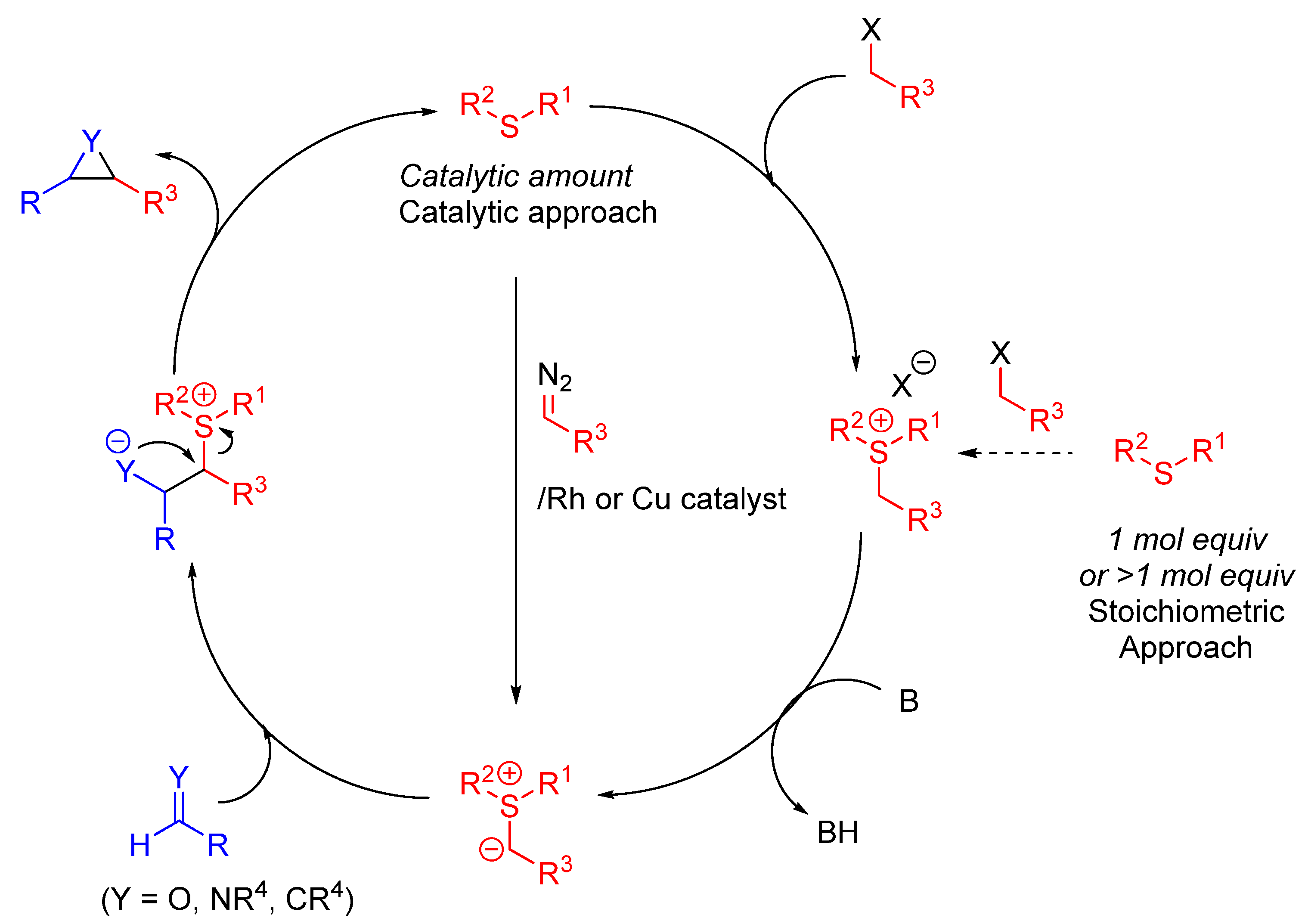
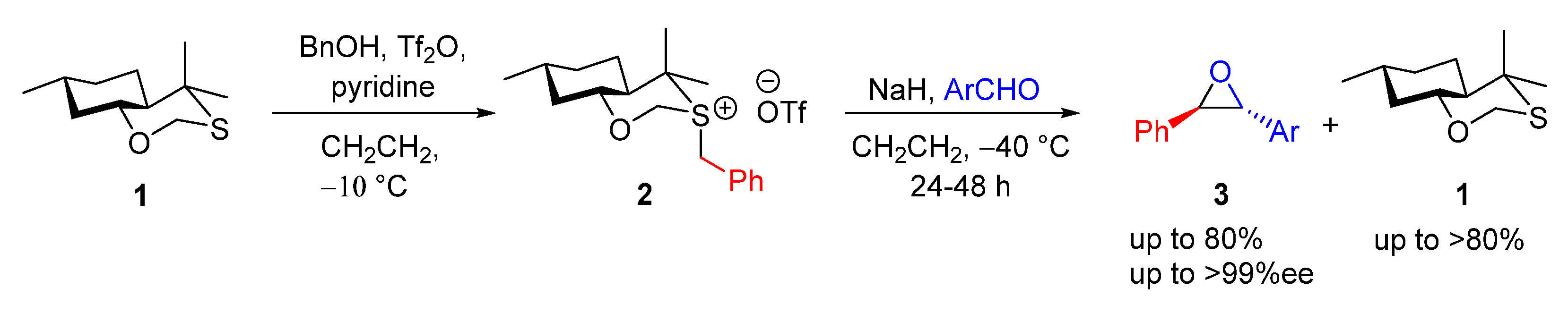
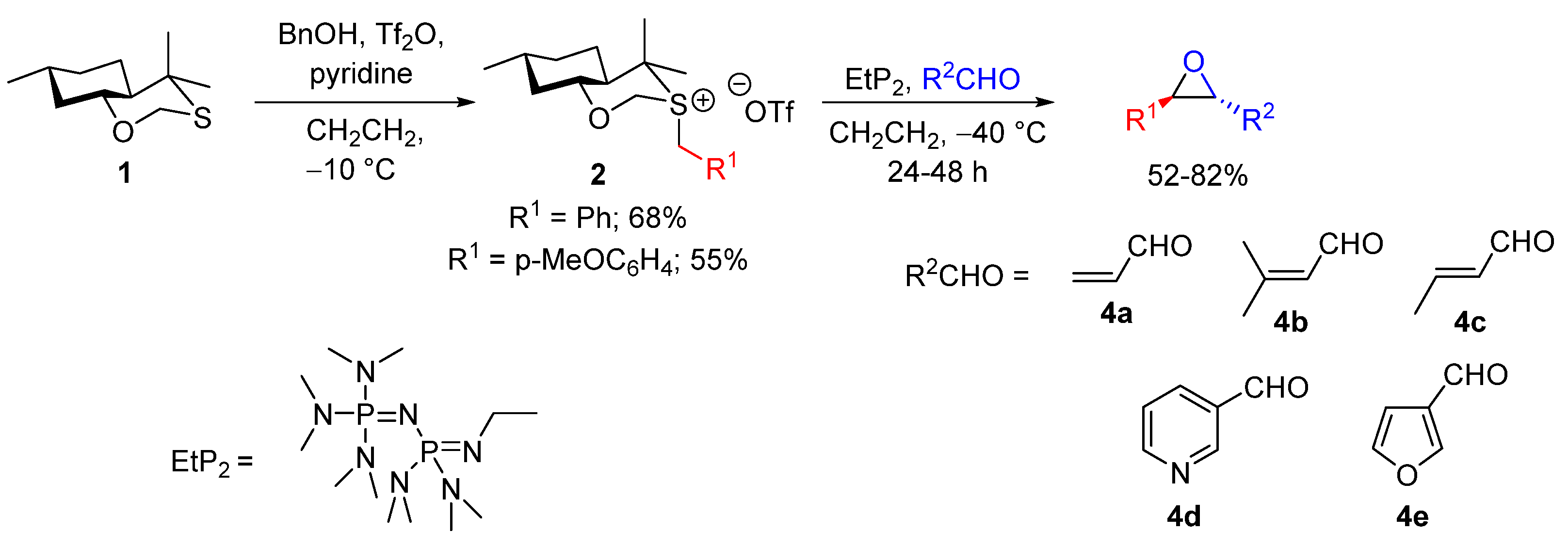
4.1.3. Catalytic Sulfonium Ylide-Mediated Epoxidation
4.2. Aziridination
4.2.1. Stoichiometric Sulfonium Ylide-Mediated Aziridination
4.2.2. Stoichiometric Allyl Sulfonium Ylide-Mediated Aziridination
4.2.3. Catalytic Sulfonium Ylide-Mediated Aziridination
4.3. Cyclopropanation
4.3.1. Sulfonium Ylide-Mediated Cyclopropanation
4.3.2. Allyl Sulfonium Ylide-Promoted Cyclopropanation
4.3.3. Catalytic Cyclopropanation
4.4. Miscellaneous Stereoselective Reactions of Sulfonium Ylides
4.4.1. Borane Homologation Reactions
4.4.2. [4 + 1] Cycloadditions
4.4.3. [4 + 1]-Cycloaddition Cascade Reactions
4.4.4. [3 + 1]-Cycloadditions
4.4.5. [3 + 3]/[1 + 4] Tandem Cycloaddition
4.4.6. Corey–Chaykovsky Cyclopropanation/Cloke−Wilson Rearrangement
4.4.7. [2,3]-Sigmatropic Rearrangements
4.4.8. [1,2]-Rearrangements
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wittig, G.; Geissler, G. Zur Reaktionsweise des Pentaphenyl-phosphors und einiger Derivate. Liebigs Ann. Chem. 1953, 580, 44–57. [Google Scholar] [CrossRef]
- Longwitz, L.; Werner, T. Recent Advances in Catalytic Wittig-Type Reactions Based on P(III)/P(V) Redox Cycling. Pure Appl. Chem. 2019, 91, 95–102. [Google Scholar] [CrossRef]
- Ingold, C.K.; Jessop, J.A. XCV.—Influence of Poles and Polar Linkings on the Course Pursued by Elimination Reactions. Part IX. Isolation of a Substance Believed to Contain a Semipolar Double Linking with Participating Carbon. J. Chem. Soc. 1930, 713–718. [Google Scholar] [CrossRef]
- Johnson, A.W.; LaCount, R.B. The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides. J. Am. Chem. Soc. 1961, 83, 417–423. [Google Scholar] [CrossRef]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar] [CrossRef]
- Li, A.-H.; Dai, L.-X.; Aggarwal, V.K. Asymmetric Ylide Reactions: Epoxidation, Cyclopropanation, Aziridination, Olefination, and Rearrangement. Chem. Rev. 1997, 97, 2341–2372. [Google Scholar] [CrossRef]
- Block, E. Reactions of Organosulfur Compounds; Academic Press: New York, NY, USA, 1978; ISBN 0121070506. [Google Scholar]
- Trost, B.M.; Melvin, L.S., Jr. Sulfur Ylides: Emerging Synthetic Intermediates; Academic Press: New York, NY, USA, 1975; ISBN 0127010602. [Google Scholar]
- Von, E.; Doering, W.; Hoffmann, A.K.; d-Orbital, R., III. Deuterium Exchange in Methyl “Onium” Salts and in Bicyclo [2.2.1] heptane-1-sulfonium Iodide. J. Am. Chem. Soc. 1955, 77, 521–526. [Google Scholar] [CrossRef]
- Von, E.; Doering, W.; Schreiber, K.C.; d-Orbital, R., II. Comparative Reactivity of Vinyldimethylsulfonium and Vinyltrimethylammonium Ions. J. Am. Chem. Soc. 1955, 77, 514–520. [Google Scholar] [CrossRef]
- Mondal, M.; Chen, S.; Kerrigan, N.J. Recent developments in vinylsulfonium and vinylsulfoxonium salt chemistry. Molecules 2018, 23, 738. [Google Scholar] [CrossRef]
- Mitchell, K.A.R. The Use of Outer d Orbitals in Bonding. Chem. Rev. 1969, 69, 157–178. [Google Scholar] [CrossRef]
- Lehn, J.-M.; Wipff, G. Stereoelectronic Properties, Stereospecificity, and Stabilization of α-Oxa and α-Thia Carbanion. J. Am. Chem. Soc. 1976, 98, 7498–7505. [Google Scholar] [CrossRef]
- Bernardi, F.; Csizmadia, I.G.; Mangini, A.; Schlegel, H.B.; Whangbo, M.H.; Wolfe, S. The Irrelevance of d-Orbital Conjugation. I. The α-Thiocarbanion. A Comparative Quantum Chemical Study of the Static and Dynamic Properties and Proton Affinities of Carbanions Adjacent to Oxygen and to Sulfur. J. Am. Chem. Soc. 1975, 97, 2209–2218. [Google Scholar] [CrossRef]
- Streitwieser, A., Jr.; Williams, J.E., Jr. Ab initio SCF-MO calculations of thiomethyl anion. Polarization in stabilization of carbanions. J. Am. Chem. Soc. 1975, 97, 191–192. [Google Scholar] [CrossRef]
- Hoffmann, R.; Howell, J.M.; Muetterties, E.L. Molecular Orbital Theory of Pentacoordinate Phosphorus. J. Am. Chem. Soc. 1972, 94, 3047–3058. [Google Scholar] [CrossRef]
- Zbang, X.-M.; Bordwell, F.G. Equilibrium Acidities and Homolytic Bond Dissociation Energies of the Acidic C-H Bonds in P-Substituted Triphenylphosphonium Cations. J. Am. Chem. Soc. 1994, 116, 968–972. [Google Scholar] [CrossRef]
- Dobado, J.A.; Martınez-Garcıa, H.; Molina, J.M.; Sundberg, M.R. Chemical Bonding in Hypervalent Molecules Revised. 3. Application of the Atoms in Molecules Theory to Y3X-CH2 (X = N, P, or As; Y = H or F) and H2X-CH2 (X = O, S.; or Se) Ylides. J. Am. Chem. Soc. 2000, 122, 1144–1149. [Google Scholar] [CrossRef]
- Ripin, D.H.; Evans, D.A. pKa Values Compilation. Available online: https://organicchemistrydata.org/hansreich/resources/pka/#pka_water_compilation_evans (accessed on 21 March 2022).
- Aggarwal, V.K.; Harvey, J.N.; Robiette, R. On the Importance of Leaving Group Ability in Reactions of Ammonium, oxonium, Phosphonium and Sulfonium Ylides. Angew. Chem. Int. Ed. 2005, 44, 5468–5471. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Thompson, A.; Jones, R.V.H. Synthesis of Sulfonium Salts by Sulfide Alkylation; An Alternative Approach. Tetrahedron Lett. 1994, 46, 8659–8660. [Google Scholar] [CrossRef]
- Kemp, D.S.; Vellaccio, F. Studies toward practical thioxanthene-derived protective groups. 9,10-Propanothioxanthylium salts. J. Org. Chem. 1981, 46, 1807–1810. [Google Scholar] [CrossRef]
- Trost, B.M.; Bogdanowicz, M.J. Preparation of cyclopropyldiphenylsulfonium and 2-methylcyclopropyldiphenylsulfonium fluoroborate and their ylides. Stereochemistry of sulfur ylides. J. Am. Chem. Soc. 1973, 95, 5298–5307. [Google Scholar] [CrossRef]
- Andersen, K.K.; Caret, R.L.; Ladd, D.L. Synthesis of optically active dialkylarylsulfonium salts from alkyl aryl sulfoxides. J. Org. Chem. 1976, 41, 3096–3100. [Google Scholar] [CrossRef]
- Kozhushkov, S.I.; Alcarazo, M. Synthetic Applications of Sulfonium Salts. Eur. J. Inorg. Chem. 2020, 26, 2486–2500. [Google Scholar] [CrossRef]
- Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev. 2019, 119, 8701–8780. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Winn, C.L. Catalytic, Asymmetric Sulfur Ylide-Mediated Epoxidation of Carbonyl Compounds: Scope, Selectivity, and Applications in Synthesis. Acc. Chem. Res. 2004, 37, 611–620. [Google Scholar] [CrossRef]
- McGarrigle, E.M.; Myers, E.L.; Illa, O.; Shaw, M.A.; Riches, S.L.; Aggarwal, V.K. Chalcogenides as Organocatalysts. Chem. Rev. 2007, 107, 5841–5883. [Google Scholar] [CrossRef]
- Davis, R.L.; Stiller, J.; Naicker, T.; Jiang, H.; Jørgensen, K.A. Asymmetric organocatalytic epoxidations: Reactions, scope, mechanisms, and applications. Angew. Chem. Int. Ed. 2014, 53, 7406–7426. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Ye, S.; Sun, X.-L. Telluronium and Sulfonium Ylides for Organic Transformation. Synlett 2005, 18, 2720–2730. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Bae, I.; Lee, H.-Y.; Richardson, J.; Williams, D.T. Sulfur-Ylide-Mediated Synthesis of Functionalized and Trisubstituted Epoxides with High Enantioselectivity; Application to the Synthesis of CDP-840. Angew. Chem. Int. Ed. 2003, 42, 3274–3278. [Google Scholar] [CrossRef]
- Solladié-Cavallo, A.; Bouérat, L.; Roje, M. Asymmetric synthesis of trans-disubstituted aryl-vinyl epoxides: A p-methoxy effect. Tetrahedron Lett. 2000, 41, 7309–7312. [Google Scholar] [CrossRef]
- Solladié-Cavallo, A.; Roje, M.; Isarno, T.; Sunjic, V.; Vinkovic, V. Pyridyl and Furyl Epoxides of More Than 99% Enantiomeric Purities: The Use of a Phosphazene Base. Eur. J. Org. Chem. 2000, 6, 1077–1080. [Google Scholar] [CrossRef]
- McGarrigle, E.M.; Aggarwal, V.K. Enantioselective Organocatalysis: Reactions and Experimental Procedures; Dalko, P.I., Ed.; Wiley-VCH: Weinheim, Germany, 2007; Chapter 10. [Google Scholar]
- Aggarwal, V.K. Comprehensive Asymmetric Catalysis II.; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: New York, NY, USA, 1999; pp. 679–693. [Google Scholar]
- Blot, V.; Brière, J.-F.; Davoust, M.; Minière, S.; Reboul, V.; Metzner, P. Developments of Asymmetric Synthesis Mediated by Chiral Sulfur Reagents. Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 1171–1182. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Hebach, C. Carboxylate-stabilized sulfur ylides (thetin salts) in asymmetric epoxidation for the synthesis of glycidic acids. Mechanism and implications. Org. Biomol. Chem. 2005, 3, 1419–1427. [Google Scholar] [CrossRef]
- Hansch, M.; Illa, O.; McGarrigle, E.M.; Aggarwal, V.K. Synthesis and application of easily recyclable thiomorpholines for use in sulfur ylide mediated asymmetric epoxidation of aldehydes. Chem. Asian J. 2008, 3, 1657–1663. [Google Scholar] [CrossRef]
- Sarabia, F.; Chammaa, S.; García-Castro, M.; Martín-Gálvez, F. A highly efficient methodology of asymmetric epoxidation based on a novel chiral sulfur ylide. Chem. Commun. 2009, 38, 5763–5765. [Google Scholar] [CrossRef]
- Illa, O.; Arshad, M.; Ros, A.; McGarrigle, E.M.; Aggarwal, V.K. Practical and Highly Selective Sulfur Ylide Mediated Asymmetric Epoxidations and Aziridinations Using an Inexpensive, Readily Available Chiral Sulfide. Applications to the Synthesis of Quinine and Quinidine. J. Am. Chem. Soc. 2010, 132, 1828–1830. [Google Scholar] [CrossRef]
- Arshad, M.; Fernández, M.A.; McGarrigle, E.M.; Aggarwal, V.K. Synthesis of quinine and quinidine using sulfur ylide-mediated asymmetric epoxidation as a key step. Tetrahedron Asymmetry 2010, 21, 1771–1776. [Google Scholar] [CrossRef]
- Illa, O.; Namutebi, M.; Saha, C.; Ostovar, M.; Chen, C.C.; Haddow, M.F.; Nocquet-Thibault, S.; Lusi, M.; McGarrigle, E.M.; Aggarwal, V.K. Practical and Highly Selective Sulfur Ylide-Mediated Asymmetric Epoxidations and Aziridinations Using a Cheap and Readily Available Chiral Sulfide: Extensive Studies To Map Out Scope, Limitations, and Rationalization of Diastereo- and Enantioselectivities. J. Am. Chem. Soc. 2013, 135, 11951–11966. [Google Scholar] [CrossRef]
- Deng, X.-M.; Cai, P.; Ye, S.; Sun, X.-L.; Liao, W.-W.; Li, K.; Tang, Y.; Wu, Y.-D.; Dai, L.-X. Enantioselective synthesis of vinylcyclopropanes and vinylepoxides mediated by camphor-derived sulfur ylides: Rationale of enantioselectivity, scope, and limitation. J. Am. Chem. Soc. 2006, 128, 9730–9740. [Google Scholar] [CrossRef]
- Wang, Q.-G.; Deng, X.-M.; Zhu, B.-H.; Ye, L.-W.; Sun, X.-L.; Li, C.-Y.; Zhu, C.-Y.; Shen, Q.; Tang, Y. Tandem Michael addition/ylide epoxidation for the synthesis of highly functionalized cyclohexadiene epoxide derivatives. J. Am. Chem. Soc. 2008, 130, 5408–5409. [Google Scholar] [CrossRef]
- Furukawa, N.; Sugihara, Y.; Fujihara, H. Camphoryl sulfide as a chiral auxiliary and a mediator for one-step synthesis of optically active 1,2-diaryloxiranes. J. Org. Chem. 1989, 54, 4222–4224. [Google Scholar] [CrossRef]
- Saito, T.; Akiab, D.; Sakairi, M.; Kanazawa, S. Preparation of a novel, camphor-derived sulfide and its evaluation as a chiral auxiliary mediator in asymmetric epoxidation via the Corey–Chaykovsky reaction. Tetrahedron Lett. 2001, 42, 57–59. [Google Scholar] [CrossRef]
- Gui, Y.; Li, J.; Guo, C.S.; Li, X.L.; Lu, Z.F.; Huang, Z.Z. A New Chiral Organosulfur Catalyst for Highly Stereoselective Synthesis of Epoxides. Adv. Synth. Catal. 2008, 350, 2483–2487. [Google Scholar] [CrossRef]
- Zanardi, J.; Leriverend, C.; Aubert, D.; Julienne, K.; Metzner, P. A Catalytic Cycle for the Asymmetric Synthesis of Epoxides Using Sulfur Ylides. J. Org. Chem. 2001, 66, 5620–5623. [Google Scholar] [CrossRef]
- Winn, C.L.; Bellenie, B.R.; Goodman, J.M. A highly enantioselective one-pot sulfur ylide epoxidation reaction. Tetrahedron Lett. 2002, 43, 5427–5430. [Google Scholar] [CrossRef]
- Miyake, Y.; Oyamada, A.; Nishibayashi, Y.; Uemura, S. Asymmetric synthesis of epoxides from aromatic aldehydes and benzyl halides catalyzed by C2 symmetric optically active sulfides having a binaphthyl skeleton. Heteroat. Chem. 2002, 13, 270–275. [Google Scholar] [CrossRef]
- Davoust, M.; Brière, J.-F.; Jaffres, P.-A.; Metzner, P. Design of Sulfides with a Locked Conformation as Promoters of Catalytic and Asymmetric Sulfonium Ylide Epoxidation. J. Org. Chem. 2005, 70, 4166–4169. [Google Scholar] [CrossRef]
- Minière, S.; Reboul, V.; Metzner, P.; Fochi, M.; Bonini, B.F. Catalytic ferrocenyl sulfides for the asymmetric transformation of aldehydes into epoxides. Tetrahedron Asymmetry 2004, 15, 3275–3280. [Google Scholar] [CrossRef]
- Wu, H.-Y.; Chang, C.-W.; Chein, R.-J. Enantioselective Synthesis of (Thiolan-2-yl)diphenylmethanol and Its Application in Asymmetric, Catalytic Sulfur Ylide-Mediated Epoxidation. J. Org. Chem. 2013, 78, 5788–5793. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.; Kavanagh, S.A.; Connon, P.B.; Connon, S.J. Catalytic (Asymmetric) Methylene Transfer to Aldehydes. Org. Lett. 2010, 12, 608–611. [Google Scholar] [CrossRef] [PubMed]
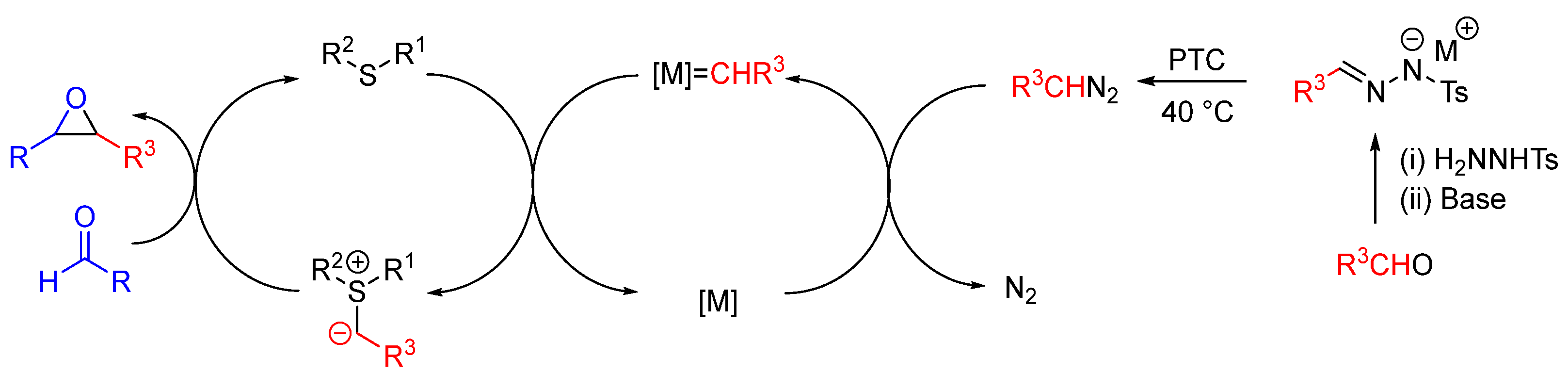
- Aggarwal, V.K.; Abdel-Rahman, H.; Jones, R.V.H.; Lee, H.Y.; Reid, B.D. Novel Catalytic Cycle for the Synthesis of Epoxides from Aldehydes and Sulfur Ylides Mediated by Catalytic Quantities of Sulfides and Rh2(OAc)4. J. Am. Chem. Soc. 1994, 116, 5973–5974. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Alonso, E.; Hynd, G.; Lydon, K.M.; Palmer, M.J.; Porcelloni, M.; Studley, J.R. Application of Chiral Sulfides to Catalytic Asymmetric Aziridination and Cyclopropanation with In Situ Generation of the Diazo Compound. Angew. Chem. Int. Ed. 2001, 40, 1433–1436. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Alonso, E.; Bae, I.; Hynd, G.; Lydon, K.M.; Palmer, M.J.; Patel, M.; Porcelloni, M.; Richardson, J.; Stenson, R.A.; et al. A New Protocol for the In Situ Generation of Aromatic, Heteroaromatic, and Unsaturated Diazo Compounds and Its Application in Catalytic and Asymmetric Epoxidation of Carbonyl Compounds. Extensive Studies To Map Out Scope and Limitations, and Rationalization of Diastereo- and Enantioselectivities. J. Am. Chem. Soc. 2003, 125, 10926–10940. [Google Scholar] [CrossRef]
- Myllymaki, V.T.; Lindvall, M.K.; Koskinen, A.M.P. Computer-assisted discovery of novel amino acid derived sulfides for enantioselective epoxidation of aldehydes. Tetrahedron 2001, 57, 4629–4635. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Arangoncillo, C.; Winn, C.L. Simple preparation of trans-epoxides via ylide intermediates. Synthesis 2005, 8, 1378–1382. [Google Scholar] [CrossRef]
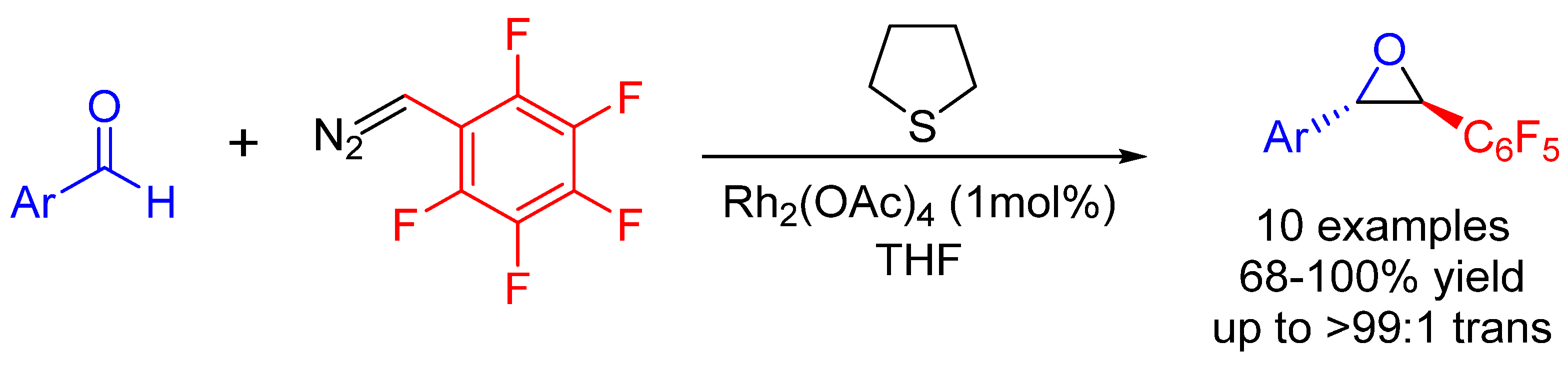
- Zhu, S.; Xing, C.; Pang, W.; Zhu, S. Rh2(OAc)4-catalyzed formation of trans-alkenes from the reaction of aldehydes with perfluorophenyl diazomethane through tellurium ylide. Tetrahedron Lett. 2006, 47, 5897–5900. [Google Scholar] [CrossRef]
- Gabriel, S. Ueber vinylamin. Ber. Dtsch. Chem. Ges. 1888, 21, 1049–1057. [Google Scholar] [CrossRef]
- Gabriel, S. Bromäthylamin.(II.). Ber. Dtsch. Chem. Ges. 1888, 21, 2664–2669. [Google Scholar] [CrossRef]
- Padwa, A. Comprehensive Heterocyclic Chemistry III; Ramsden, A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Chapter 1; pp. 1–104. [Google Scholar]
- Singh, G.S.; D’hooghe, M.; Kimpe, N.D. Synthesis and Reactivity of C-Heteroatom-Substituted Aziridines. Chem. Rev. 2007, 107, 2080–2135. [Google Scholar] [CrossRef]
- Sweeney, J.B. Aziridines and Epoxides in Organic Synthesis; Yudin, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; Chapter 4; pp. 117–144. [Google Scholar]
- Aggarwal, V.K.; Badine, M.; Moorthie, V. Aziridines and Epoxides in Organic Synthesis; Yudin, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; Chapter 1; pp. 1–35. [Google Scholar]
- Zhou, P.; Chen, B.-C.; Davis, F.A. Aziridines and Epoxides in Organic Synthesis; Yudin, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; Chapter 3; pp. 73–115. [Google Scholar]
- Padwa, A.; Murphree, S.; Gribble, G.W. Progress in Heterocyclic Chemistry; Gribble, G.W., Gilchrist, T.L., Eds.; Elsevier Science: Oxford, UK, 2000; Volume 15, pp. 75–99. [Google Scholar]
- Padwa, A.; Murphree, S.S. Progress in Heterocyclic Chemistry; Gribble, G.W., Gilchrist, T.L., Eds.; Elsevier Science: Oxford, UK, 2000; Volume 12, Chapter 4.1; p. 57. [Google Scholar]
- Sweeney, J.B. Aziridines: Epoxides’ ugly cousins? Chem. Soc. Rev. 2002, 31, 247–258. [Google Scholar] [CrossRef]
- Aires-de-Sousa, J.; Prabhakar, S.; Lobo, A.M.; Rosa, A.M.; Gomes, M.J.S.; Corvo, M.C.; Williams, D.J.; White, A.J.P. Asymmetric synthesis of N-aryl aziridines. Tetrahedron Asymmetry 2002, 12, 3349–3365. [Google Scholar] [CrossRef]
- Hu, X.E. Nucleophilic ring opening of aziridines. Tetrahedron 2004, 60, 2701–2743. [Google Scholar] [CrossRef]
- Pineschi, M. Asymmetric Ring-Opening of Epoxides and Aziridines with Carbon Nucleophiles. Eur. J. Org. Chem. 2006, 22, 4979–4988. [Google Scholar] [CrossRef]
- Florio, S.; Luisi, R. Aziridinyl Anions: Generation, Reactivity, and Use in Modern Synthetic Chemistry. Chem. Rev. 2010, 110, 5128–5157. [Google Scholar] [CrossRef]
- Lu, P. Recent developments in regioselective ring opening of aziridines. Tetrahedron 2010, 66, 2549–2560. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in asymmetric aziridination. Tetrahedron 2010, 66, 1509–1555. [Google Scholar] [CrossRef]
- Osborn, H.M.I.; Sweeney, J.B. The asymmetric synthesis of aziridines. Tetrahedron Asymmetry 1997, 8, 1693–1715. [Google Scholar] [CrossRef]
- Müller, P.; Fruit, C. Enantioselective catalytic aziridinations and asymmetric nitrene insertions into CH bonds. Chem. Rev. 2003, 103, 2905–2920. [Google Scholar] [CrossRef]
- Möβner, C.; Bolm, C. Transition Metals for Organic Synthesis, 2nd ed.; Beller, M., Bolm, C., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp. 389–402. [Google Scholar]
- Aggarwal, V.K.; McGarrigle, E.M.; Shaw, M.A. Science of Synthesis; Georg Thieme Verlag: Stuttgart, Germany, 2010; Volume 37, pp. 311–347. [Google Scholar]
- Muchalski, H.; Johnston, J.N. Science of Synthesis; Georg Thieme Verlag: Stuttgart, Germany, 2011; Volume 1, pp. 155–184. [Google Scholar]
- Karila, D.; Dodd, R.H. Recent Progress in Iminoiodane-Mediated Aziridination of Olefins. Curr. Org. Chem. 2011, 15, 1509–1538. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Asymmetric Aziridination. Adv. Synth. Catal. 2014, 356, 1899–1935. [Google Scholar] [CrossRef]
- Dokli, I.; Matanovic, I.; Hamersak, Z. Sulfur Ylide Promoted Synthesis of N-Protected Aziridines: A Combined Experimental and Computational Approach. Chem. Eur. J. 2010, 16, 11744–11752. [Google Scholar] [CrossRef]
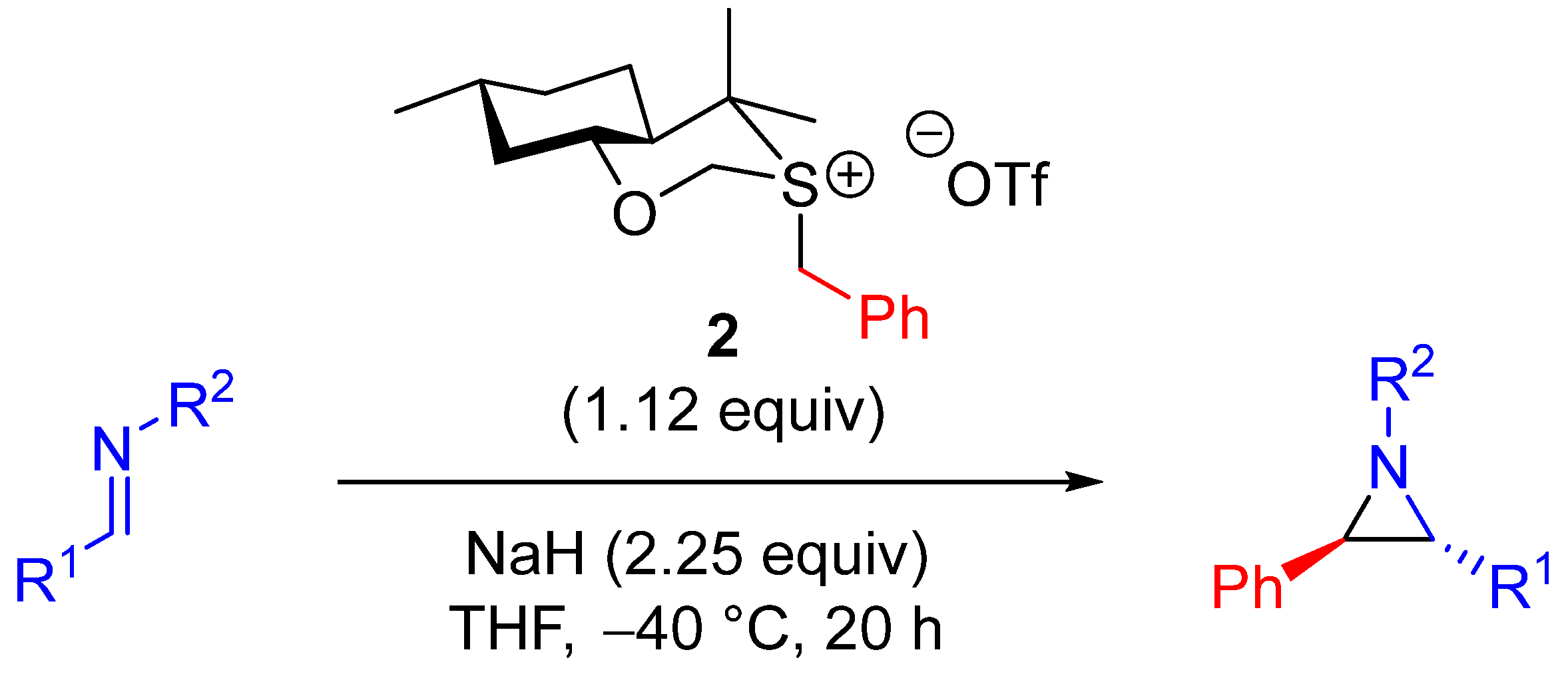
- Kavanagh, S.A.; Piccinini, A.; Connon, S.J. The asymmetric synthesis of terminal aziridines by methylene transfer from sulfonium ylides to imines. Org. Biomol. Chem. 2013, 11, 3535–3540. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K. Catalytic Asymmetric Epoxidation and Aziridination Mediated by Sulfur Ylides. Evolution of a Project. Synlett 1998, 4, 329–336. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Ferrara, M.; O’Brien, C.J.; Thompson, A.; Jones, R.V.H.; Fieldhouse, R. Scope and limitations in sulfur ylide mediated catalytic asymmetric aziridination of imines: Use of phenyldiazomethane, diazoesters and diazoacetamides. J. Chem. Soc. Perkin Trans. 1 2001, 14, 1635–1643. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Stenson, R.A.; Jones, R.V.H.; Fieldhouse, R.; Blacker, J. A novel procedure for the synthesis of aziridines: Application of Simmons–Smith reagents to aziridination. Tetrahedron Lett. 2001, 42, 1587–1589. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Coogan, M.P.; Stenson, R.A.; Jones, R.V.H.; Fieldhouse, R.; Blacker, J. Developments in the Simmons−Smith-Mediated Epoxidation Reaction. Eur. J. Org. Chem. 2002, 2, 319–326. [Google Scholar] [CrossRef]
- Xue, Z.; Dee, V.M.; Hope-Weeks, L.J.; Whittlesey, B.R.; Mayer, M.F. Asymmetric aziridination of N-tert-butanesulfinyl imines with phenyldiazomethane via sulfur ylides. ARKIVOC 2010, 7, 65–80. [Google Scholar] [CrossRef]
- Saito, T.; Sakairi, M.; Akiba, D. Enantioselective synthesis of aziridines from imines and alkyl halides using a camphor-derived chiral sulfide mediator via the imino Corey–Chaykovsky reaction. Tetrahedron Lett. 2001, 42, 5451–5454. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Smith, H.W.; Hynd, G.; Jones, R.V.H.; Fieldhouse, R.; Spey, S.E. Catalytic cyclopropanation of electron deficient alkenes mediated by chiral and achiral sulfides: Scope and limitations in reactions involving phenyldiazomethane and ethyl diazoacetate. J. Chem. Soc. Perkin Trans. 1 2000, 19, 3267–3276. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Grange, E. Asymmetric Sulfonium Ylide Mediated Cyclopropanation: Stereocontrolled Synthesis of (+)-LY354740. Chem. Eur. J. 2006, 12, 568–575. [Google Scholar] [CrossRef]
- Hudlicky, T.; Rulin, F.; Lovelace, T.C.; Reed, J.W. Studies in Natural Product Chemistry, Stereoselective Synthesis; Attaur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 1989; Volume 3, Part B; p. 3. [Google Scholar]
- Hudlicky, T.; Reed, J.W.; Trost, B.M. Rearrangements of Vinylcyclopropanes and Related Systems in Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1991; Volume 5, pp. 899–970. [Google Scholar]
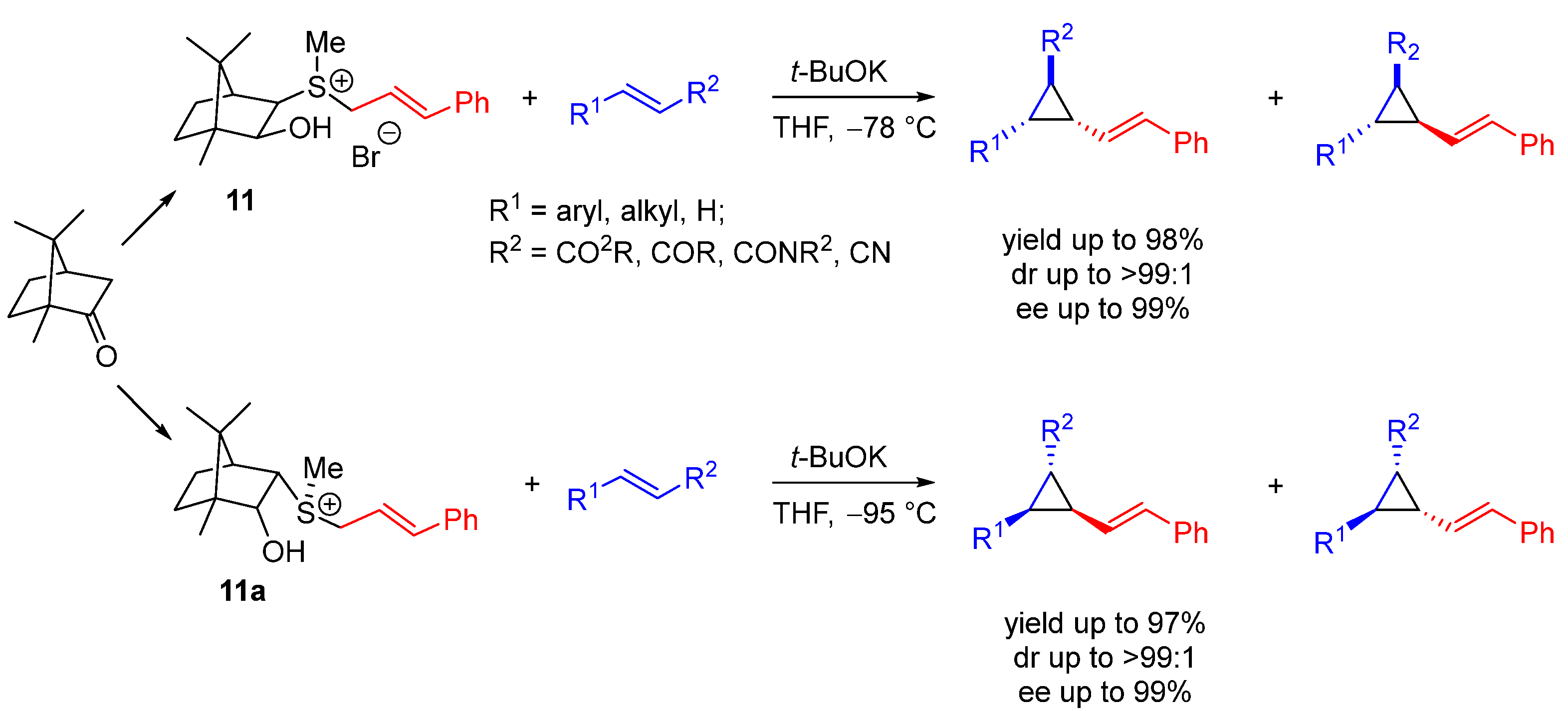
- Ye, S.; Huang, Z.-Z.; Xia, C.-A.; Tang, Y.; Dai, L.-X. A Novel Chiral Sulfonium Ylide: Highly Enantioselective Synthesis of Vinylcyclopropanes. J. Am. Chem. Soc. 2002, 124, 2432–2433. [Google Scholar] [CrossRef]
- Li, A.-H.; Dai, L.-X.; Hou, X.-L.; Huang, Y.-Z.; Li, F.-W. Preparation of Enantiomerically Enriched (2R,3R)- or (2S,3S)-trans-2,3-Diaryloxiranes via Camphor-Derived Sulfonium Ylides. J. Org. Chem. 1996, 61, 489–493. [Google Scholar] [CrossRef]
- Lee, K.Y.; Kim, S.C.; Kim, J.N. Synthesis of Cyclopropane Derivatives Starting from the Baylis-Hillman Adducts Using Sulfur Ylide Chemistry. Bull. Korean Chem. Soc. 2006, 27, 319–321. [Google Scholar] [CrossRef]
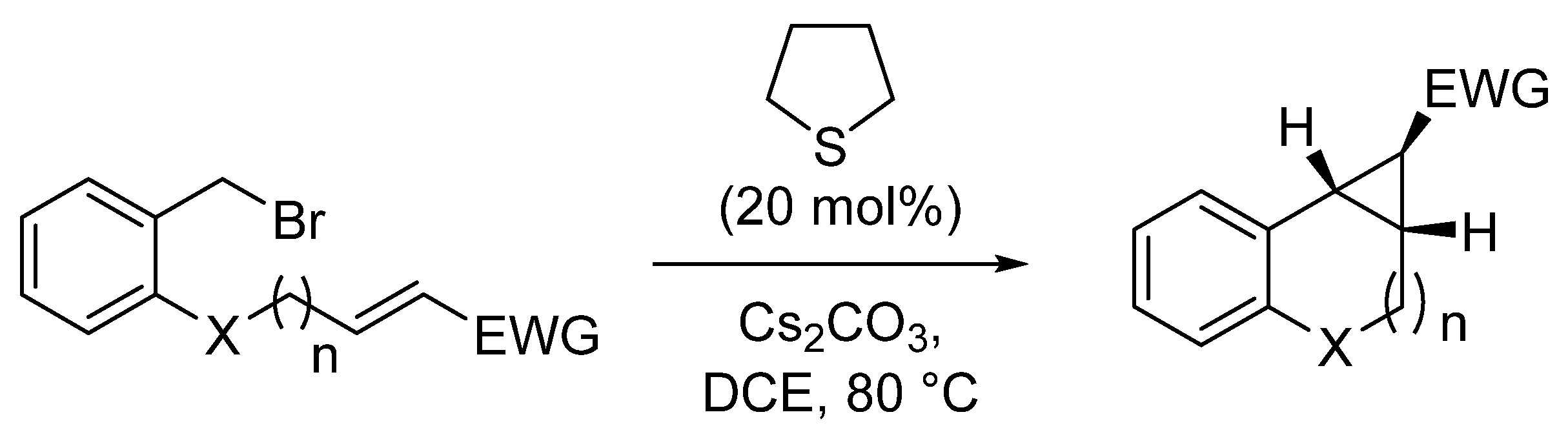
- Ye, L.-W.; Sun, X.-L.; Li, C.-Y.; Tang, Y. Tetrahydrothiophene-Catalyzed Synthesis of Benzo[n.1.0] Bicycloalkanes. J. Org. Chem. 2007, 72, 1335–1340. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Smith, H.W.; Jones, R.V.H.; Fieldhouse, R. Catalytic asymmetric cyclopropanation of electron deficient alkenes mediated by chiral sulfides. Chem. Commun. 1997, 18, 1785–1786. [Google Scholar] [CrossRef]
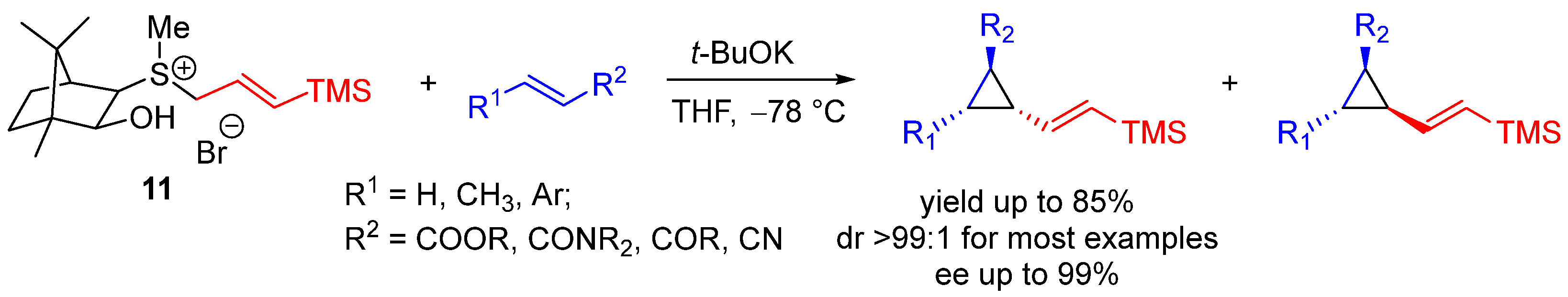
- Lu, L.-Q.; Li, T.-R.; Wang, Q.; Xiao, W.-J. Beyond sulfide-centric catalysis: Recent advances in the catalytic cyclization reactions of sulfur ylides. Chem. Soc. Rev. 2017, 46, 4135–4149. [Google Scholar] [CrossRef]
- Kunz, R.K.; MacMillan, D.W.C. Enantioselective Organocatalytic Cyclopropanations. The Identification of a New Class of Iminium Catalyst Based upon Directed Electrostatic Activation. J. Am. Chem. Soc. 2005, 127, 3240–3241. [Google Scholar] [CrossRef]
- Hartikka, A.; Arvidsson, P.I. Tetrazolic Acid Functionalized Dihydroindol: Rational Design of a Highly Selective Cyclopropanation Organocatalyst. J. Org. Chem. 2007, 72, 5874–5877. [Google Scholar] [CrossRef]
- Hartikka, A.; Ślósarczyk, A.T.; Arvidsson, P.I. Application of novel sulfonamides in enantioselective organocatalyzed cyclopropanation. Tetrahedron Asymmetry 2007, 18, 1403–1409. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.-H.; Dong, S.-X.; Lin, L.-L.; Feng, X.-M. Asymmetric Organocatalytic Cyclopropanation of Cinnamone Derivatives with Stabilized Sulfonium Ylides. J. Org. Chem. 2013, 78, 6322–6327. [Google Scholar] [CrossRef]
- Biswas, A.; Sarkar, S.D.; Tebben, L.; Studer, A. Enantioselective cyclopropanation of enals by oxidative N-heterocyclic carbene catalysis. Chem. Commun. 2012, 48, 5190–5192. [Google Scholar] [CrossRef]
- Mamai, A.; Madalengoitia, J.S. Lewis acid mediated diastereoselective and enantioselective cyclopropanation of Michael acceptors with sulfur ylides. Tetrahedron Lett. 2000, 41, 9009–9014. [Google Scholar] [CrossRef]
- Huang, X.; Klimczyk, S.; Veiros, L.F.; Maulide, N. Stereoselective intramolecular cyclopropanation through catalytic olefin activation. Chem. Sci. 2013, 4, 1105–1110. [Google Scholar] [CrossRef]
- Klimczyk, S.; Huang, X.; Kahlig, H.; Veiros, L.F.; Maulide, N. Stereoselective Gold(I) Domino Catalysis of Allylic Isomerization and Olefin Cyclopropanation: Mechanistic Studies. J. Org. Chem. 2015, 80, 5719–5729. [Google Scholar] [CrossRef]
- Sabbatani, J.; Huang, X.; Veiros, L.F.; Maulide, N. Gold-Catalyzed Intermolecular Synthesis of Alkylidenecyclopropanes through Catalytic Allene Activation. Chem. Eur. J. 2014, 20, 10636–10639. [Google Scholar] [CrossRef]
- Klimczyk, S.; Misale, A.; Huang, X.; Maulide, N. Dimeric TADDOL Phosphoramidites in Asymmetric Catalysis: Domino Deracemization and Cyclopropanation of Sulfonium Ylides. Angew. Chem. Int. Ed. 2015, 54, 10365–10369. [Google Scholar] [CrossRef]
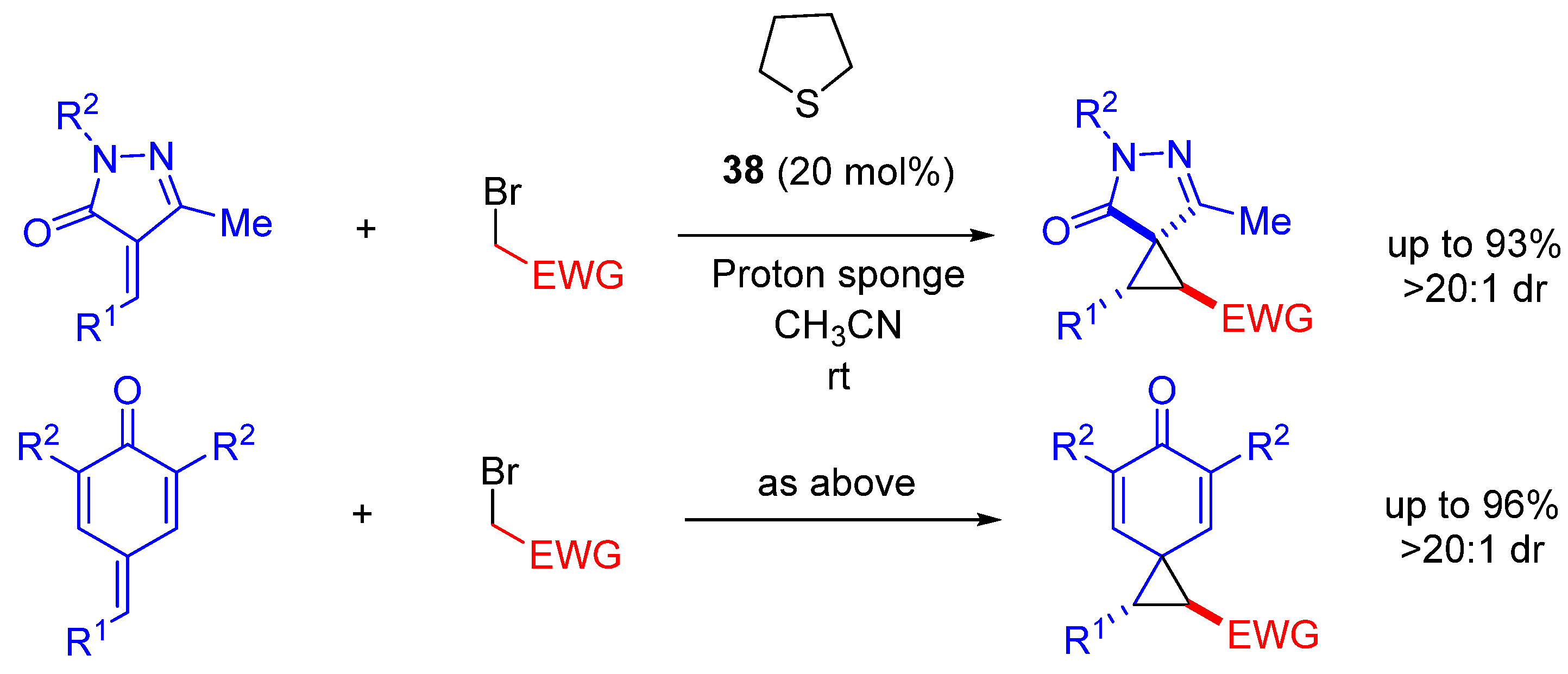
- Su, Y.; He, H.; Zhao, Y.; Li, Q.; Feng, Y.; Cao, G.; Huang, D.; Wang, K.-H.; Huo, C.; Hu, Y. Sulfide-Catalyzed Diastereoselective Spirocyclopropanation: Constructing Spiro-cyclopropanyl-pyrazolones From α-Arylidenepyrazolones. Asian J. Org. Chem. 2021, 10, 1778–1785. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, Y.; Chang, B.; Ling, Q.; Feng, Y.; Zhao, X.; Huang, D.; Wang, K.-H.; Huo, C.; Hu, Y. Diastereoselective synthesis of spirocyclopropanyl-cyclohexadienones via direct sulfide-catalyzed [2 + 1] annulation of para-quinone methides with bromides. Org. Biomol. Chem. 2020, 18, 4257–4266. [Google Scholar] [CrossRef]
- Crudden, C.M.; Edwards, D. Catalytic Asymmetric Hydroboration: Recent Advances and Applications in Carbon−Carbon Bond-Forming Reactions. Eur. J. Org. Chem. 2003, 2003, 4695–4712. [Google Scholar] [CrossRef]
- Kaufmann, D.E.; Matteson, D.S. Science of Synthesis: Boron Compounds; Georg Thieme Verlag: Stuttgart, Germany, 2004; Volume 6. [Google Scholar]
- Hall, D.G. Boronic Acids. Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 1–85. [Google Scholar]
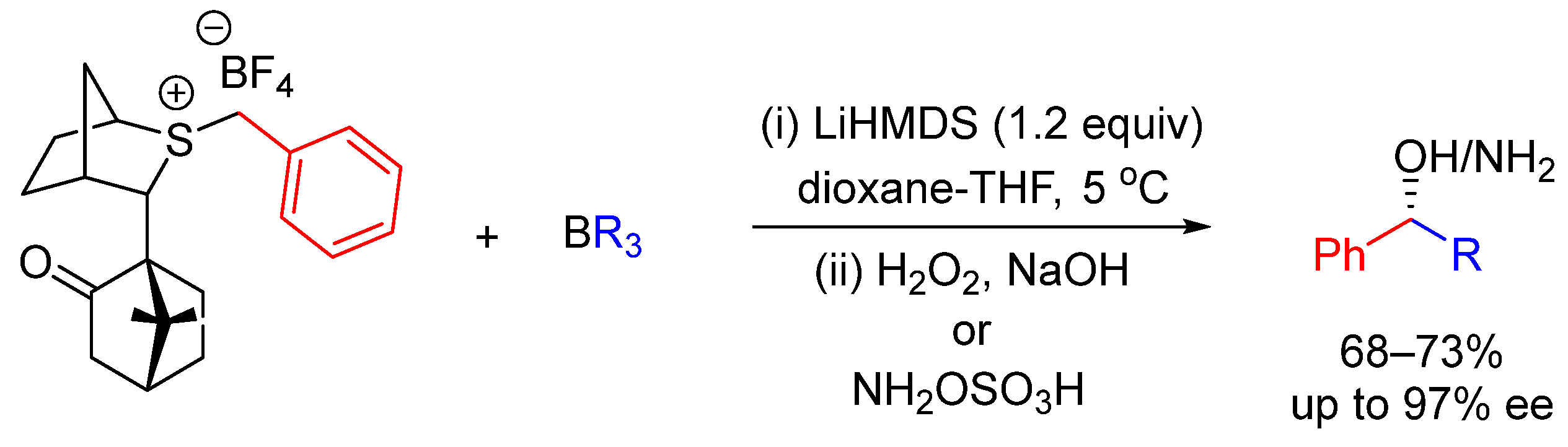
- Aggarwal, V.K.; Fang, G.Y.; Schmidt, A.T. Synthesis and Applications of Chiral Organoboranes Generated from Sulfonium Ylides. J. Am. Chem. Soc. 2005, 127, 1642–1643. [Google Scholar] [CrossRef]
- Fang, G.Y.; Wallner, O.A.; Blasio, N.D.; Ginesta, X.; Harvey, J.N.; Aggarwal, V.K. Asymmetric Sulfur Ylide Reactions with Boranes: Scope and Limitations, Mechanism and Understanding. J. Am. Chem. Soc. 2007, 129, 14632–14639. [Google Scholar] [CrossRef]
- Fang, G.Y.; Aggarwal, V.K. Asymmetric Synthesis of α-Substituted Allyl Boranes and Their Application in the Synthesis of Iso-agatharesinol. Angew. Chem. Int. Ed. 2007, 46, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.; Robiette, R.; Fang, G.Y.; Knowles, S.L.; Woodrow, M.D.; Harvey, J.N.; Aggarwal, V.K. Reactions of silyl-stabilized sulfur ylides with organoboranes: Enantioselectivity, mechanism, and understanding. Org. Biomol. Chem. 2008, 6, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-Q.; Xiao, W.-J. Catalytic Asymmetric Synthesis of Chiral Dihydrobenzofurans through a Formal [4 + 1] Annulation Reaction of Sulfur Ylides and In Situ Generated ortho-Quinone Methides. Eur. J. Org. Chem. 2017, 87, 233–236. [Google Scholar] [CrossRef]
- Chen, J.-R.; Dong, W.-R.; Candy, M.; Pan, F.-F.; Jorres, M.; Bolm, C. Enantioselective Synthesis of Dihydropyrazoles by Formal [4 + 1] Cycloaddition of in Situ-Derived Azoalkenes and Sulfur Ylides. J. Am. Chem. Soc. 2012, 134, 6924–6927. [Google Scholar] [CrossRef]
- Li, T.-R.; Tan, F.; Lu, L.-Q.; Wei, Y.; Wang, Y.-N.; Liu, Y.-Y.; Yang, Q.-Q.; Chen, J.-R.; Shi, D.-Q.; Xiao, W.-J. Asymmetric trapping of zwitterionic intermediates by sulphur ylides in a palladium-catalyzed decarboxylation-cycloaddition sequence. Nat. Commun. 2014, 5, 5500. [Google Scholar] [CrossRef]
- Guo, C.; Janssen-Muller, D.; Fleige, M.; Lerchen, A.; Daniliuc, C.G.; Glorius, F. Mechanistic Studies on a Cooperative NHC Organocatalysis/Palladium Catalysis System: Uncovering Significant Lessons for Mixed Chiral Pd (NHC) (PR3) Catalyst Design. J. Am. Chem. Soc. 2017, 139, 4443–4451. [Google Scholar] [CrossRef]
- Wang, Q.; Qi, X.-T.; Lu, L.-Q.; Li, T.-R.; Yuan, Z.-G.; Zhang, K.; Li, B.-J.; Lan, Y.; Xiao, W.-J. Iron-Catalyzed Decarboxylative (4 + 1) Cycloadditions: Exploiting the Reactivity of Ambident Iron-Stabilized Intermediates. Angew. Chem. Int. Ed. 2016, 55, 2840–2844. [Google Scholar] [CrossRef]
- Wang, Q.; Li, T.-R.; Lu, L.-Q.; Li, M.-M.; Zhang, K.; Xiao, W.-J. Catalytic Asymmetric [4 + 1] Annulation of Sulfur Ylides with Copper−Allenylidene Intermediates. J. Am. Chem. Soc. 2016, 138, 8360–8363. [Google Scholar] [CrossRef]
- Gong, P.; Wang, J.; Yao, W.-B.; Xie, X.-S.; Xie, J.-W. Diastereoselective and Enantioselective Formal [4 + 1] Ylide Annulation Leading to Optically Active Isoxazoline N-Oxides. Adv. Synth. Catal. 2022, 364, 1185–1199. [Google Scholar] [CrossRef]
- Lu, L.-Q.; Cao, Y.-J.; Liu, X.-P.; An, J.; Yao, C.-J.; Ming, Z.-H.; Xiao, W.-J. A New Entry to Cascade Organocatalysis: Reactions of Stable Sulfur Ylides and Nitroolefins Sequentially Catalyzed by Thiourea and DMAP. J. Am. Chem. Soc. 2008, 130, 6946–6948. [Google Scholar] [CrossRef]
- Lu, L.-Q.; Li, F.; An, J.; Cheng, Y.; Chen, J.-R.; Xiao, W.-J. Hydrogen-Bond-Mediated Asymmetric Cascade Reaction of Stable Sulfur Ylides with Nitroolefins: Scope, Application and Mechanism. Chem. Eur. J. 2012, 18, 4073–4079. [Google Scholar] [CrossRef]
- Lu, L.-Q.; Li, F.; An, J.; Zhang, J.-J.; An, X.-L.; Hua, Q.-L.; Xiao, W.-J. Construction of Fused Heterocyclic Architectures by Formal [4 + 1]/[3 + 2] Cycloaddition Cascade of Sulfur Ylides and Nitroolefins. Angew. Chem. Int. Ed. 2009, 48, 9542–9545. [Google Scholar] [CrossRef]
- Deng, Y.; Massey, L.A.; Zavalij, P.Y.; Doyle, M.P. Catalytic Asymmetric [3 + 1]-Cycloaddition Reaction of Ylides with Electrophilic Metallo-Enolcarbene Intermediates. Angew. Chem. Int. Ed. 2017, 56, 7479–7483. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Wang, Z.; Hui, X.-P. Efficiently diastereoselective synthesis of functionalized hydro-carbazoles by base-mediated tandem annulation of 1-(2-amino-aryl)prop-2-en-1-ones and sulfur ylide. Org. Chem. Front. 2020, 7, 1469–1473. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, N.; Cai, W.; Huang, Y. Asymmetric Sequential Corey−Chaykovsky Cyclopropanation/ Cloke−Wilson Rearrangement for the Synthesis of 2,3- Dihydrofurans. Org. Lett. 2021, 23, 8755–8760. [Google Scholar] [CrossRef]
- Zhang, Z.; Sheng, Z.; Yu, W.; Wu, G.; Zhang, R.; Chu, W.-D.M.; Zhang, Y.; Wang, J. Catalytic Asymmetric Trifluoromethylthiolation via Enantioselective [2,3]-Sigmatropic Rearrangement of Sulfonium Ylides. Nat. Chem. 2017, 9, 970–976. [Google Scholar] [CrossRef]
- Lin, X.; Tang, Y.; Yang, W.; Tan, F.; Lin, L.; Liu, X.; Feng, X. Chiral Nickel(II) Complex Catalyzed Enantioselective Doyle−Kirmse Reaction of α-Diazo Pyrazoleamides. J. Am. Chem. Soc. 2018, 140, 3299–3305. [Google Scholar] [CrossRef]
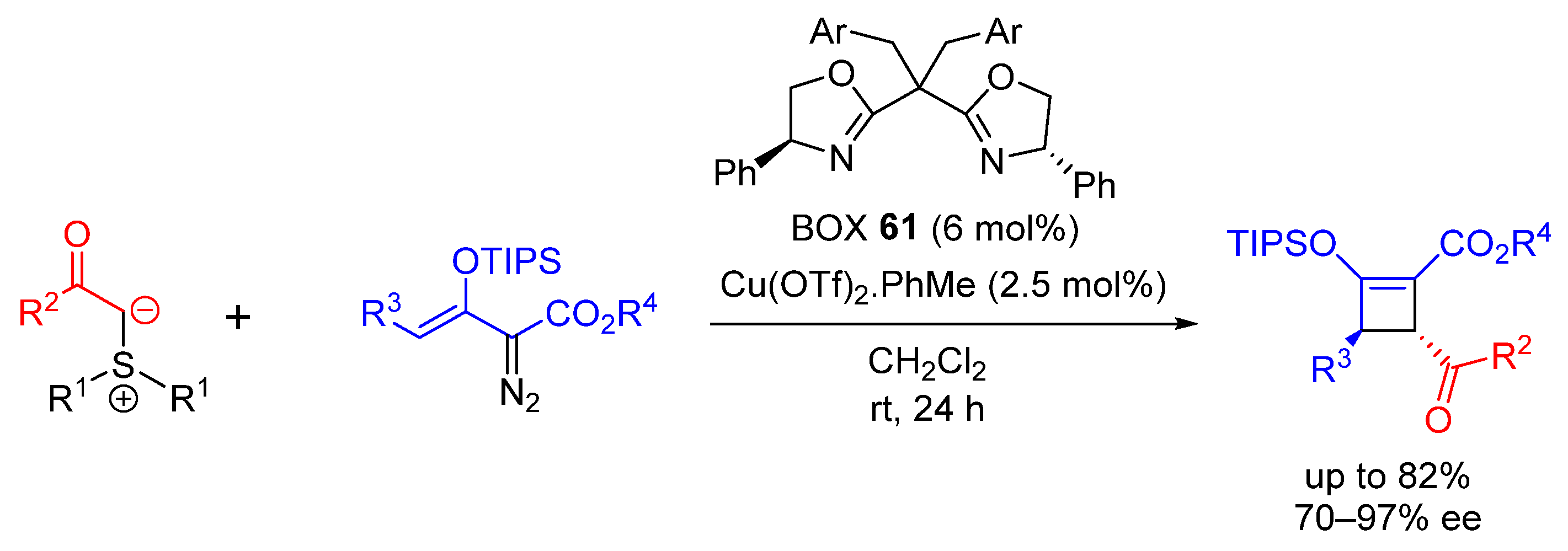
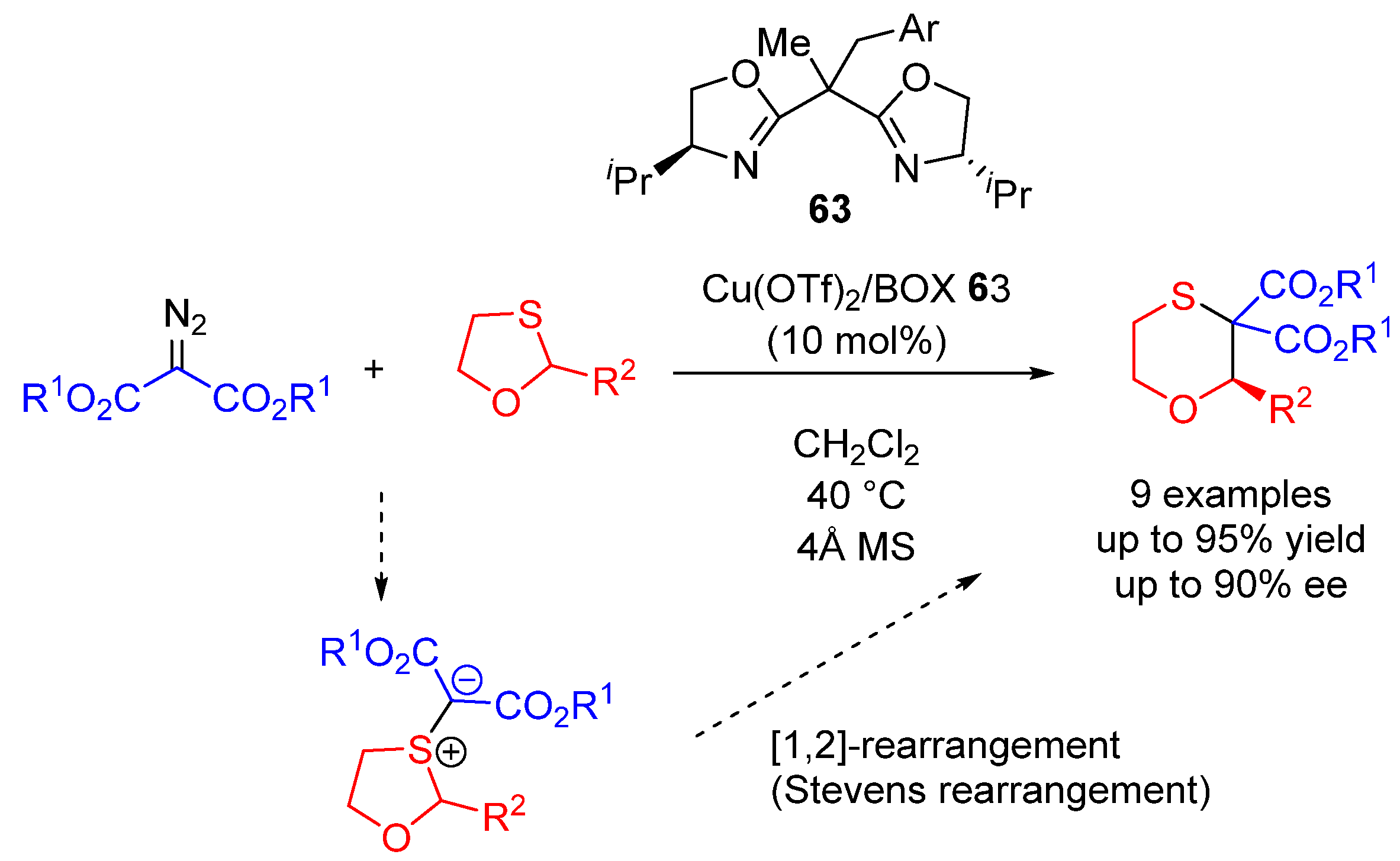
- Qu, J.-P.; Xu, Z.-H.; Zhou, J.; Cao, C.-L.; Sun, X.-L.; Dai, L.-X.; Tang, Y. Ligand-Accelerated Asymmetric [1,2]-Stevens Rearrangment of Sulfur Ylides via Decomposition of Diazomalonates Catalyzed by Chiral Bisoxazoline/Copper Complex. Adv. Synth. Catal. 2009, 351, 308–312. [Google Scholar] [CrossRef]





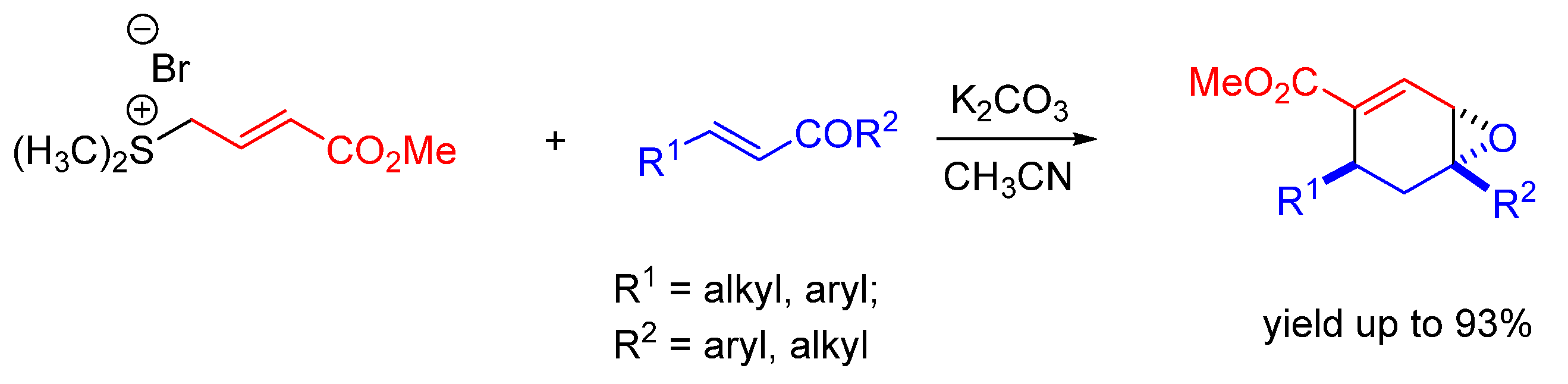
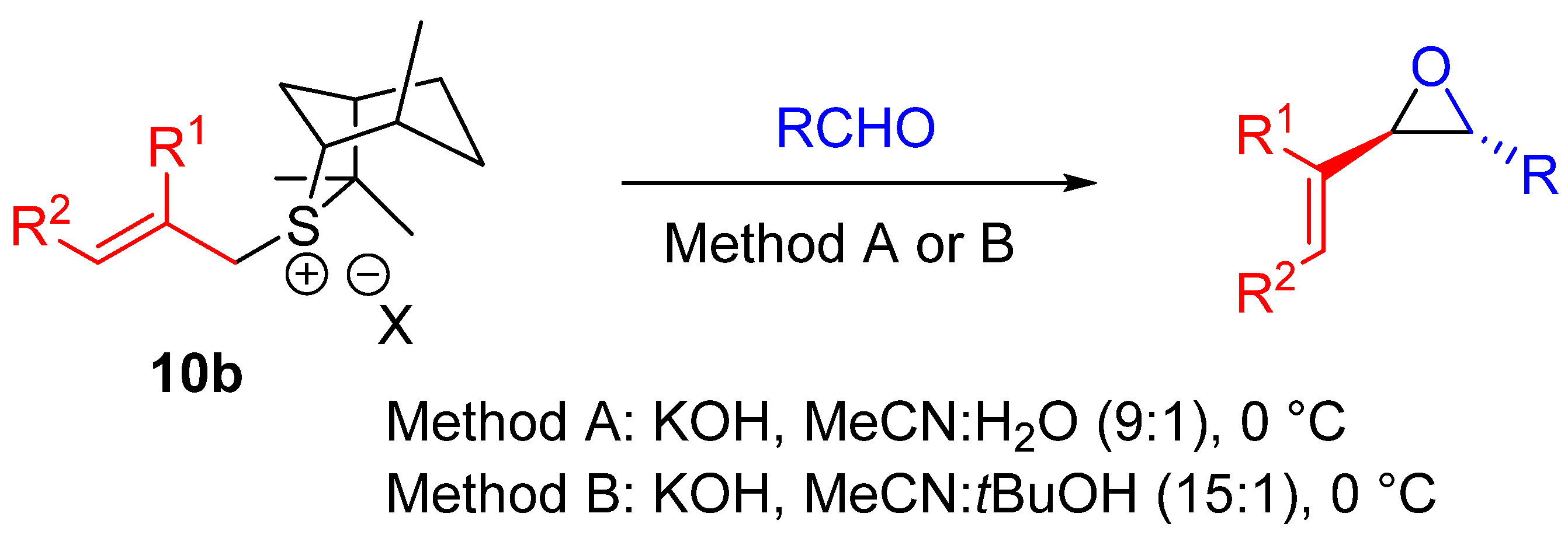

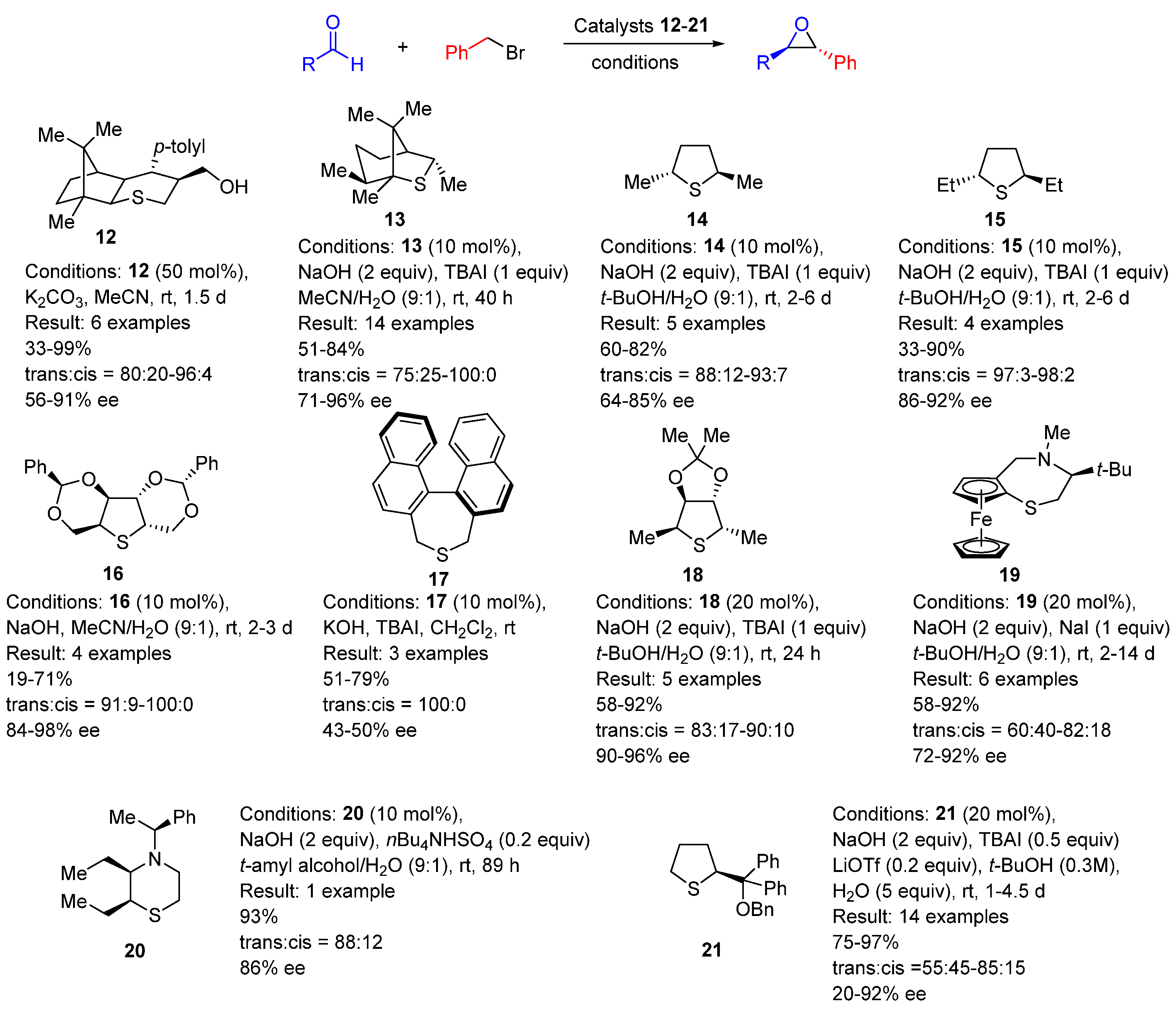
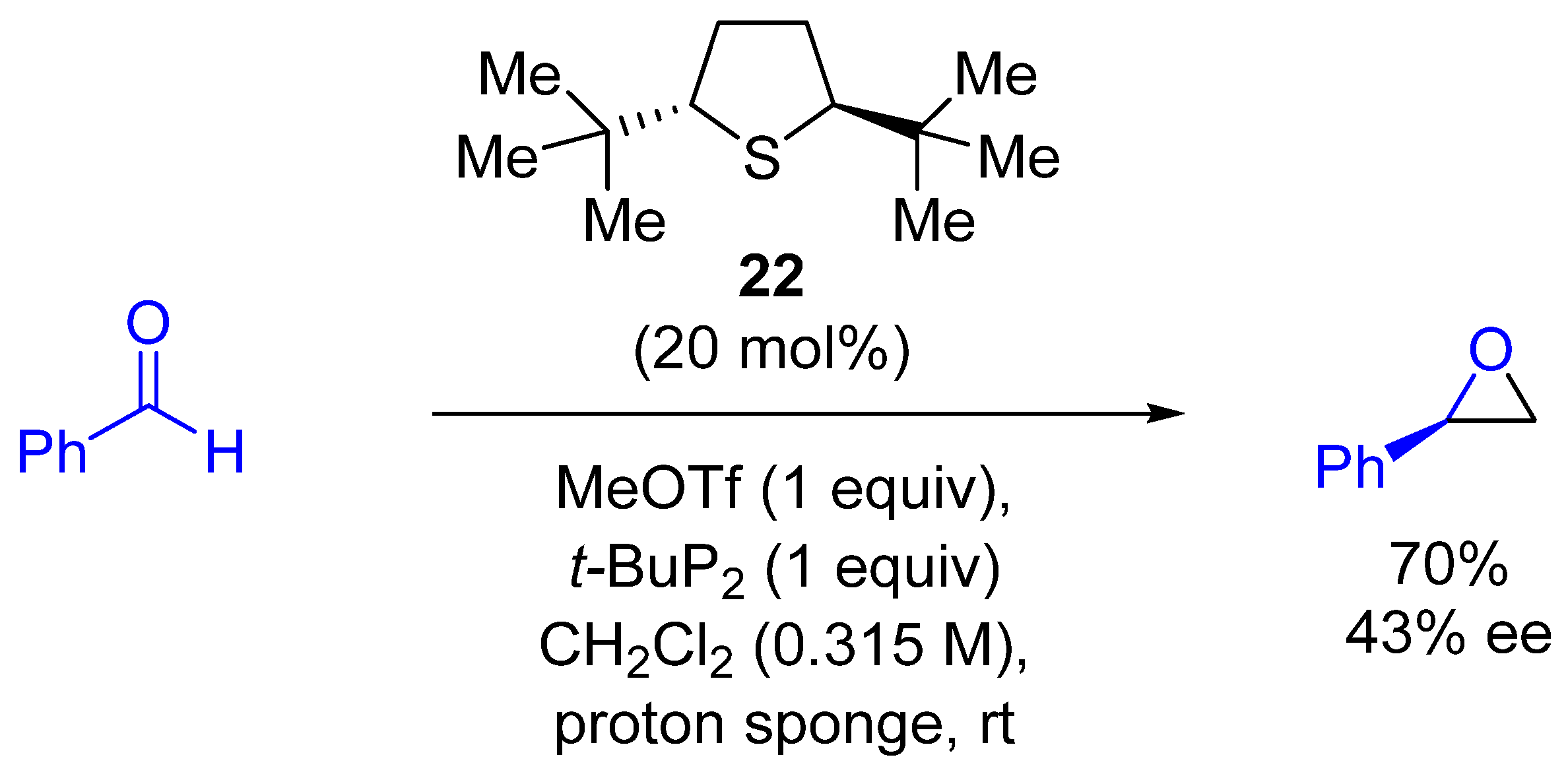
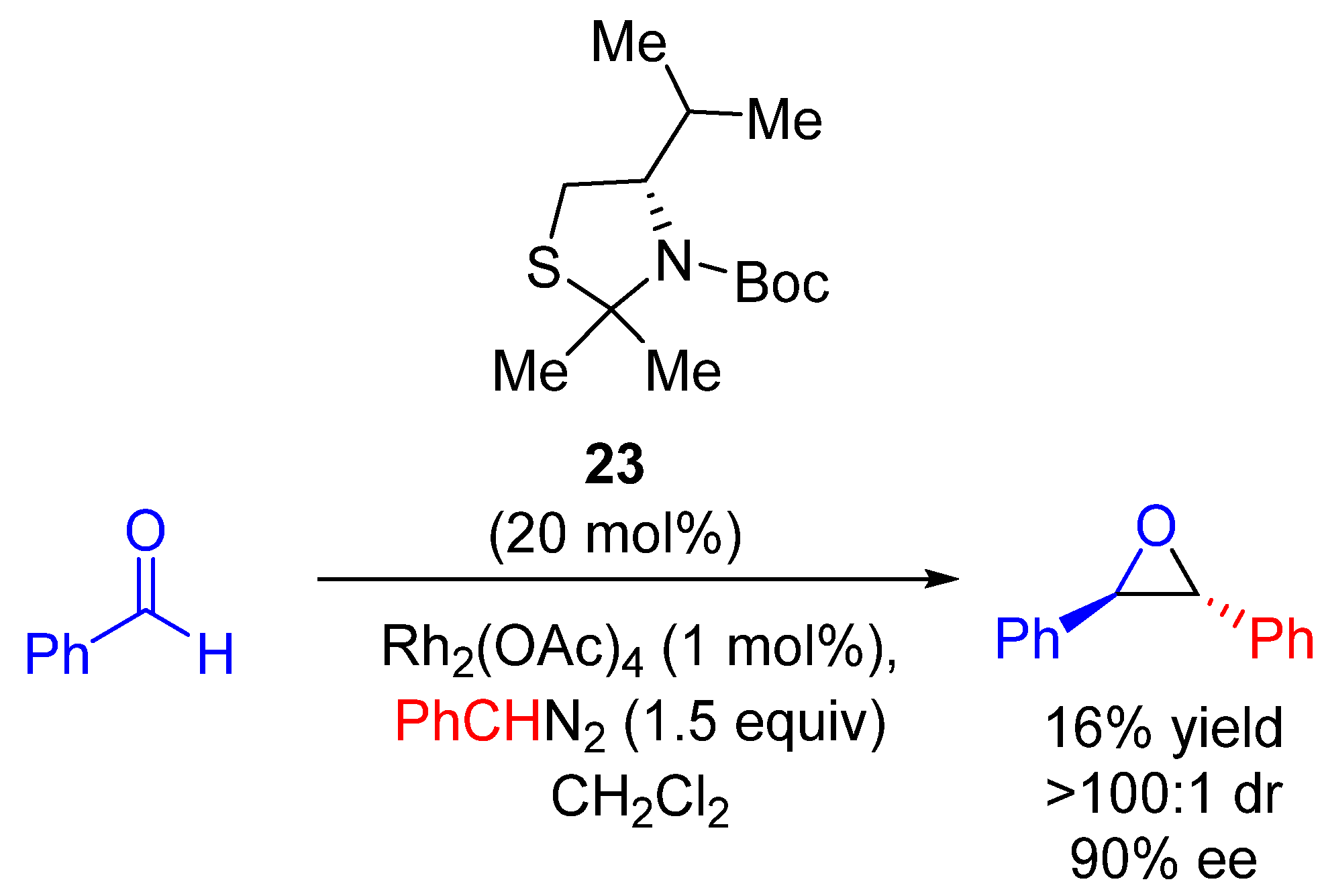


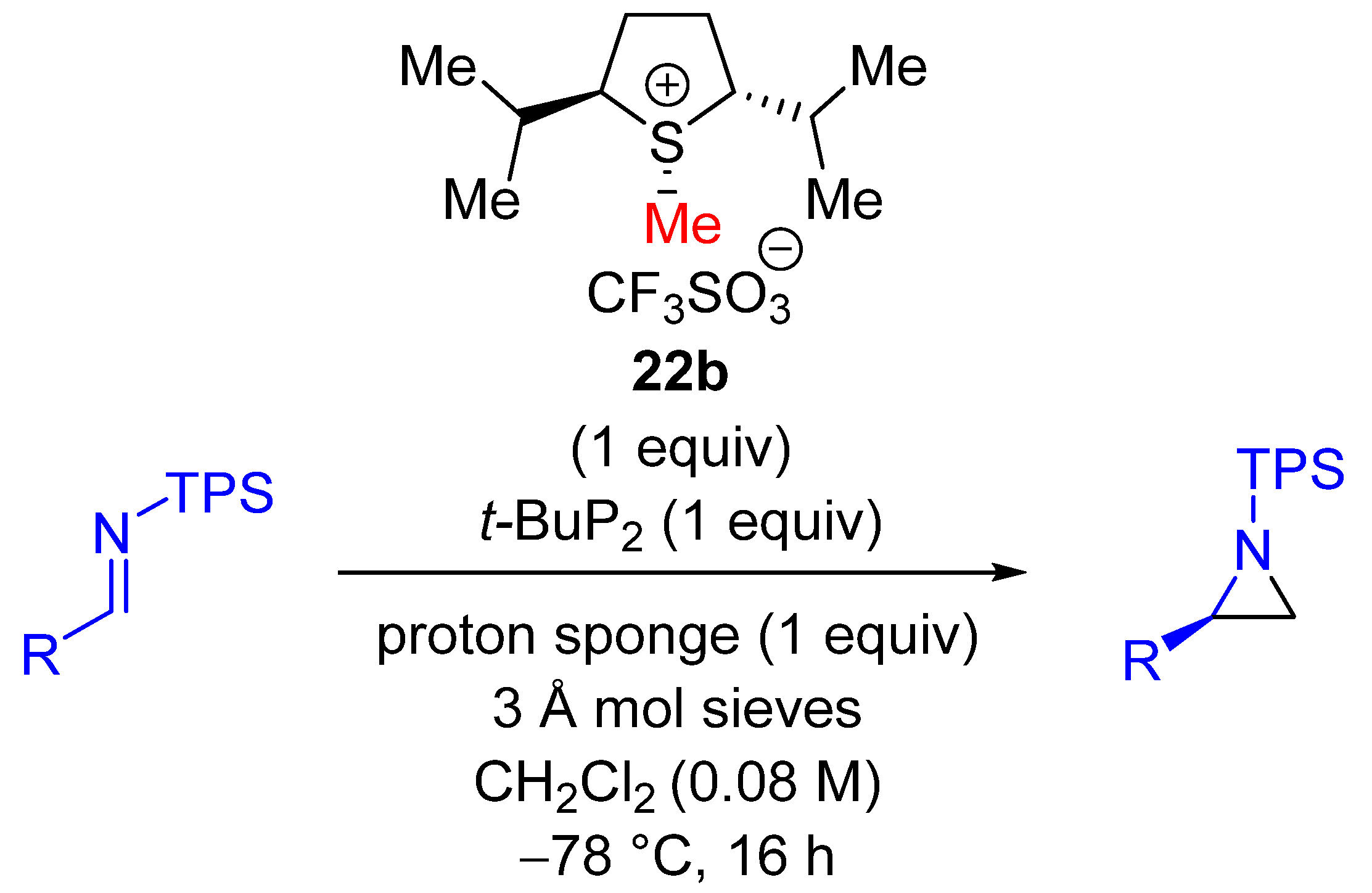
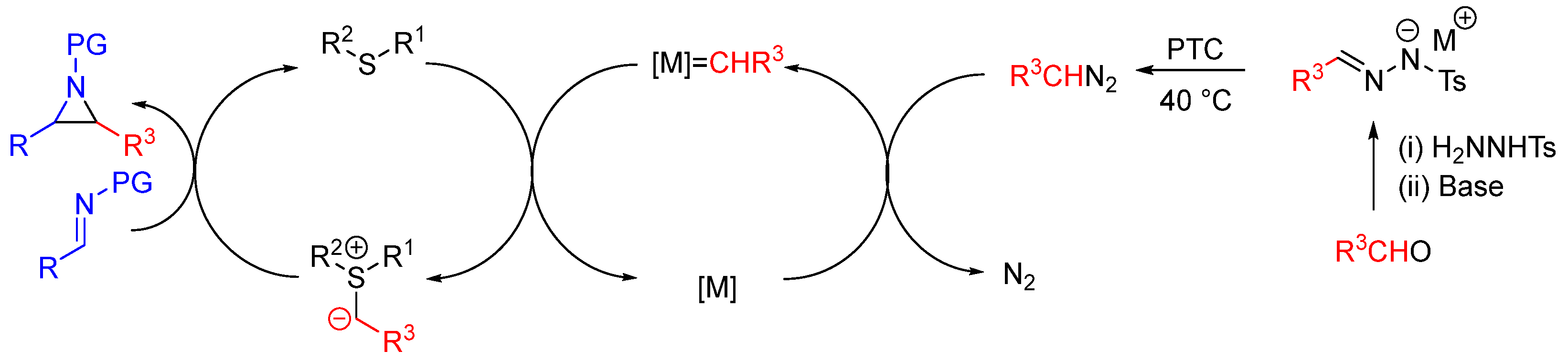

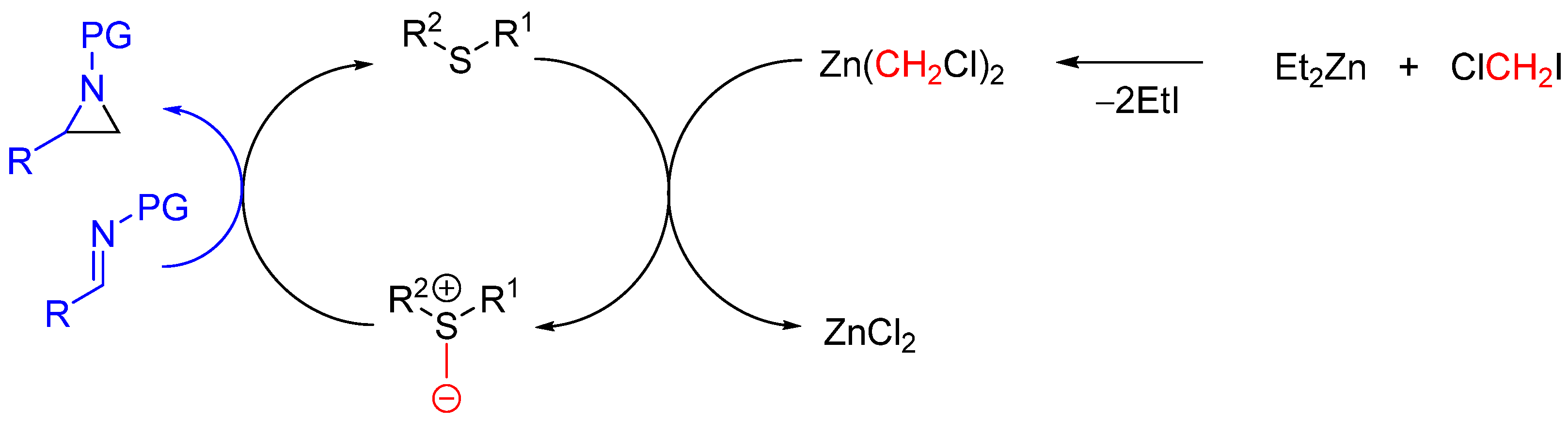
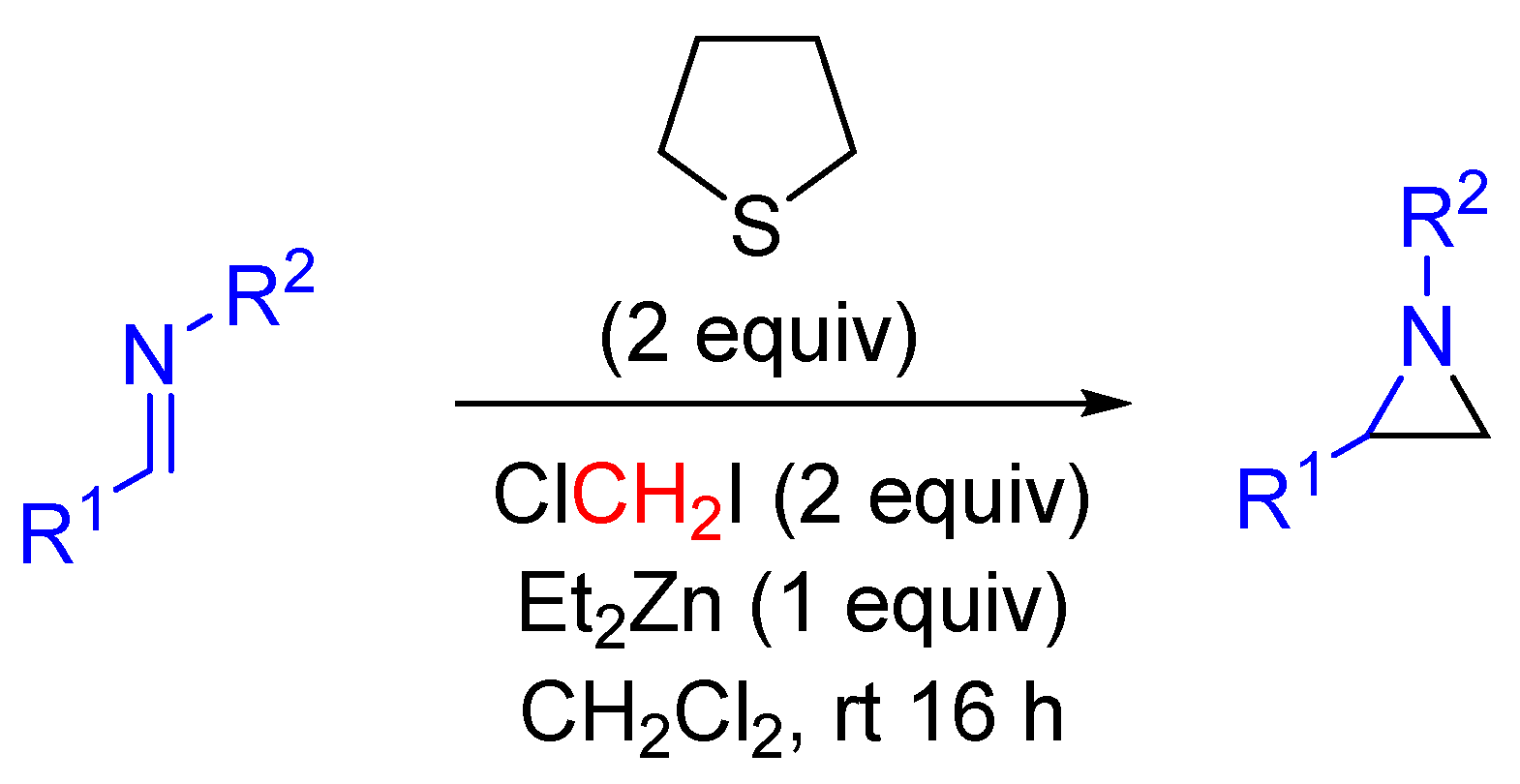
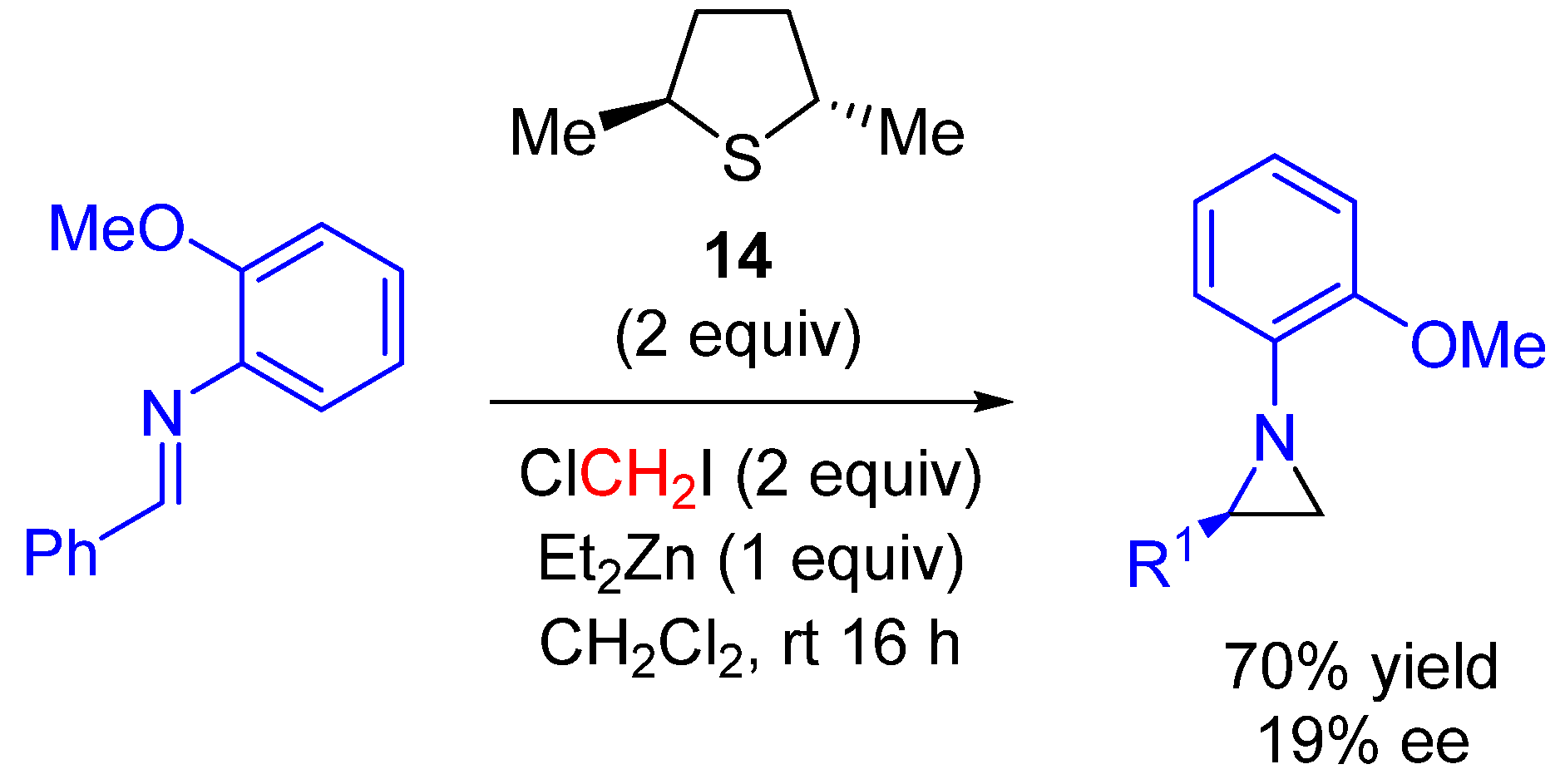
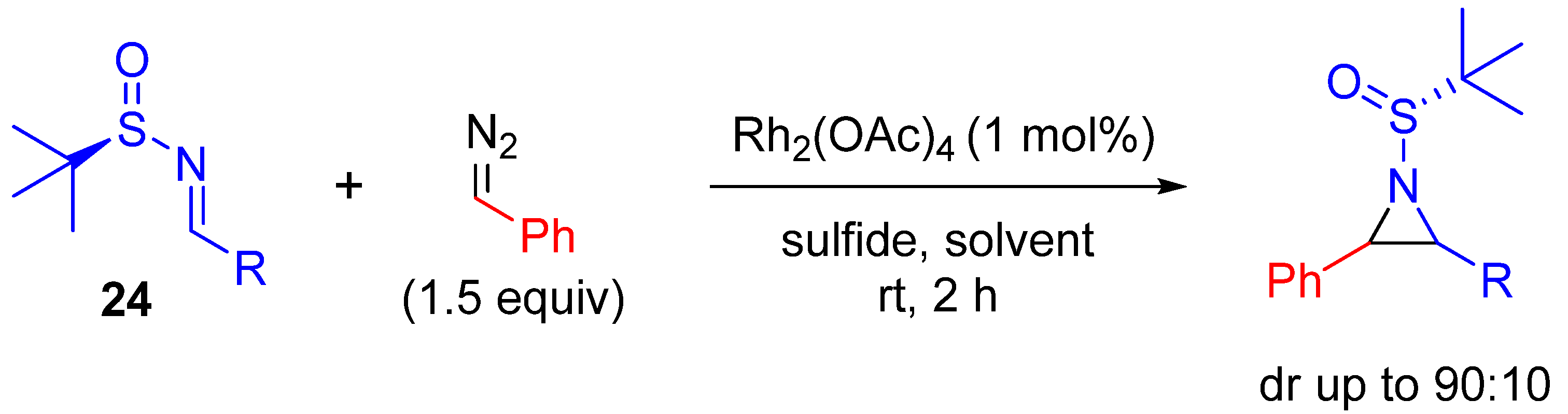




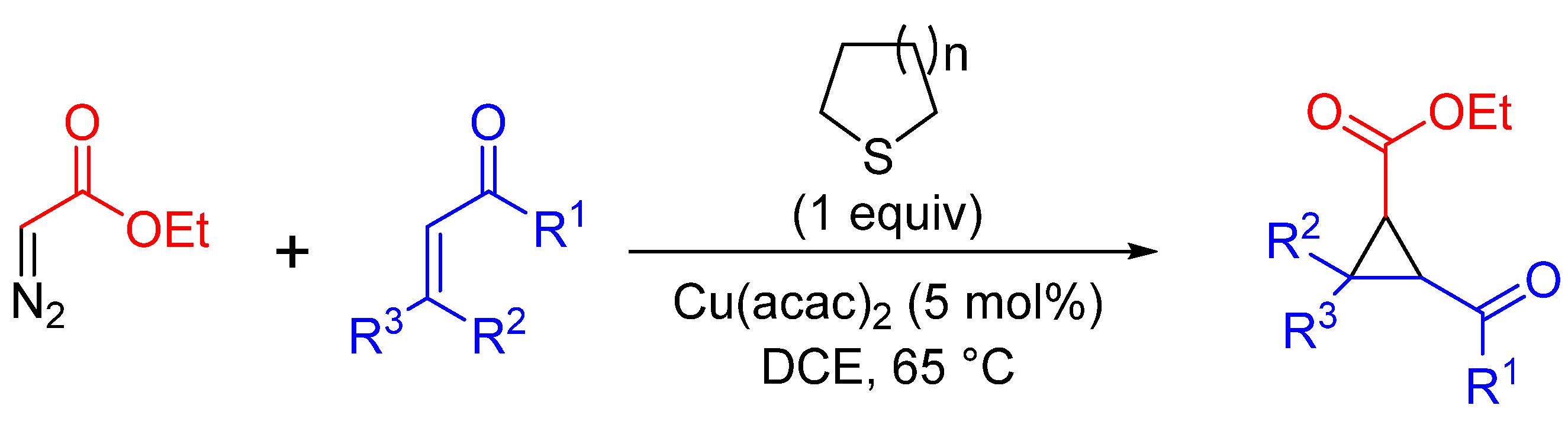
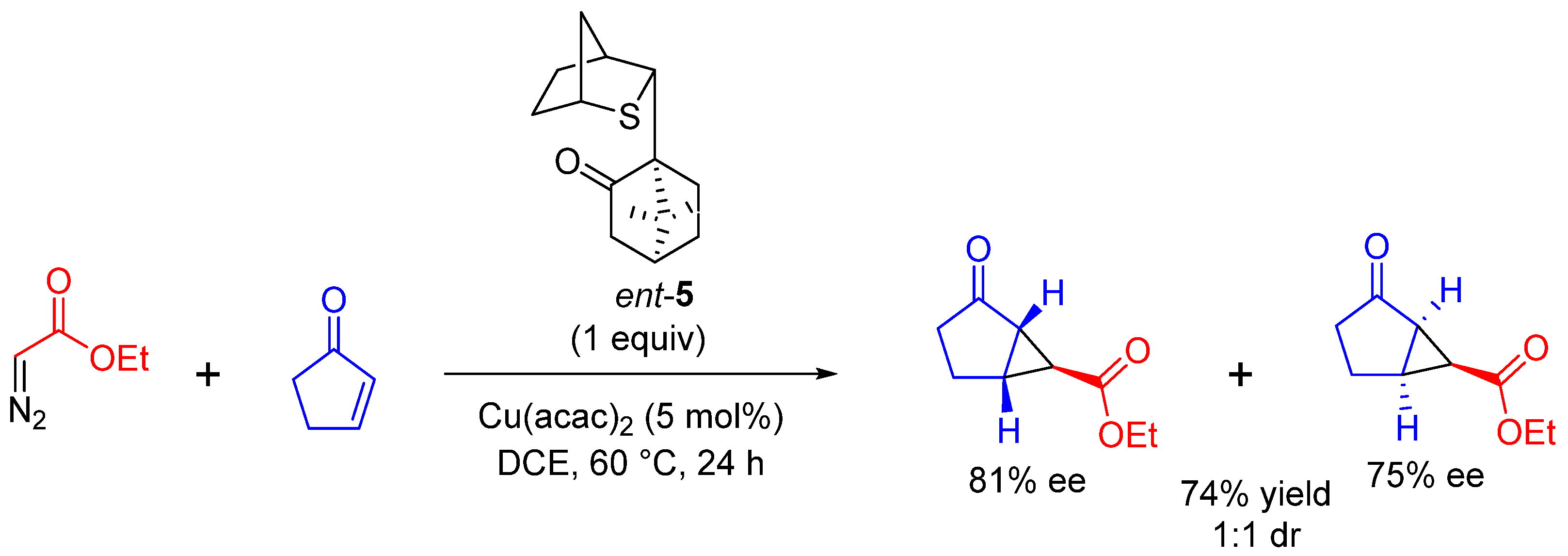

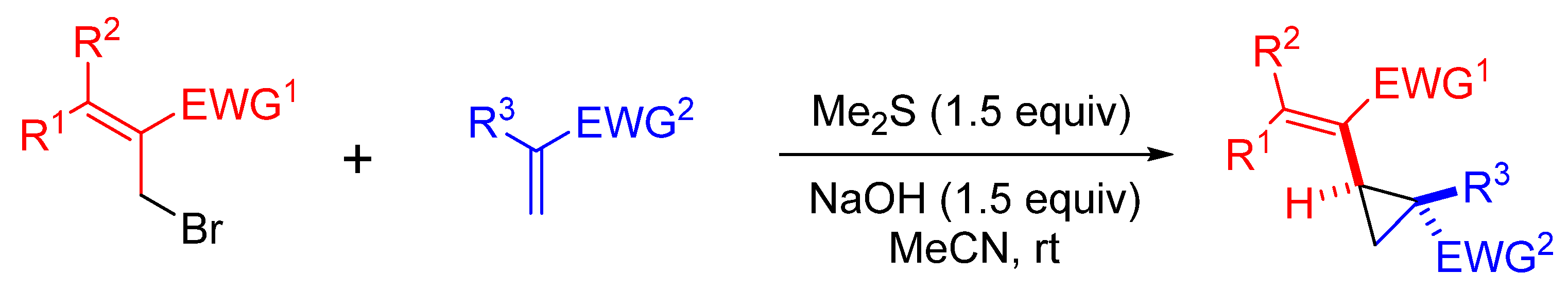
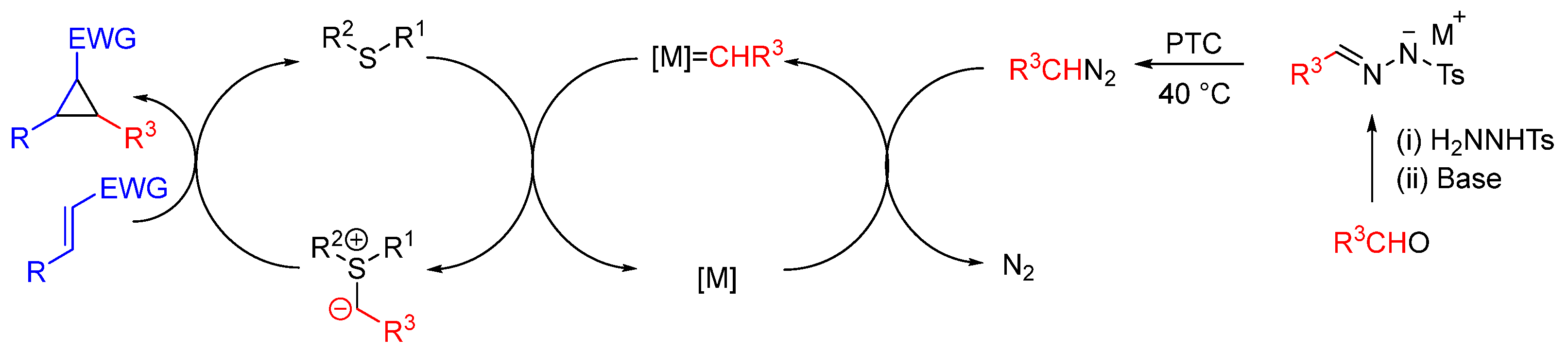

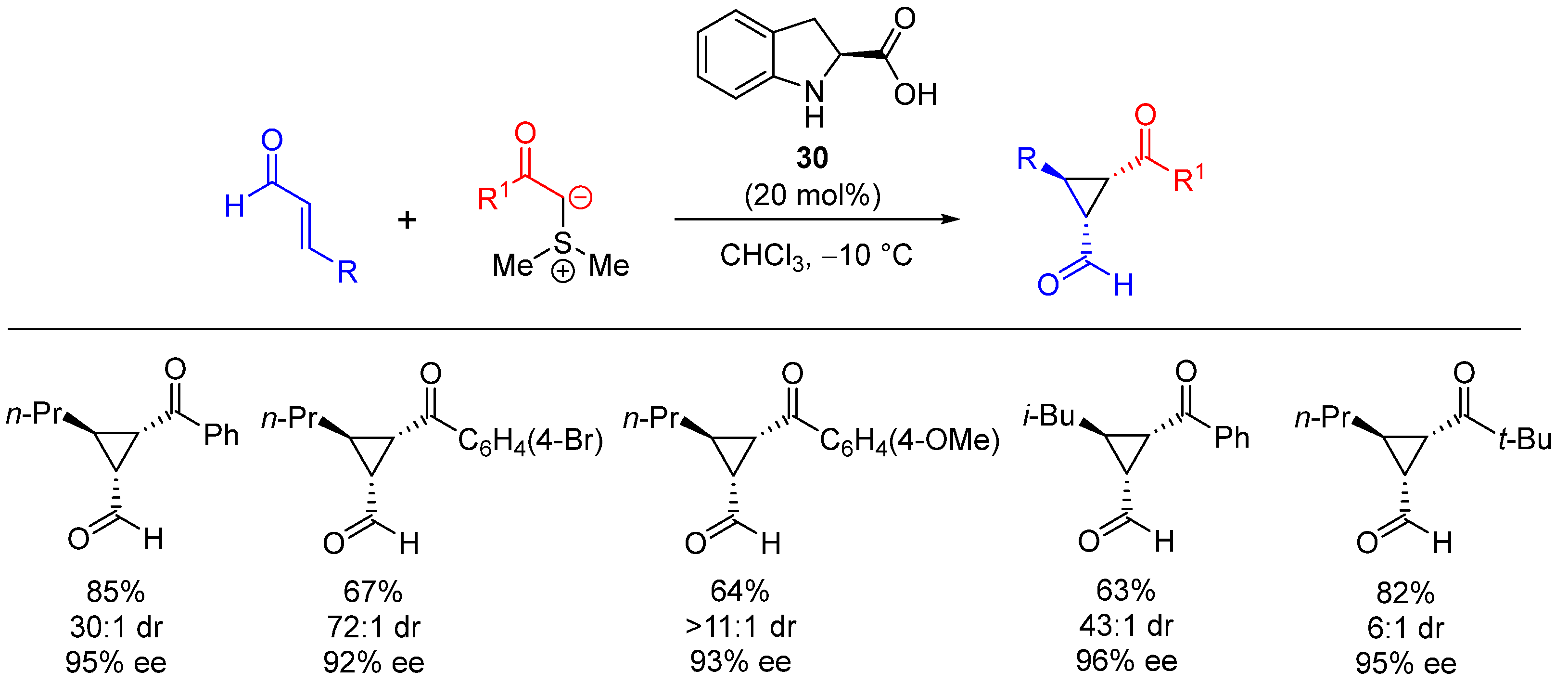
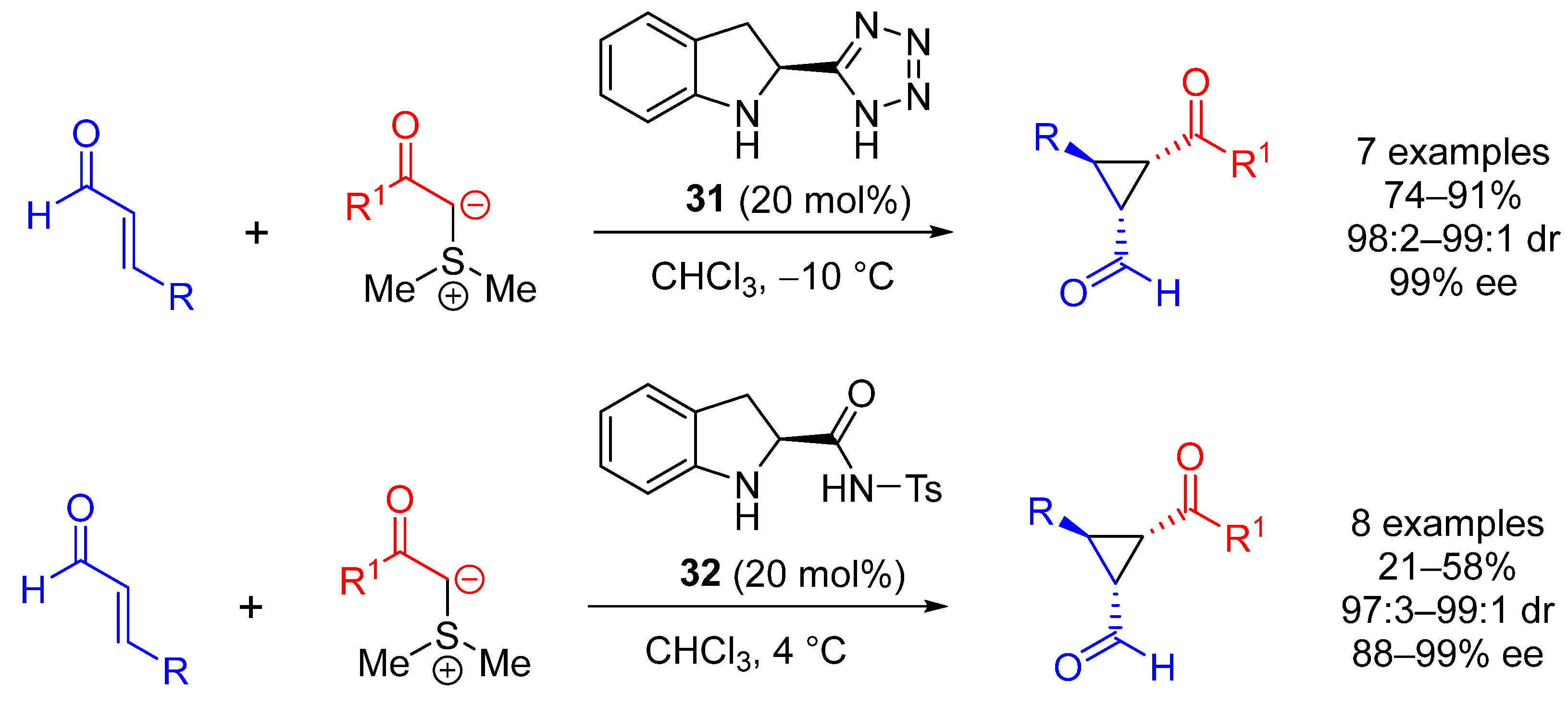

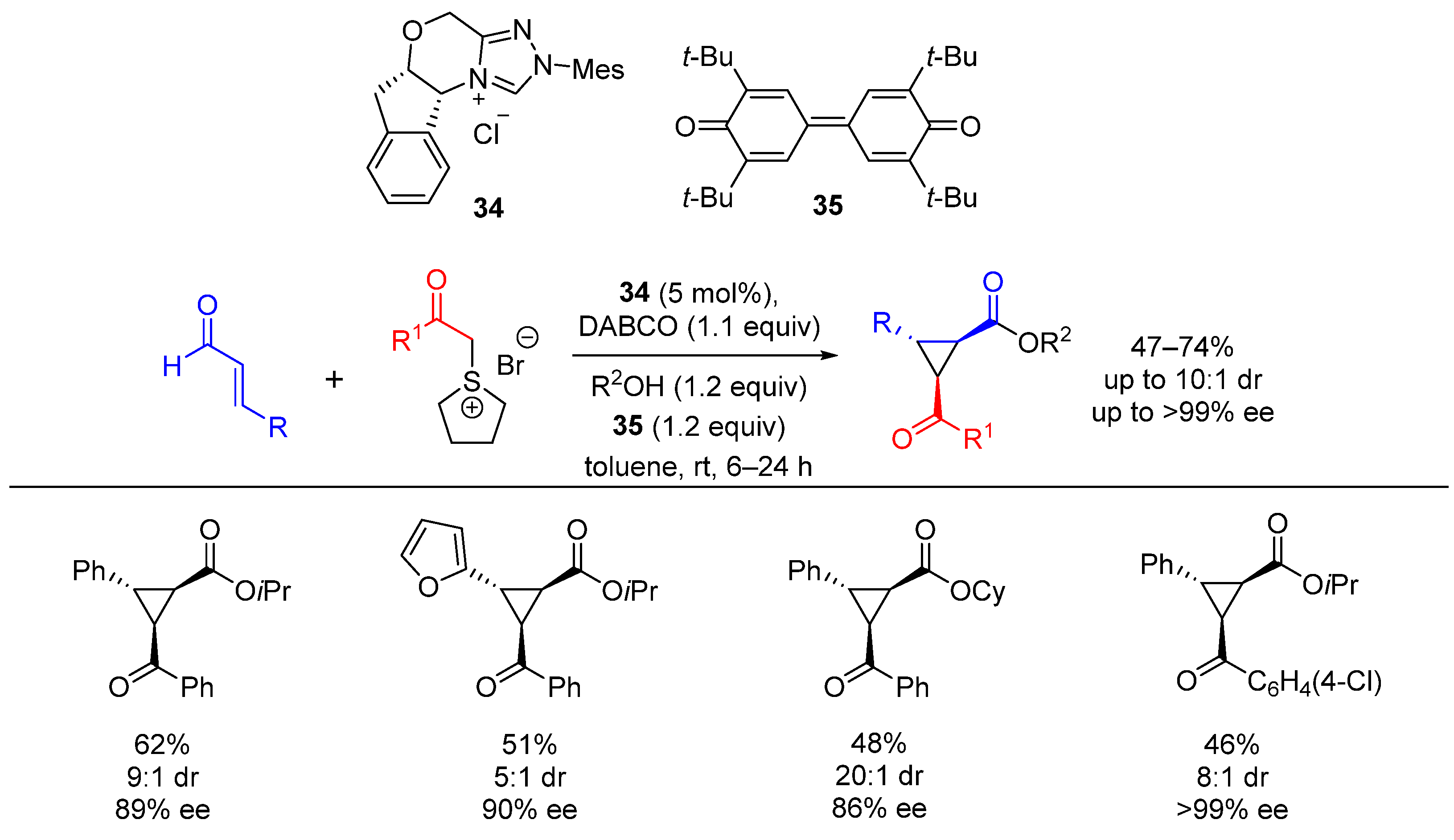

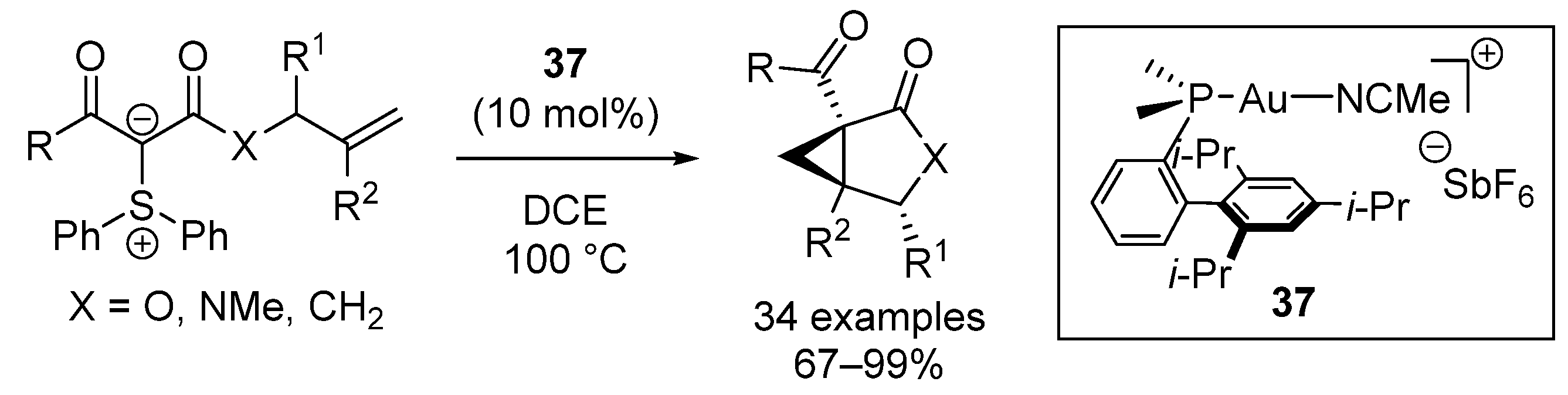
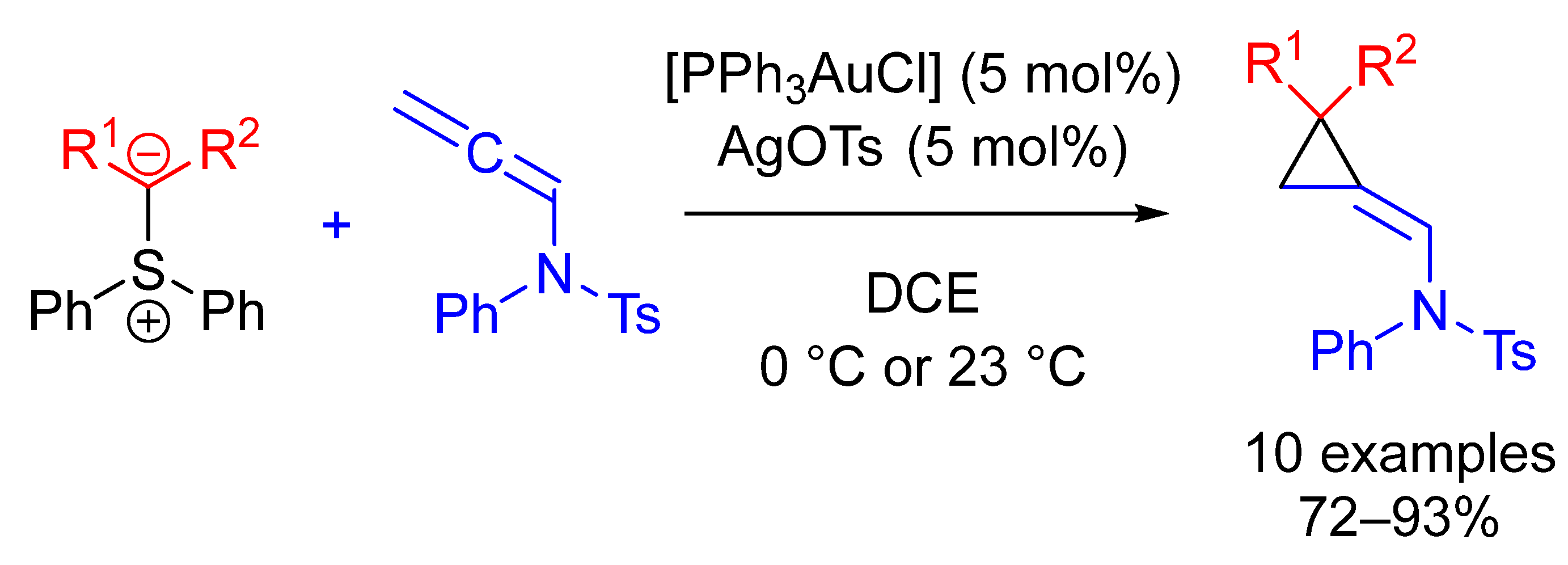
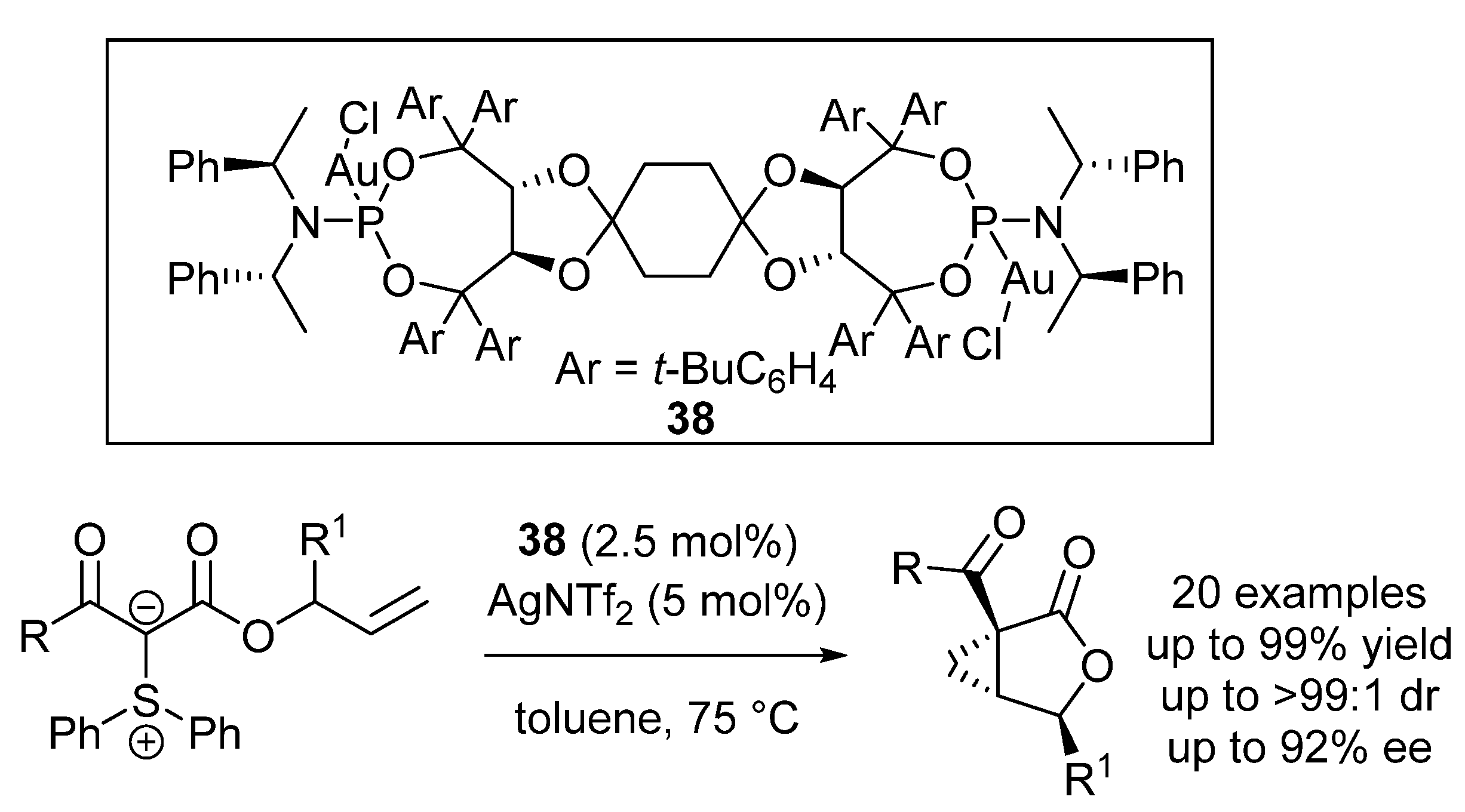
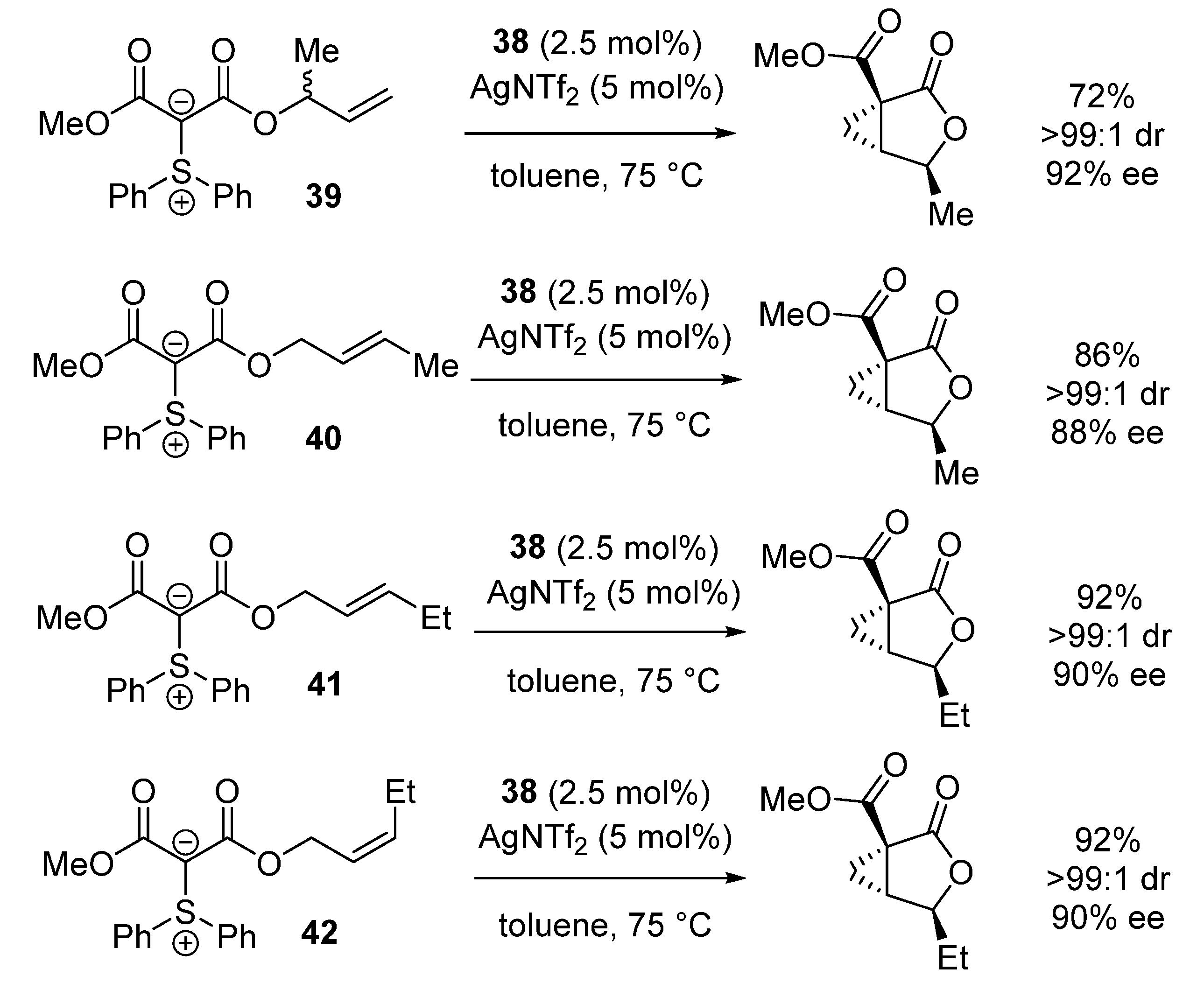


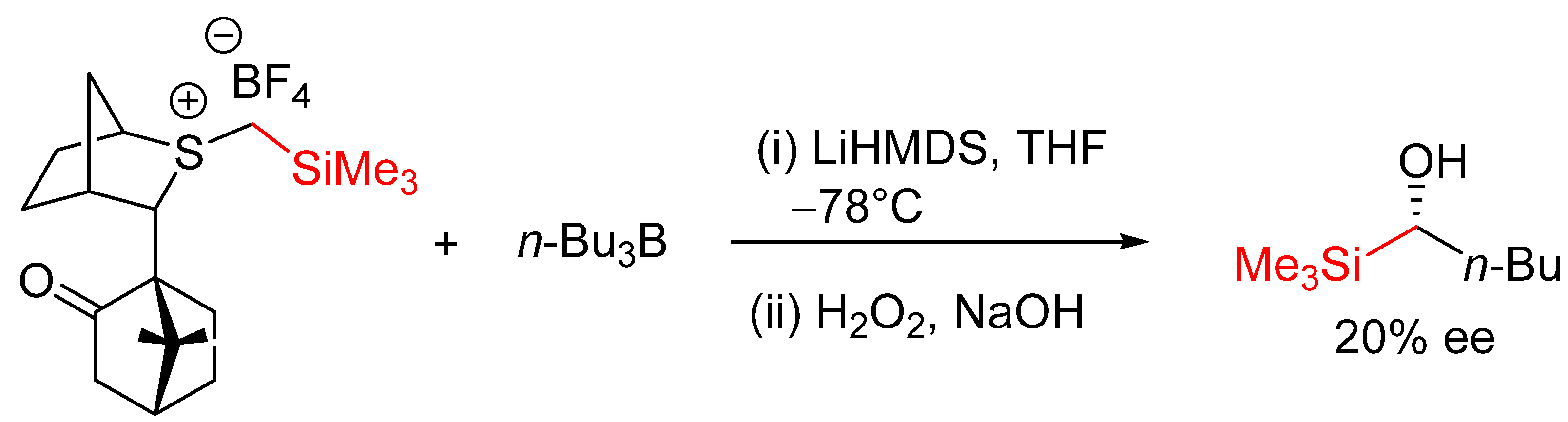
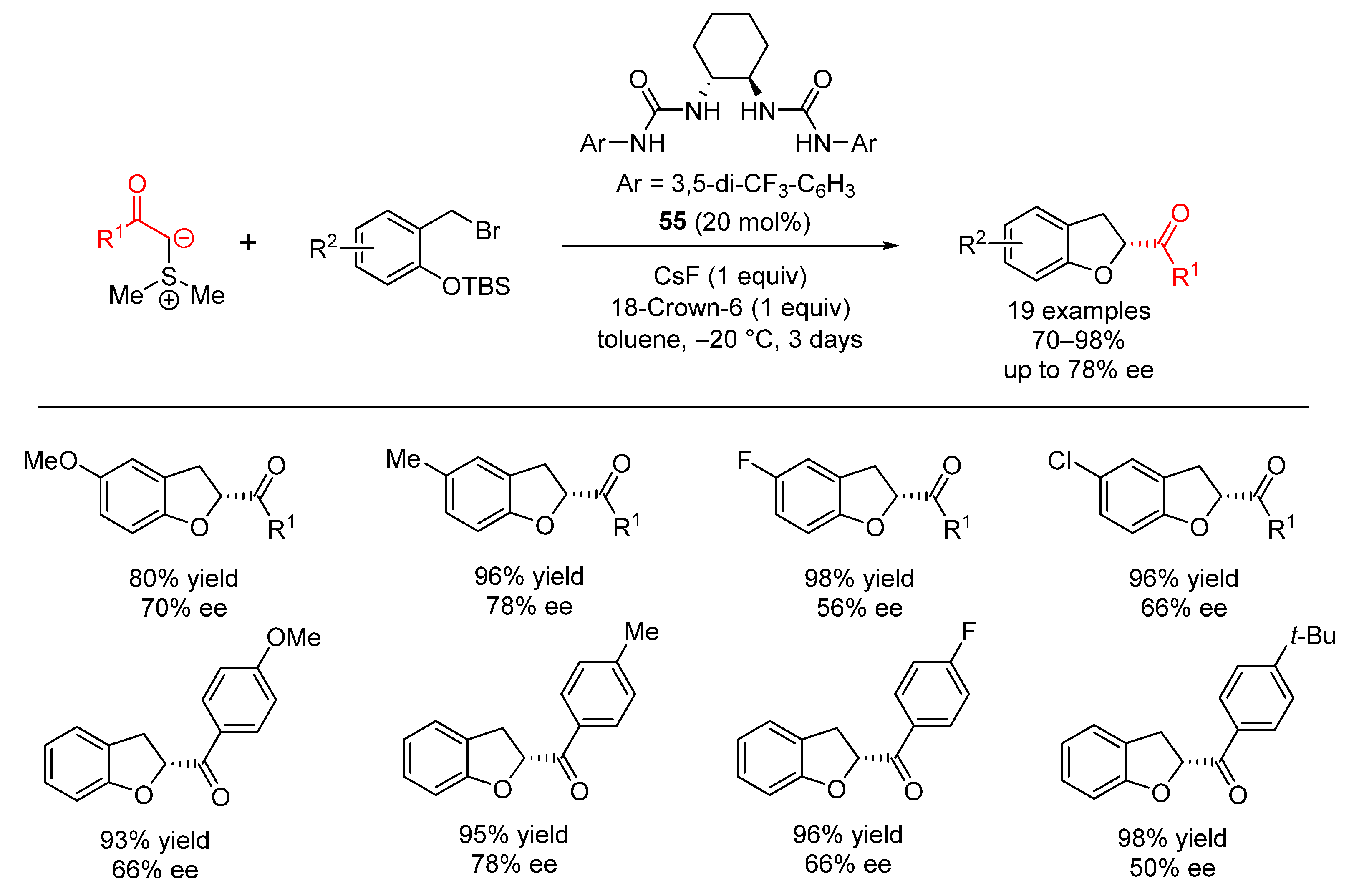
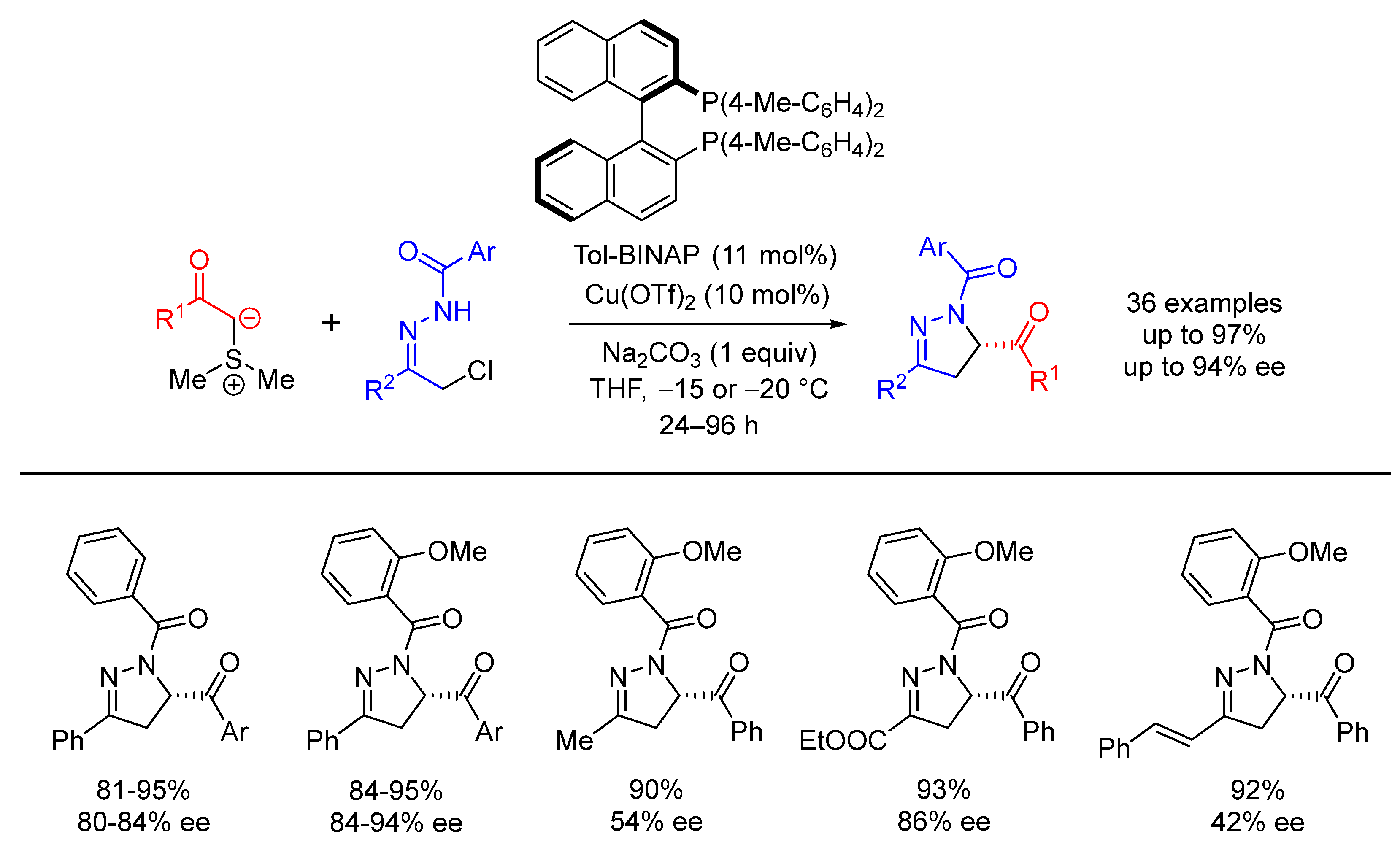
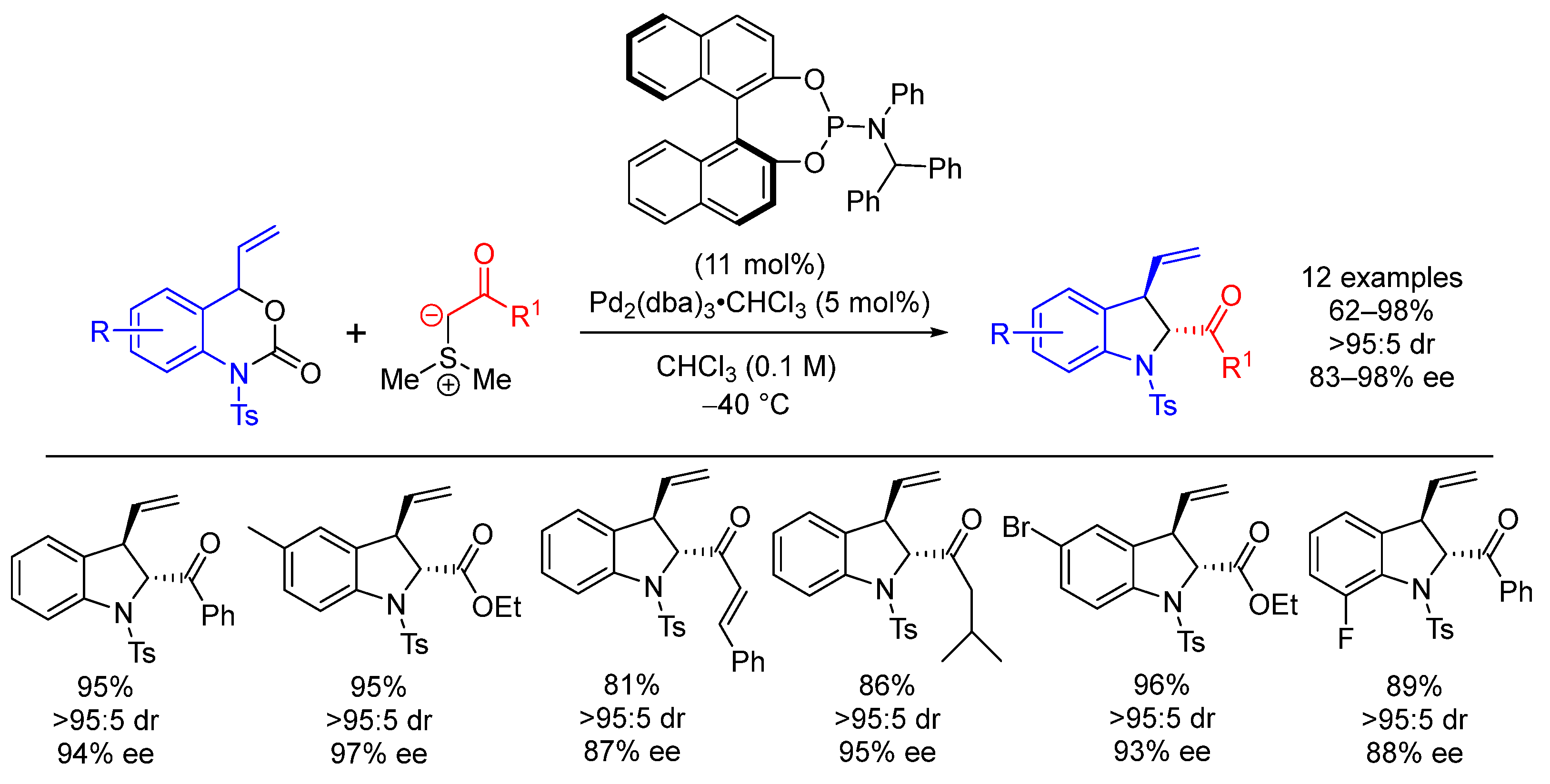
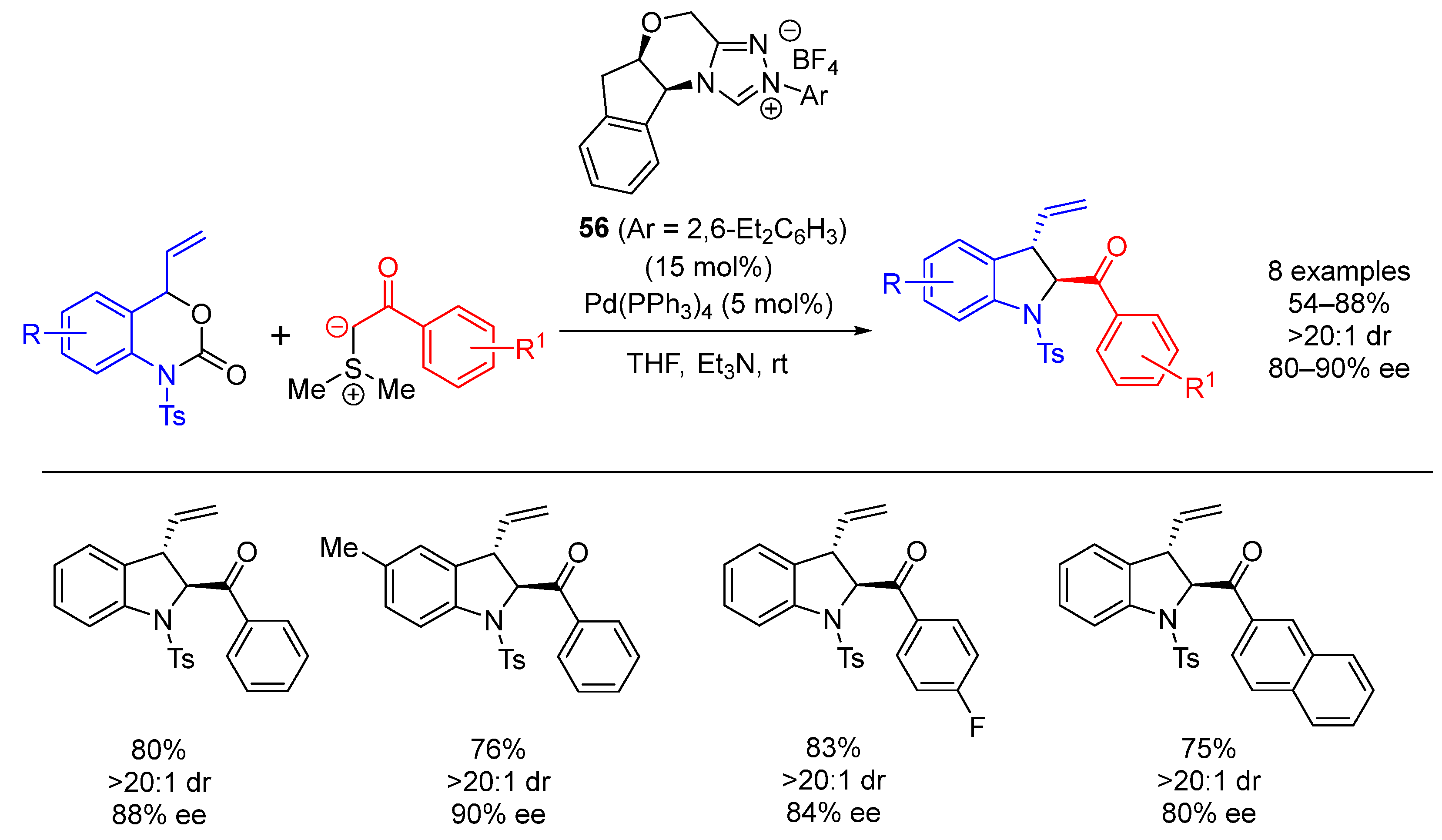
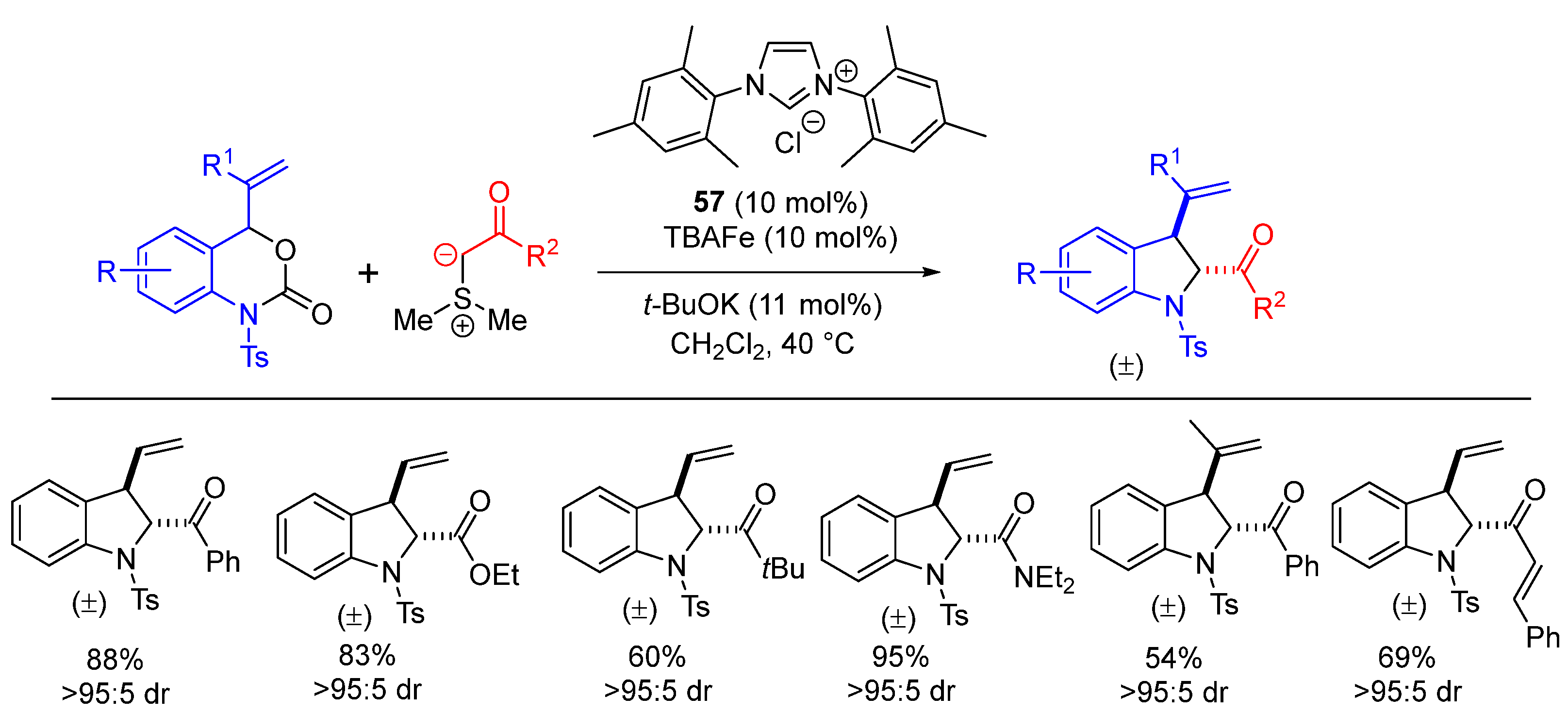
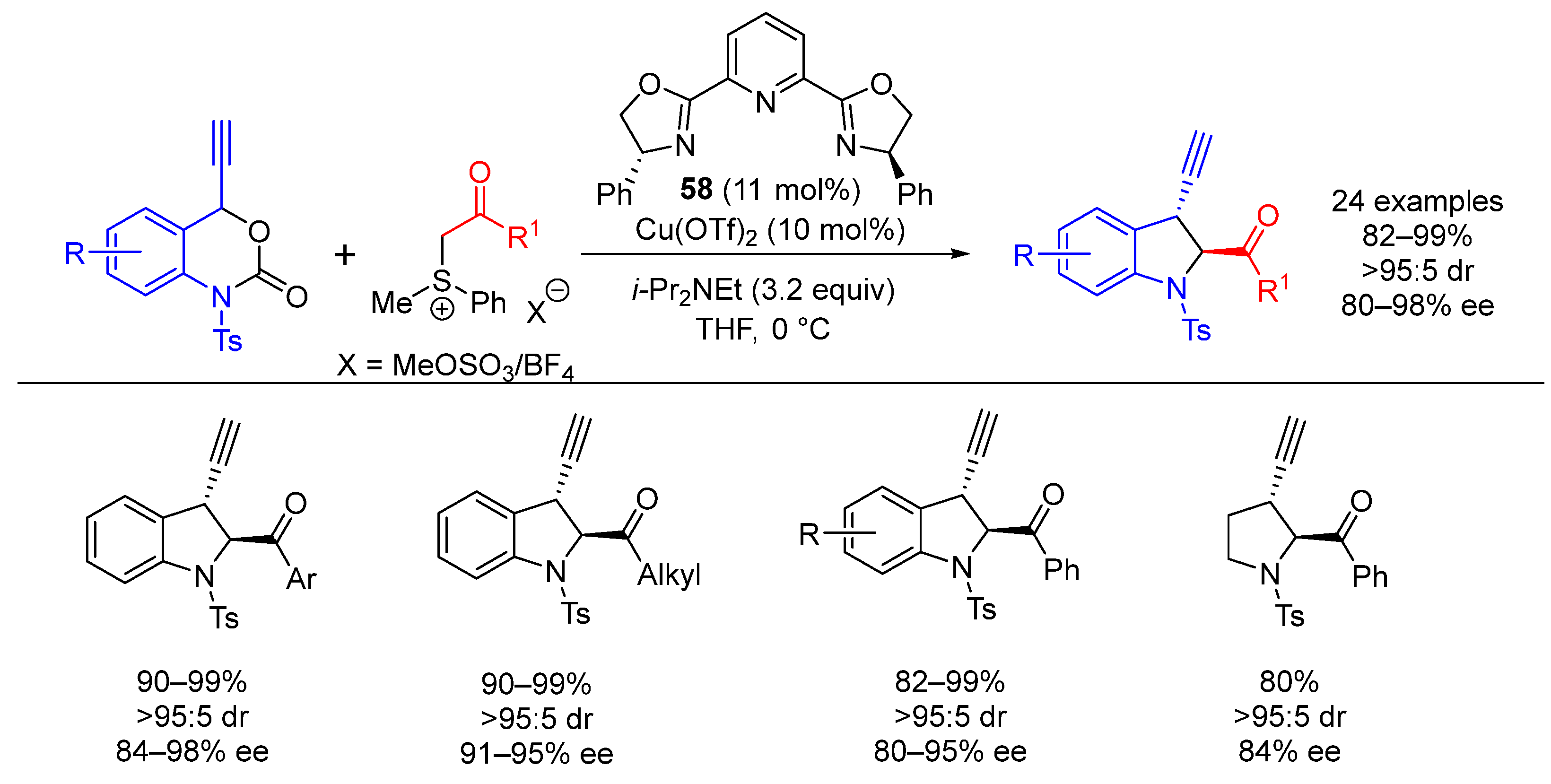


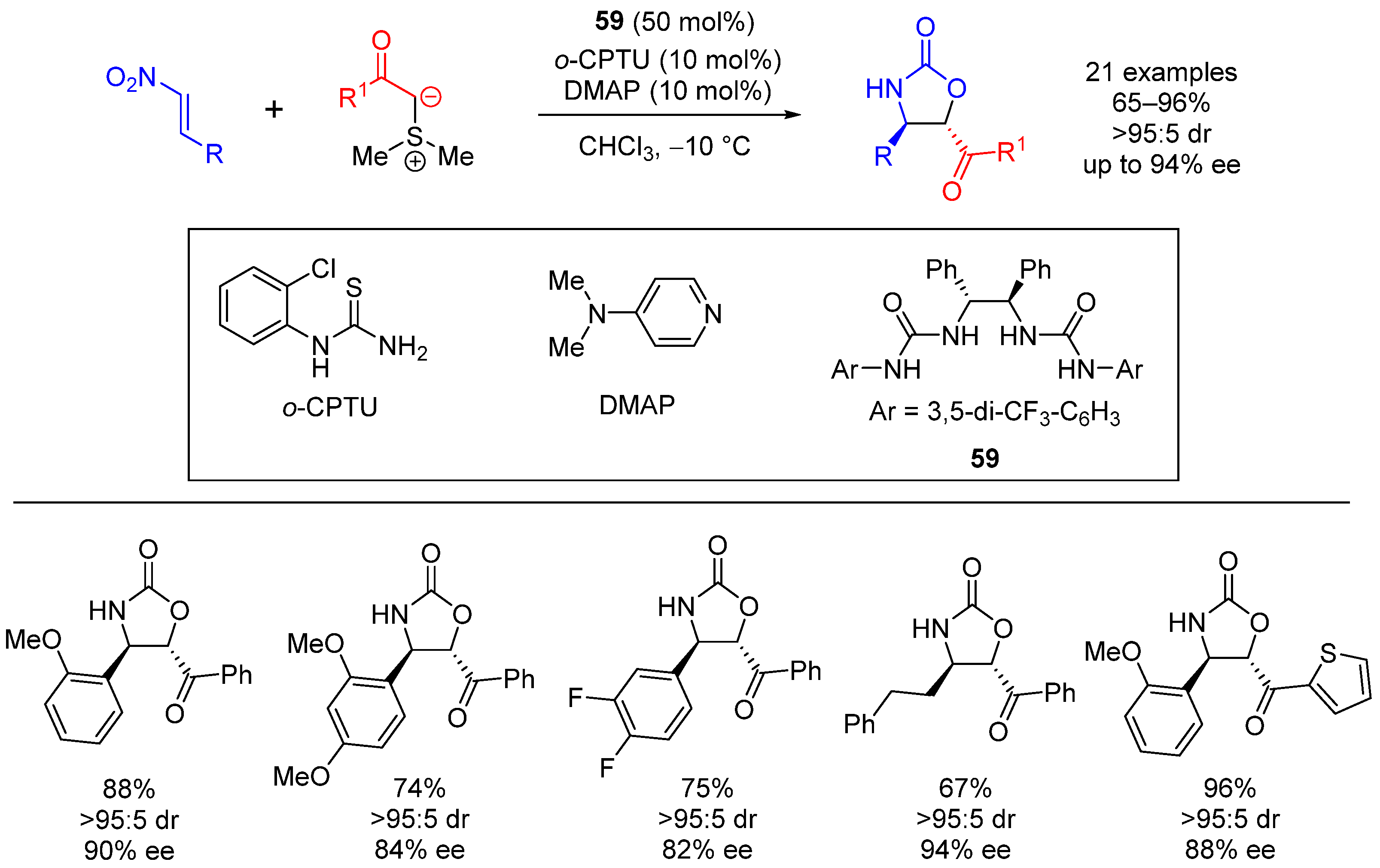
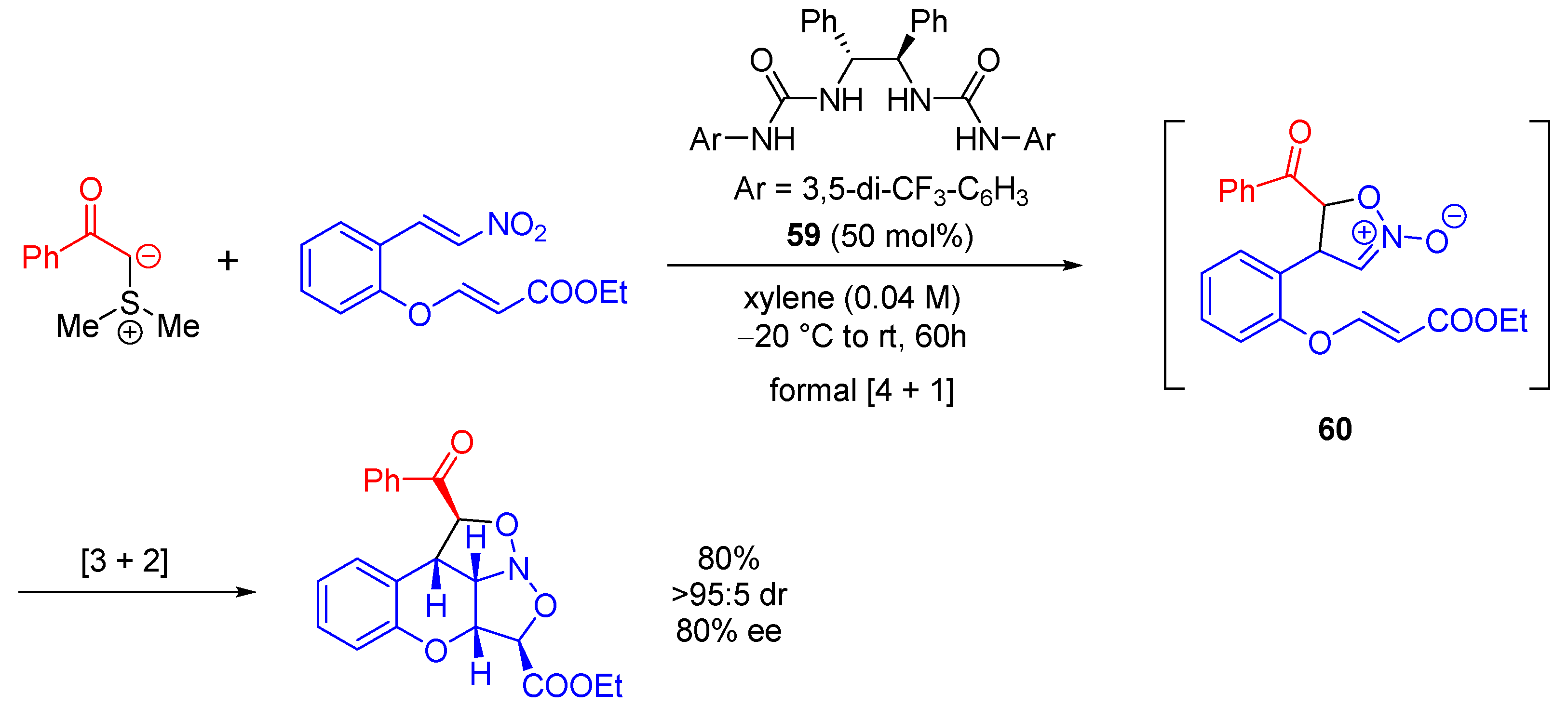





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R1 (Ylide) | R2CHO | Epoxide: Epoxycyclopropane: Cyclopropane. | trans:cis (Epoxide) | ee (%), trans-epoxide |
|---|---|---|---|---|---|
| 1 | Ph | 4a | 77:11:12 | 100:0 | 97 |
| 2 | p-MeOC6H4 | 4a | 100:0:0 | 77:23 | 95 |
| 3 | Ph | 4b | 100:0:0 | 97:3 | >99 |
| 4 | Ph | 4c | 100:0:0 | 97:3 | >99 |
| 5 | Ph | 4d | 100:0 | 97 | |
| 6 | Ph | 4e | 100:0 | >99 |
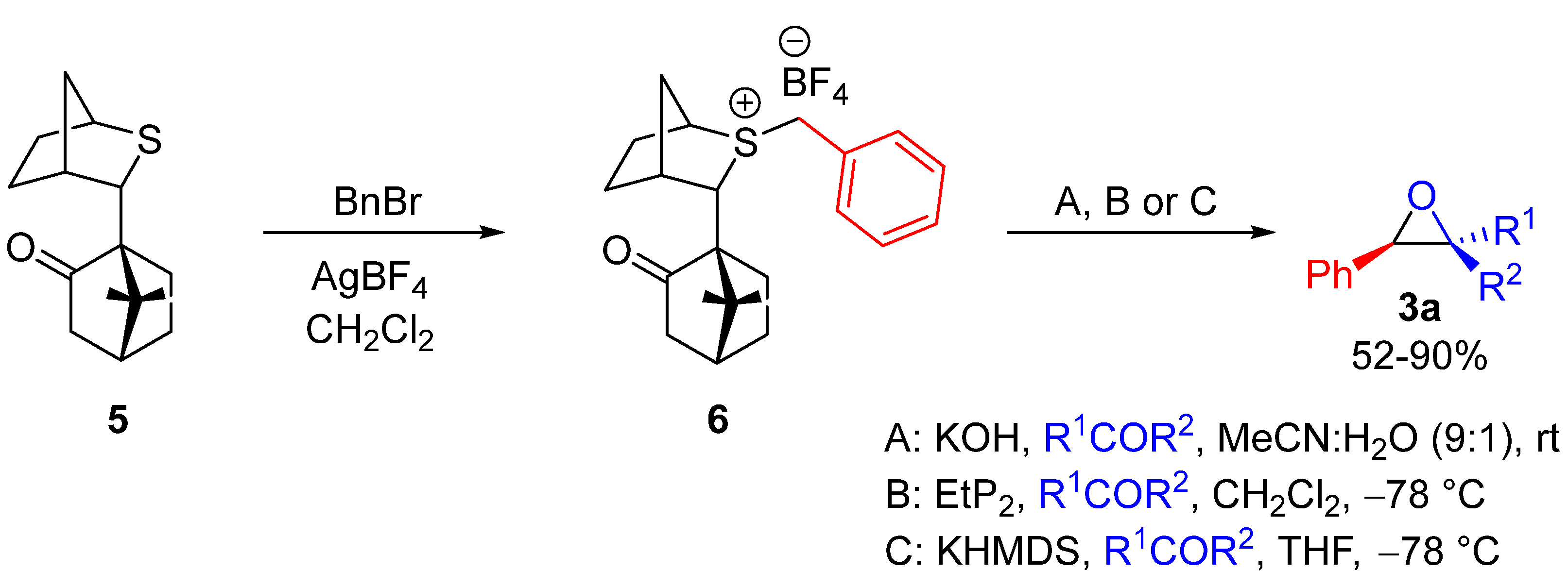
| Entry | R1COR2 | Method | Yield of 3a (%) | trans:cis | ee trans (%) |
|---|---|---|---|---|---|
| 1 | PhCHO | A | 75 | 98:2 | 98 |
| 2 | 2-PyrCHO | B | 88 | 98:2 | 99 |
| 3 | n-BuCHO | C | 87 | 90:10 | >99 |
| 4 | CH2=C(Me)CHO | B | 52 | >99:1 | 95 |
| 5 | I-MeCH=CH2CHO | B | 90 | >99:1 | 95 |
| 6 | Cyclohexanone | B | 85 | -- | 92 |
| 7 | p-NO2PhCOMe | B | 73 | >1:99 | 71 |
| 8 | PhCOMe | B | 77 | 33:67 | 93 |
| Entry | X | R1 | R2 | R | Method | Yield (%) | dr (trans:cis) | ee (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | BF4 | H | Ph | Ph | A | 65 | 80:20 | 70 |
| 2 | BF4 | Me | Ph | Ph | A | 97 | >95:5 | 98 |
| 3 | OTf | Me | H | Ph | A | 80 | >95:5 | 98 |
| 4 | BF4 | Me | Ph | Cy | B | 77 | >95:5 | 96 |
| 5 | OTf | Me | H | Cy | B | 77 | >95:5 | 94 |
| 6 | OTf | H | H | Ph | A | 57 | 75:25 | 40 |
| Entry | R1 | R2 | Yield (%) | trans:cis | trans ee (%) |
|---|---|---|---|---|---|
| 1 | Ph | Boc | 60 | 90:10 | 97 |
| 2 | PMP | Boc | 31 | 91:9 | 96 |
| 3 | 1-Napthyl | Boc | 75 | 98:2 | 96 |
| 4 | tert-Butyl | SES | 62 | 0:100 | -- (98 cis) |
| 5 | tert-Butyl | Ts | 68 | 0:100 | -- (97 cis) |
| 6 | 9-Anthryl | SES | 53 | 91:9 | 98 |
| Entry | R | Yield (%) | trans:cis | trans ee (%) |
|---|---|---|---|---|
| 1 | Ph | 72 | 85:15 | 98 |
| 2 | p-MeC6H4 | 63 | 86:14 | 98 |
| 3 | p-ClC6H4 | 65 | 75:25 | 98 |
| 4 | p-MeOC6H4 | 80 | 83:17 | 98 |
| 5 | (E)-PhCH=CH | 78 | >99:1 | 96 |
| 6 | (E)-TMSCH=CH | 78 | 87:13 | 98 |
| Entry | R | Yield (%) | ee (%) |
|---|---|---|---|
| 1 | Ph | 91 | 23 |
| 2 | p-ClC6H4 | 88 | 18 |
| 3 | p-MeOC6H4 | 92 | 25 |
| 4 | o-ClC6H4 | 87 | 30 |
| 5 | 2,4,6-trimethyl-C6H2 | 87 | 25 |
| 6 | 1-Naphthyl | 92 | 23 |
| 7 | Cinnamyl | 92 | 18 |
| 8 | c-Hex | 91 | 20 |
| Entry | R | X | Yield (%) | cis:trans | cis ee (%) |
|---|---|---|---|---|---|
| 1 | p-MeOC6H4 | OEt | 53 | 3:2 | 45 |
| 2 | Ph | OEt | 80 | 3:1 | 58 |
| 3 | p-ClC6H4 | OEt | 72 | 5:1 | 54 |
| 4 | p-NO2C6H4 | OEt | 83 | 12:1 | 56 |
| 5 | Cy | OEt | 76 | 11:1 | 44 |
| 6 | Ph | NEt2 | 98 | 1:1 | 30 |
| Entry | R1 | R2 | Yield (%) |
|---|---|---|---|
| 1 | Ph | Ts | 68 |
| 2 | p-NO2C6H4 | Ts | 66 |
| 3 | p-AcOC6H4 | Ts | 68 |
| 4 | Cy | Ts | 72 |
| 5 | Ph | SES | 72 |
| 6 | Ph | p-MeOC6H4 | <3 |
| 7 | Ph | o-MeOC6H4 | 79 |
| Entry | R1 | R2 | R3 | Method | trans:cis | Yield (%) | trans ee (%) |
|---|---|---|---|---|---|---|---|
| 1 | Me | Ph | Ts | A | 78:22 | 76 | 98 |
| 2 | Me | H | Ts | A | 83:17 | 98 | 98 |
| 3 | H | H | Ts | A | 85:15 | 73 | 88 |
| 4 | H | Ph | Ts | A | 80:20 | 81 | 90 |
| 5 | Me | H | Ph2PO | B | 84:16 | 84 | 98 |
| 6 | H | H | Ph2PO | B | 86:14 | 83 | 82 |
| Entry | R1 | R2 | R3 | Yield (%) | trans:cis | trans ee (%) |
|---|---|---|---|---|---|---|
| 1 | Ph | Ph | Ts | >99 | 75:25 | 92 |
| 2 | p-Tol | Ph | Ts | 87 | 74:26 | 89 |
| 3 | p-NO2Ph | Ph | Ts | >99 | 65:35 | 98 |
| 4 | Ph | p-Tol | Ts | >99 | 79:21 | 89 |
| 5 | Ph | p-MeOPh | Ts | 94 | 63:37 | 86 |
| 6 | Ph | p-ClPh | Ts | 86 | 78:22 | 92 |
| 7 | Ph | Ph | PhSO2 | 84 | 76:24 | 92 |
| 8 | Ph | Ph | MeSO2 | 79 | 67:33 | 92 |
| Entry | R1 | R2 | Yield (%) | trans:cis | trans ee (%) |
|---|---|---|---|---|---|
| 1 | p-MeOC6H4 | SES | 60 | 2.5:1 | 92 |
| 2 | Ph | SES | 75 | 2.5:1 | 94 |
| 3 a | Ph | SES | 66 | 2.5:1 | 95 |
| 4 | p-ClC6H4 | SES | 82 | 2:1 | 98 |
| 5 | Cy | SES | 50 | 2.5:1 | 98 |
| 6 | (E)-PhCH=CH | SES | 59 | 8:1 | 94 |
| 7 | 3-Furfuryl | Ts | 72 | 8:1 | 95 |
| 8 | t-Bu | Ts | 53 | 2:1 | 73 |
| 9 | Ph | Ts | 68 | 2.5:1 | 98 |
| 10 | Ph | SO2-β-C10H7 | 70 | 3:1 | 97 |
| 11 | Ph | TcBoc | 71 | 6:1 | 90 |
| Entry | R1 | R2 | R3 | n | Yield (%) | dr |
|---|---|---|---|---|---|---|
| 1 | Ph | H | Ph | 2 | 72 | 4:2:1 |
| 2 | Me | H | H | 2 | 64 | >95:5 |
| 3 | OEt | H | COOEt | 2 | 68 | >95:5 |
| 4 | Me | Me | Me | 2 | 5 | >95:5 |
| 5 | -(CH2)2- | H | 1 | 81 | 1:1 |
| Entry | R1 | R2 | EWG1 | R3 | EWG2 | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | Ph | H | COOMe | H | COMe | 12 | 45 |
| 2 | p-MeC6H4 | H | COOMe | H | COMe | 12 | 47 |
| 3 | p-ClC6H4 | H | COOMe | H | COMe | 13 | 49 |
| 4 | p-MeC6H4 | H | COOMe | Cl | CN | 20 | 57 |
| 5 | H | Ph | CN | H | COMe | 12 | 48 |
| Entry | X | n | EWG | Yield (%) |
|---|---|---|---|---|
| 1 | O | 1 | COOMe | 76 |
| 2 | CH2 | 0 | COOMe | 53 a |
| 3 | CH2 | 1 | COOEt | 63 |
| 4 | CH2 | 1 | CHO | 64 |
| 5 | CH2 | 1 | COPh | 64 a |
| Entry | R1 | R2 | Sulfide | Yield (%) | 28:29 | 28 ee (%) |
|---|---|---|---|---|---|---|
| 1 | Ph | Ph | 25 | 92 | 4:1 | -- |
| 2 | Ph | Ph | THT | 40 | 1:1 | -- |
| 3 | Ph | Ph | 26 | 38 | 4:1 | 97 |
| 4 | Ph | Ph | 5 | 30 a | 5:1 | 89 |
| 5 | Ph | Ph | 27 | 73 | 4:1 | 91 |
| 6 | Ph | Me | 27 | 5 | -- | -- |
| 7 | Me | Ph | 27 | 50 | 4:1 | 90 |
| 8 | H | OEt | 27 | 10 | 7:1 | -- |
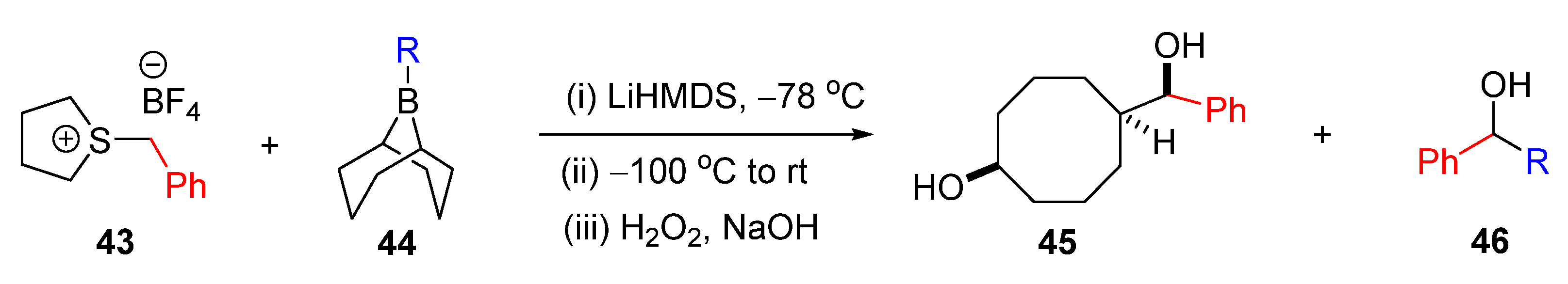
| Entry | R | Yield (%) 45 | Yield (%) 46 |
|---|---|---|---|
| 1 | Hexyl | 56 | 41 |
| 2 | Allyl | 51 | 39 |
| 3 | Benzyl | 51 | 35 |
| 4 | i-Pr | Trace | 77 |
| 5 | Cyclopropyl | 89 | Trace |
| 6 | Ph | Trace | 94 |
| 7 | 1-Hexenyl | Trace | 21 |
| 8 | 1-Hexynyl | 92 | Trace |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mondal, M.; Connolly, S.; Chen, S.; Mitra, S.; Kerrigan, N.J. Recent Developments in Stereoselective Reactions of Sulfonium Ylides. Organics 2022, 3, 320-363. https://doi.org/10.3390/org3030024
Mondal M, Connolly S, Chen S, Mitra S, Kerrigan NJ. Recent Developments in Stereoselective Reactions of Sulfonium Ylides. Organics. 2022; 3(3):320-363. https://doi.org/10.3390/org3030024
Chicago/Turabian StyleMondal, Mukulesh, Sophie Connolly, Shi Chen, Shubhanjan Mitra, and Nessan J. Kerrigan. 2022. "Recent Developments in Stereoselective Reactions of Sulfonium Ylides" Organics 3, no. 3: 320-363. https://doi.org/10.3390/org3030024